

Abstract
This study was conducted to analyze a comprehensive panel of single nucleotide polymorphisms (SNP) in DNA repair genes to determine the relationship between polymorphisms and the survival outcome of patients with early stage non‐small‐cell lung cancer (NSCLC). Three hundred and ten consecutive patients with surgically resected NSCLC were enrolled. Forty‐eight SNP in 27 DNA repair genes were genotyped and their associations with overall survival (OS) and disease‐free survival (DFS) were analyzed. Individually, six SNP exhibited significant associations with survival outcome. When the six SNP were combined, OS and DFS decreased as the number of bad genotypes increased (P trend < 0.0001 for both). Patients with three, and four or five bad genotypes had a significantly worse OS and DFS compared with those carrying zero or one bad genotypes (adjusted hazard ratio [aHR] for OS = 3.53, 95% confidence interval [CI] = 1.25–9.97, P = 0.02, and aHR for DFS = 3.31, 95% CI = 1.41–7.76, P = 0.006; and aHR for OS = 5.47, 95% CI = 1.87–16.00, P = 0.002, and aHR for DFS = 4.42, 95% CI = 1.82–10.74, P = 0.001, respectively). These findings suggest that the six SNP identified can be used as prognostic markers for patients with surgically resected early stage NSCLC. (Cancer Sci 2010; 101: 2436–2442)
Lung cancer, predominantly non‐small‐cell lung cancer (NSCLC), is the most common cause of death worldwide.( 1 ) Despite advances made in the diagnosis and treatment of NSCLC in the last few decades, the prognosis for lung cancer is still very poor, with a 5‐year survival rate of ∼15%. In addition, even among patients with resectable NSCLC, the majority still die from disease recurrence. Although the TNM staging system is the best prognostic index for operable NSCLC, patients with the same pathological stage of disease display marked variability in recurrence and survival.( 2 ) Therefore, research is currently in progress to identify the molecular markers that are associated with the prognosis of patients with lung cancer. Single nucleotide polymorphisms (SNP) are the most common form of human genetic variations. Several studies have demonstrated that some of these SNP affect the expression or activities of enzymes, and are therefore associated with the risk of cancer.( 3 , 4 ) In addition, genetic polymorphisms have become increasingly studied as potential prognostic markers in a variety of cancers, including lung cancer.( 5 , 6 , 7 )
DNA repair systems are essential for the maintenance of genome integrity, and therefore the deregulation of DNA repair genes can dramatically increase the risk of cancer.( 8 ) In humans, more than 125 genes are involved in the DNA damage response and several kinds of DNA repair pathways exist, such as base excision repair, nucleotide excision repair, mismatch repair and double strand break repair.( 9 ) Although the effects of DNA repair gene polymorphisms on the risk of cancer have been widely investigated, only a few studies have evaluated the prognostic significance of DNA repair gene polymorphisms in patients with cancer. We have hypothesized that individual variation in DNA repair capacity conferred by DNA repair gene polymorphisms influences the survival outcome of patients with lung cancer. To test this hypothesis, we analyzed a comprehensive panel of polymorphisms in DNA repair genes to determine the relationship between polymorphisms and the survival outcome of patients with early stage NSCLC.
Materials and Methods
Patients. The study included all patients (n = 310) with stage I, II or IIIA (micro‐invasive N2) NSCLC who underwent curative surgical resection at the Kyungpook National University Hospital (Daegu, Korea) between September 1998 and August 2006. Patients who underwent chemotherapy or radiotherapy prior to surgery were excluded to avoid the effects on DNA. All of the patients included in this study were ethnic Koreans. The histological type of lung cancers was as follows: 189 cases of squamous cell carcinomas (SCC); 116 cases of adenocarcinomas; and five cases of large cell carcinomas. The pathological staging of tumors, which was determined according to the International System for Staging Lung Cancer,( 10 ) was as follows: stage I, 177 patients; stage II, 46 patients; and stage IIIA, 87 patients. Written informed consent was obtained from all patients prior to surgery and this study was approved by the Institutional Review Board of the Kyungpook National University Hospital.
Polymorphism selection and genotyping. We searched the public database and related literature to identify potