

Abstract
Romidepsin (FK228) is a potent histone deacetylase (HDAC) inhibitor, which has a potent anticancer activity, but its molecular mechanism is unknown. We investigated the mechanism of FK228‐induced apoptosis in the human leukemia cell line HL‐60 and its hydrogen peroxide (H2O2)‐resistant sub‐clone, HP100, and the human colon cancer cell line Caco‐2. Cytotoxicity and DNA ladder formation induced by FK228 could be detected in HL‐60 cells after a 24‐h incubation, whereas they could not be detected in HP100 cells. Trichostatin A (TSA), an HDAC inhibitor, induced DNA ladder formation in both HL‐60 and HP100 cells. In contrast, FK228 inhibited HDAC activity in both HL‐60 and HP100 cells to a similar extent. These findings suggest that FK228‐induced apoptosis involves H2O2‐mediated pathways and that TSA‐induced apoptosis does not. Flow cytometry revealed H2O2 formation and a change in mitochondrial membrane potential (Δψm) in FK228‐treated cells. FK228 also induced apoptosis in Caco‐2 cells, which was prevented by N‐acetyl‐cysteine, suggesting that reactive oxygen species participate in apoptosis in various types of tumor cells. Interestingly, in a cell‐free system, FK228 generated superoxide (O2 −) in the presence of glutathione, suggesting that H2O2 is derived from dismutation of O2 − produced through redox‐cycle of FK228. Therefore, in addition to HDAC inhibition, H2O2 generated from FK228 may participate in its apoptotic effect. (Cancer Sci 2010;)
Romidepsin (FK228), a potent histone deacetylase (HDAC) inhibitor known as depsipeptide, is a naturally occurring polypeptide which has been isolated from Chromobacterium violaceum. ( 1 ) This drug has a potent anticancer activity against human tumor xenografts and murine tumors,( 2 ) and is therefore expected to be a novel and promising anticancer drug, currently under clinical evaluation in the USA.( 3 , 4 ) FK228 induces caspase‐dependent apoptosis via the mitochondrial pathway in small‐cell lung cancer cells,( 5 ) and induces the activation of the proapoptotic Bcl‐2 family protein Bid upstream of mitochondrial membrane dysfunction.( 6 ) FK228‐induced cell cycle arrest, apoptosis, and histone hyperacetylation in lung carcinoma cells are associated with increase in p21 expression.( 7 )
It has been reported that other HDAC inhibitors also induced apoptosis through reactive oxygen species (ROS) generation.( 8 , 9 , 10 , 11 , 12 ) FK228 and PS‐341, a proteasome inhibitor, synergistically induce apoptosis in gastrointestinal adenocarcinoma cells depending on ROS‐mediated signals.( 13 ) Recently, it is reported that FK228 increases ROS in urinary bladder cancer cells.( 14 ) However, the mechanism of apoptosis and ROS generation induced by FK228 has not well been clarified. We have investigated the mechanism of apoptosis induction by various anticancer drugs through the generation of ROS, such as hydrogen peroxide (H2O2) and superoxide (O2 −).( 15 , 16 , 17 , 18 , 19 , 20 ) FK228 contains an intramolecular disulfide bond, raising the idea that ROS may be generated by the redox‐cycling reaction of FK228 and participate in apoptosis.
In this study, we investigated the mechanism of FK228‐induced apoptosis in HL‐60 cells and HP100 cells, H2O2‐resistant cells derived from HL‐60. To examine the cytotoxic effect of FK228 on solid tumor, we also used Caco‐2 cells, derived from colon cancer. The mechanism of apoptosis was analyzed by examining cytotoxicity, DNA ladder formation, H2O2 generation, mitochondrial membrane potential (Δψm) change, and HDAC activity. In addition, we examined O2 − production induced by FK228 in a cell‐free system.
Materials and Methods
Chemicals. FK228 was provided by Astellas Pharmaceutical (Tokyo, Japan). Trichostatin A (TSA) was purchased from Wako Chemical (Osaka, Japan). Proteinase K was obtained from Merck (Darmstadt, Germany). Fluorescent probes, 5‐(and‐6)‐chloromethyl‐2′,7′‐dichlorodihydrofluorescein diacetate acetyl ester (CM‐H2DCFDA) and 3,3′‐dihexyloxacarbocyanine iodide (DiOC6[3]) were purchased from Molecular Probes (Eugene, OR, USA). Glutathione (GSH) was from Kohjin (Tokyo, Japan). Diethylenetriamine‐N,N,N′,N′′,N′′‐pentaacetic acid (DTPA) and Hoechst 33342 solution (1 mg/mL) were from Dojin Chemicals (Kumamoto, Japan). Superoxide dismutase (SOD; 3000 U/mg from bovine erythrocytes), cytochrome c and N‐acetyl‐cysteine (NAC) were from Sigma Chemical (St. Louis, MO, USA).
Cell culture and treatment with FK228 and TSA. HP100 cells were derived from human leukemia HL‐60 cells by repeated exposure to H2O2, followed by outgrowth of viable cells. HP100 cells were approximately 340‐fold more resistant to H2O2 than HL‐60 cells.( 21 ) Catalase activity of HP100 cells is 18 times higher than that of HL‐60 cells.( 22 ) HL‐60 and HP100 cells were grown in RPMI‐1640 supplemented with 6% FBS at 37°C under 5% CO2 in a humidified atmosphere. The cells (0.5 × 106 or 1 × 106 cells/mL) were then treated with the indicated concentrations of FK228 and TSA.
Human colon cancer Caco‐2 cells were cultured in DMEM – high glucose supplemented with 10% FBS and non‐essential amino acids (1% v/v) at 37°C under 5% CO2 in a humidified atmosphere. The cells (0.2 × 106 cells/mL) were seeded in 12‐well plate for 24 h before treatment.
Determination of cytotoxicity induced by FK228 and TSA in HL‐60 and HP100 cells. A lactate dehydrogenase (LDH) activity assay kit (CytoTox‐ONE Homogeneous Membrane Integrity Assay; Promega, Madison, WI, USA) was used to measure cytotoxicity in cultures. 5.0 × 104 cells were seeded into each well of 96‐well culture plates. After the treatment with FK228 or TSA for 24 h, cytotoxicity was analyzed according to the manufacturer’s instructions.
Detection of apoptosis induced by FK228 and TSA in HL‐60 and HP100 cells. For analysis of DNA ladder formation after the cells were treated with FK228 and TSA, cells were washed twice with PBS. Cells (2 × 106 cells), resuspended in 1 mL cytoplasm extraction buffer (10 mM Tris [pH 7.5], 150 mM NaCl, 5 mM MgCl2, and 0.5% Triton X‐100), were centrifuged at 1000 g for 5 min at 4°C. The pellet was resuspended in lysis buffer (10 mM Tris [pH 7.5], 400 mM NaCl, and 1 mM EDTA) and centrifuged at 12 000 g for 10 min at 4°C. The supernatant was then treated with 0.2 mg/mL RNase overnight at room temperature, followed by treatment with proteinase K as described previously.( 23 ) DNA ladder formation was analyzed by conventional electrophoresis.
Flow cytometric detection of peroxide and Δψm in cells treated with FK228. To evaluate cellular peroxide levels after the cells were treated with FK228, the treated cells were incubated with 5 μM CM‐H2DCFDA for 30 min at 37°C.( 24 ) To assess changes in Δψm, FK228‐treated cells were incubated with 40 nM DiOC6(3) for 15 min at 37°C.( 25 ) Cells were then washed twice with PBS. Following resuspension in PBS, the cells were analyzed on a flow cytometer (FACScan; Becton Dickinson, San Jose, CA, USA). Dead cells and debris were excluded from the analysis. The data were analyzed using the data analysis program, Cell Quest (Becton Dickinson).
HDAC enzymatic assay. HDAC fluorescent activity assays using a Fluor‐de‐Lys HDAC fluorometric cellular activity assay kit (Enzo Life Sciences, Plymouth Meeting, PA, USA) were performed according to the manufacturer’s instructions, and a Gemini XPS microplate spectrofluorometer (Molecular Devices, Sunnyvale, CA, USA) was used with excitation at 360 nm and emission at 460 nm.
Assessment of apoptosis in Caco‐2 cells. Cells were exposed to 10 nM FK 228 for 48 h, and were washed twice with PBS and fixed with 1% glutaraldehyde for 12 h. The fixed cells were then washed again with PBS and the nucleus was stained with 100 μg/mL of Hoechst 33342 for 5 min. The cells were examined under an inverted fluorescence microscope (Axiovert 200; Carl Zeiss, Göttingen, Germany).
Detection of O2 − derived from FK228 in the presence of GSH. To detect O2 − generation from FK228, 200 μM cytochrome c was added to the reaction mixture, which contained 50 μM FK228 and 1 mM GSH in 10 mM sodium phosphate buffer (pH 7.8) containing 2.5 μM DTPA. A maximum absorption at 550 nm due to ferrocytochrome c formed by ferricytochrome c reduction was measured at 37°C with a UV‐visible spectrophotometer (UV‐2500PC; Shimadzu, Kyoto, Japan). The actual amount of O2 − generation was calculated by subtracting absorbance with 100 U/mL SOD from that without SOD at 550 nm ( = 21.1 × 103/M/cm).( 26 )
Results
Cytotoxicity in HL‐60 and HP100 cells induced by FK228 and TSA. We have determined cytotoxicity in the cells treated with FK228 and TSA using an LDH release assay. This assay is commonly used for testing the cytotoxicity of various experimental compounds. As observed in Figure 1(a), HL‐60 cells were remarkably more sensitive to FK228 in comparison to HP100 cells. The cytotoxicity at 10 nM FK228 was about 20% in HL‐60 cells, whereas the cytotoxicity at 500 nM FK228 was only approximately 10% in HP100 cells. Therefore, HL‐60 cells were at least 50‐fold more sensitive to FK228 than HP100 cells. On the other hand, the cytotoxicity of TSA was detectable at no less than 500 nM in both HL‐60 and HP100 cells (Fig. 1b).
Figure 1.
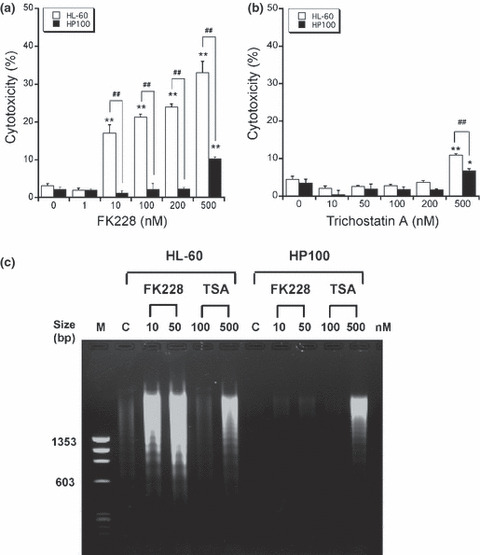
Detection of cytotoxicity and DNA ladder formation in HL‐60 and HP100 cells treated with romidepsin (FK228) and trichostatin A (TSA). HL‐60 and HP100 cells (0.5 × 106 cells/mL) were treated with FK228 and TSA at 37°C for 24 h. After treatments with FK228 (a) and TSA (b), cytotoxicity was analyzed by the LDH activity assay kit (CytoTox‐ONE Homogeneous Membrane Integrity Assay; Promega, Madison, WI, USA) according to the manufacturer’s instructions. The data are expressed as means ± SD (n = 3). *P < 0.05 and **P < 0.01, versus control; ##P < 0.01, HL‐60 versus HP100 by Student’s t‐test. (c) The cells were lysed, and DNA was extracted and analyzed by conventional electrophoresis. Marker lane: size marker DNA (φX174/Hae III digest).
DNA ladder formation in HL‐60 and HP100 cells treated with FK228 and TSA. We have analyzed DNA ladder formation, which is characteristic for apoptosis, in the cells treated with FK228 and TSA using conventional electrophoresis (Fig. 1c). The DNA ladder formation by FK228 could be detected at 10 and 50 nM in HL‐60 cells after a 24‐h incubation, whereas it could not be detected in HP100 cells. It was detectable at 500 nM TSA in both HL‐60 and HP100 cells to a similar extent after a 24‐h incubation, but in a different manner from FK228.
Generation of peroxide and change of Δψm in HL‐60 and HP100 cells treated with FK228. As shown in Figure 2, peroxide generation in HL‐60 cells was observed at 4 h, and significantly increased at 6 and 16 h after the treatment with FK228, while its generation in HP100 cells was not observed at 4 and 6 h, and increased at 16 h. Figure 3 shows change in Δψm in HL‐60 and HP100 cells treated with FK228, to examine whether FK228 induces mitochondrial permeability transition. Δψm was apparently increased in HL‐60 at 5 and 10 nM. Only slight increase in Δψm was observed in HP100 at these concentrations in comparison with HL‐60 cells.
Figure 2.
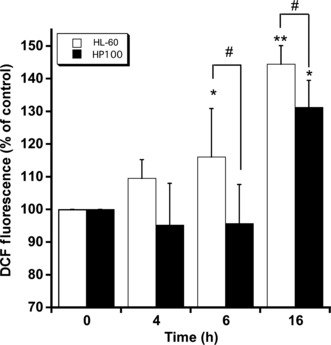
Generation of hydrogen peroxide (H2O2) in HL‐60 and HP100 cells treated with romidepsin (FK228). HL‐60 and HP100 cells (1 × 106 cells/mL) were treated with 10 nM FK228 at 37°C for the indicated times. After the treatment, the cells were incubated with 5 μM 5‐(and‐6)‐chloromethyl‐2′,7′‐dichlorodihydrofluorescein diacetate acetyl ester (CM‐H2DCFDA) for 30 min at 37°C. The cells were analyzed with a flow cytometer (FACScan; Becton Dickinson, San Jose, CA, USA). The data are expressed as means ± SD (n = 3). *P < 0.05 and **P < 0.01, versus control; #P < 0.05, HL‐60 versus HP100 by Student’s t‐test.
Figure 3.
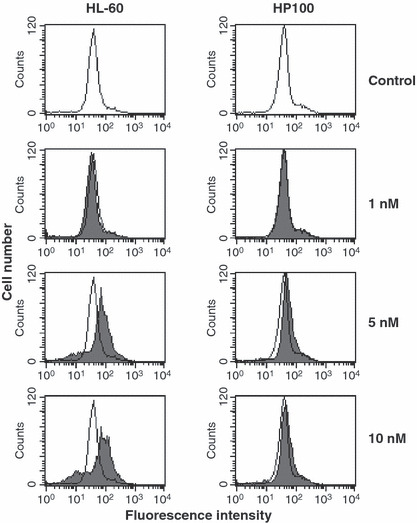
Change of mitochondorial membrane potential in HL‐60 and HP100 cells treated with romidepsin (FK228). HL‐60 and HP100 cells (1 × 106 cells/mL) were treated with FK228 at 37°C for 24 h. After the treatment, the cells were incubated with 40 nM 3,3′‐dihexyloxacarbocyanine iodide DiOC6(3) for 15 min at 37°C. The cells were analyzed with a flow cytometer (FACScan; Becton Dickinson, San Jose, CA, USA). Open peaks, control; shaded peaks, FK228‐treated cells.
HDAC activity in HL‐60 and HP100 cells treated with FK228. Inhibition of HDAC activity is a main anticancer action of FK228. Figure 4 shows HDAC activity in HL‐60 and HP100 cells treated with FK228 for 4 h. FK228 significantly inhibited HDAC cellular activities in both HL‐60 and HP100 cells to a similar extent.
Figure 4.
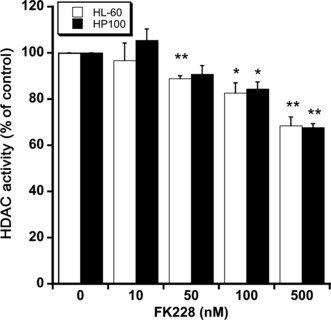
Histone deacetylase (HDAC) activity in HL‐60 and HP100 cells treated with romidepsin (FK228). HL‐60 and HP100 cells (0.5 × 106 cells/mL) were treated with FK228 at 37°C for 4 h. HDAC fluorescent activity assays using a Fluor‐de‐Lys HDAC fluorometric cellular activity assay kit (Enzo Life Sciences, Plymouth Meeting, PA, USA) were performed according to the manufacturer’s instructions. The data are expressed as means ± SD (n = 3). *P < 0.05 and **P < 0.01, versus control by Student’s t‐test.
Inhibitory effect of NAC on apoptosis in Caco‐2 cells treated with FK228. We investigated the effect of NAC, an antioxidant, on FK228‐induced apoptosis in Caco‐2 cells by morphologic analysis in order to examine whether ROS generation participates in apoptosis in a solid cancer cell line. Apoptosis was observed in FK228‐treated Caco‐2 cells, and was significantly inhibited by NAC (Fig. 5).
Figure 5.
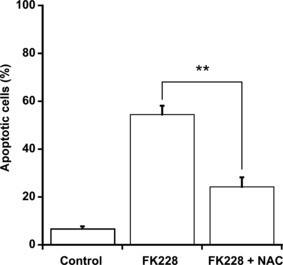
Inhibitory effect of N‐acetyl‐cysteine (NAC) on apoptosis in Caco‐2 cells treated with romidepsin (FK228). Caco‐2 cells (0.2 × 106 cells/mL) were seeded for 24 h, and then were treated with 10 nM FK228 for 48 h. Where indicated, the cells were pretreated with 10 mM NAC for 30 min. Percentage of apoptotic cells was determined by Hoechst 33342 staining. The data are expressed as means ± SD (n = 3). **P < 0.01, by Student’s t‐test.
Detection of O2 − derived from FK228 in the presence of GSH. Figure 6 shows O2 − production by FK228 in the presence and absence of GSH in a cell‐free system. O2 − was generated by FK228 reduction in the presence of GSH. When SOD was added, the amount of cytochrome c reduction was decreased, suggesting the generation of O2 −. From these results, O2 − production by 50 μM FK228 was about 25 μM in the presence of 1 mM GSH. GSH alone produced O2 −, but its amount was smaller than that in FK228 + GSH. FK228 did not produce O2 − in the absence of GSH.
Figure 6.
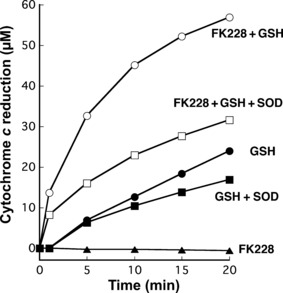
Time course of cytochrome c reduction during the incubation of romidepsin (FK228) in the presence and absence of glutathione (GSH) in a cell‐free system. The reaction mixture contained 200 μM cytochrome c, 50 μM FK228 in the presence and absence of 1 mM GSH in 10 mM sodium phosphate buffer (pH 7.8) containing 2.5 μM diethylenetriamine‐N,N,N′,N′′,N′′‐pentaacetic acid (DTPA). A maximum absorption at 550 nm was measured at 37°C with a UV‐visible spectrophotometer. The actual amount of O2 − generation was calculated by subtracting absorbance with 100 U/mL superoxide dismutase (SOD) from that without SOD at 550 nm ( = 21.1 × 103/M/cm).
Discussion
HDAC inhibitors are a novel and promising class of chemotherapeutic agent that can induce apoptosis and differentiation, and inhibit cell cycle progression.( 27 , 28 , 29 ) The present study showed that FK228 induced apoptosis in HL‐60 cells and did not in its H2O2‐resistant clone, HP100 cells. The apparent cytotoxicity and DNA ladder formation could be detected at above 10 nM in HL‐60 cells after a 24 h incubation, whereas it could not be detected under these conditions in HP100 cells. TSA induced cytotoxicity and apoptosis at 500 nM in both HL‐60 and HP100 cells. In HP100 cells treated with FK228, H2O2 generation and Δψm increase were decreased, compared with those of HL‐60 cells. It has been reported that HP100 cells are about 340‐fold more resistant to H2O2 than the parent cells, HL‐60.( 21 , 22 ) In contrast, FK228 inhibited HDAC activity, in both HL‐60 and HP100 cells to a similar extent, suggesting that an inhibitory effect of FK228 on HDAC activity is independent of H2O2 generation. Therefore, these findings can be explained by the involvement of H2O2 generation in the FK228‐induced apoptotic pathway, whereas TSA induces apoptosis independently of H2O2 generation. The independence of H2O2 generation agrees with previous results that TSA showed no influence on ROS generation in HL‐60 and human pancreatic cancer cell lines.( 30 , 31 ) Meanwhile, our data showing that NAC prevented FK228‐induced apoptosis in Caco‐2 cells confirm the possibility that ROS generation is involved in FK228‐induced apoptosis in solid cancer cell lines.
It has been reported that various HDAC inhibitors (e.g. suberoylanilide hydroxamic acid [SAHA], MS‐275, LAQ824, NCS3852 [5‐nitroso‐8‐quinolinol], sodium butyrate) induced apoptosis through ROS generation.( 8 , 9 , 10 , 11 , 12 ) SAHA induced apoptosis mediated by the cleavage of Bid, followed by disruption of the mitochondrial membrane, and production of ROS.( 8 ) FK228 can also induce Bid cleavage,( 6 ) and therefore this finding raises the possibility that FK228 generates ROS through Bid cleavage and the mitochondria‐mediated pathway. Sodium butyrate induced apoptosis mediated by p21, followed by production of ROS.( 12 ) FK228 also induced apoptosis to be associated with increase in p21;( 7 ) therefore, it is possible that FK228 generates ROS through increase in p21. Alternatively, FK228 may mediate indirect ROS generation through NAD(P)H oxidase and xanthine oxidase activation.( 15 , 16 ) However, in this study, inhibitors of NAD(P)H oxidase and xanthine oxidase did not inhibit FK228‐induced DNA ladder formation (data not shown), suggesting that indirect ROS generation via these enzymes does not participate in apoptosis.
However, interestingly, in this study, we firstly demonstrated that O2 − was generated in the presence of FK228 and GSH in a cell free system, suggesting that the reduced FK228 reacted with O2 to generate O2 − and subsequently H2O2. Therefore, the redox‐cycle of FK228 should be considered as a primary source of H2O2, since reduction of an internal sulfide bond results in the formation of two free sulfhydryl groups, which can be reoxidized in the presence of molecular oxygen to form O2 − (Fig. 7). H2O2 is considered to cause apoptosis through mitochondrial dysfunction.( 32 , 33 ) On the other hand, the reduction of an intramolecular disulfide bond of FK228 greatly enhances its HDAC inhibitory activity in cellular reducing condition involving GSH,( 34 ) and this mechanism would serve as an alternative apoptotic pathway. In conclusion, in addition to HDAC inhibition, H2O2 derived from O2 − generated by FK228, followed by the mitochondrial dysfunction, plays an important role in apoptosis.
Figure 7.
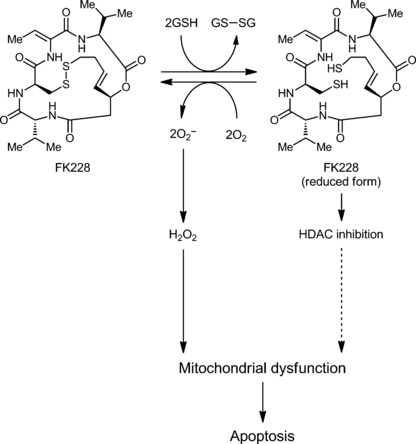
Proposed mechanisms of romidepsin (FK228)‐induced apo‐ptosis via hydrogen peroxide (H2O2) generation.
Acknowledgments
This work was supported by Grants‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan and a Kinjo Gakuin University Special Research Subsidy.
References
- 1. Ueda H, Nakajima H, Hori Y et al. FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No 968. I, Taxonomy, fermentation, isolation, physico‐chemical and biological properties, and antitumor activity. J Antibiot (Tokyo) 1994; 47: 301–10. [DOI] [PubMed] [Google Scholar]
- 2. Ueda H, Manda T, Matsumoto S et al. FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No. 968. III. Antitumor activities on experimental tumors in mice. J Antibiot (Tokyo) 1994; 47: 315–23. [DOI] [PubMed] [Google Scholar]
- 3. Schrump DS, Fischette MR, Nguyen DM et al. Clinical and molecular responses in lung cancer patients receiving Romidepsin. Clin Cancer Res 2008; 14: 188–98. [DOI] [PubMed] [Google Scholar]
- 4. Piekarz RL, Frye R, Turner M et al. Phase II multi‐institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T‐cell lymphoma. J Clin Oncol 2009; 27: 5410–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Doi S, Soda H, Oka M et al. The histone deacetylase inhibitor FR901228 induces caspase‐dependent apoptosis via the mitochondrial pathway in small cell lung cancer cells. Mol Cancer Ther 2004; 3: 1397–1402. [PubMed] [Google Scholar]
- 6. Peart MJ, Tainton KM, Ruefli AA et al. Novel mechanisms of apoptosis induced by histone deacetylase inhibitors. Cancer Res 2003; 63: 4460–71. [PubMed] [Google Scholar]
- 7. Vinodhkumar R, Song YS, Devaki T. Romidepsin (depsipeptide) induced cell cycle arrest, apoptosis and histone hyperacetylation in lung carcinoma cells (A549) are associated with increase in p21 and hypophosphorylated retinoblastoma proteins expression. Biomed Pharmacother 2008; 62: 85–93. [DOI] [PubMed] [Google Scholar]
- 8. Ruefli AA, Ausserlechner MJ, Bernhard D et al. The histone deacetylase inhibitor and chemotherapeutic agent suberoylanilide hydroxamic acid (SAHA) induces a cell‐death pathway characterized by cleavage of Bid and production of reactive oxygen species. Proc Natl Acad Sci U S A 2001; 98: 10833–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lucas DM, Davis ME, Parthun MR et al. The histone deacetylase inhibitor MS‐275 induces caspase‐dependent apoptosis in B‐cell chronic lymphocytic leukemia cells. Leukemia 2004; 18: 1207–14. [DOI] [PubMed] [Google Scholar]
- 10. Rosato RR, Almenara JA, Maggio SC et al. Potentiation of the lethality of the histone deacetylase inhibitor LAQ824 by the cyclin‐dependent kinase inhibitor roscovitine in human leukemia cells. Mol Cancer Ther 2005; 4: 1772–85. [DOI] [PubMed] [Google Scholar]
- 11. Martirosyan A, Leonard S, Shi X, Griffith B, Gannett P, Strobl J. Actions of a histone deacetylase inhibitor NSC3852 (5‐nitroso‐8‐quinolinol) link reactive oxygen species to cell differentiation and apoptosis in MCF‐7 human mammary tumor cells. J Pharmacol Exp Ther 2006; 317: 546–52. [DOI] [PubMed] [Google Scholar]
- 12. Inoue T, Kato K, Kato H et al. Level of reactive oxygen species induced by p21Waf1/CIP1 is critical for the determination of cell fate. Cancer Sci 2009; 100: 1275–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Adachi M, Zhang Y, Zhao X et al. Synergistic effect of histone deacetylase inhibitors FK228 and m‐carboxycinnamic acid bis‐hydroxamide with proteasome inhibitors PSI and PS‐341 against gastrointestinal adenocarcinoma cells. Clin Cancer Res 2004; 10: 3853–62. [DOI] [PubMed] [Google Scholar]
- 14. Choudhary S, Wang HC. Role of reactive oxygen species in proapoptotic ability of oncogenic H‐Ras to increase human bladder cancer cell susceptibility to histone deacetylase inhibitor for caspase induction. J Cancer Res Clin Oncol 2009; 135: 1601–13. [DOI] [PubMed] [Google Scholar]
- 15. Mizutani H, Tada‐Oikawa S, Hiraku Y, Oikawa S, Kojima M, Kawanishi S. Mechanism of apoptosis induced by a new topoisomerase inhibitor through the generation of hydrogen peroxide. J Biol Chem 2002; 277: 30684–9. [DOI] [PubMed] [Google Scholar]
- 16. Mizutani H, Tada‐Oikawa S, Hiraku Y, Kojima M, Kawanishi S. Mechanism of apoptosis induced by doxorubicin through the generation of hydrogen peroxide. Life Sci 2005; 76: 1439–53. [DOI] [PubMed] [Google Scholar]
- 17. Tada‐Oikawa S, Oikawa S, Kawanishi M, Yamada M, Kawanishi S. Generation of hydrogen peroxide precedes loss of mitochondrial membrane potential during DNA alkylation‐induced apoptosis. FEBS Lett 1999; 442: 65–9. [DOI] [PubMed] [Google Scholar]
- 18. Murata M, Suzuki T, Midorikawa K, Oikawa S, Kawanishi S. Oxidative DNA damage induced by a hydroperoxide derivative of cyclophosphamide. Free Radic Biol Med 2004; 37: 793–802. [DOI] [PubMed] [Google Scholar]
- 19. Kawanishi S, Hiraku Y. Amplification of anticancer drug‐induced DNA damage and apoptosis by DNA‐binding compounds. Curr Med Chem Anticancer Agents 2004; 4: 415–9. [DOI] [PubMed] [Google Scholar]
- 20. Iwamoto T, Hiraku Y, Kojima M, Kawanishi S. Amplification of C1027‐induced DNA cleavage and apoptosis by a quinacrine‐netropsin hybrid molecule in tumor cell lines. Arch Biochem Biophys 2005; 434: 232–40. [DOI] [PubMed] [Google Scholar]
- 21. Kasugai I, Yamada M. Adaptation of human leukemia HL‐60 cells to hydrogen peroxide as oxidative stress. Leuk Res 1989; 13: 757–62. [DOI] [PubMed] [Google Scholar]
- 22. Kasugai I, Yamada M. High production of catalase in hydrogen peroxide‐resistant human leukemia HL‐60 cell lines. Leuk Res 1992; 16: 173–9. [DOI] [PubMed] [Google Scholar]
- 23. Hiraku Y, Kawanishi S. DNA damage and apoptosis induced by benzene metabolites. Cancer Res 1996; 56: 5172–8. [PubMed] [Google Scholar]
- 24. Xiang J, Chao DT, Korsmeyer SJ. BAX‐induced cell death may not require interleukin 1β‐converting enzyme‐like proteases. Proc Natl Acad Sci USA 1996; 93: 14559–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Decaudin D, Geley S, Hirsch T et al. Bcl‐2 and Bcl‐XL antagonize the mitochondrial dysfunction preceding nuclear apoptosis induced by chemotherapeutic agents. Cancer Res 1997; 57: 62–7. [PubMed] [Google Scholar]
- 26. Murata M, Moriya K, Inoue S, Kawanishi S. Oxidative damage to cellular and isolated DNA by metabolites of a fungicide ortho‐phenylphenol. Carcinogenesis 1999; 20: 851–7. [DOI] [PubMed] [Google Scholar]
- 27. Emanuele S, Lauricella M, Tesoriere G. Histone deacetylase inhibitors: apoptotic effects and clinical implications. Int J Oncol 2008; 33: 637–46. [PubMed] [Google Scholar]
- 28. Marks PA, Xu WS. Histone deacetylase inhibitors: potential in cancer therapy. J Cell Biochem 2009; 107: 600–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol 2009; 27: 5459–68. [DOI] [PubMed] [Google Scholar]
- 30. Chen J, Bai H, Wang C, Kang J. Trichostatin A improves the anticancer activity of low concentrations of curcumin in human leukemia cells. Pharmazie 2006; 61: 710–6. [PubMed] [Google Scholar]
- 31. Donadelli M, Costanzo C, Beghelli S et al. Synergistic inhibition of pancreatic adenocarcinoma cell growth by trichostatin A and gemcitabine. Biochim Biophys Acta 2007; 1773: 1095–106. [DOI] [PubMed] [Google Scholar]
- 32. Kroemer G, Zamzami N, Susin SA. Mitochondrial control of apoptosis. Immunol Today 1997; 18: 44–51. [DOI] [PubMed] [Google Scholar]
- 33. Chandra J, Samali A, Orrenius S. Triggering and modulation of apoptosis by oxidative stress. Free Radic Biol Med 2000; 29: 323–33. [DOI] [PubMed] [Google Scholar]
- 34. Furumai R, Matsuyama A, Kobashi N et al. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res 2002; 62: 4916–21. [PubMed] [Google Scholar]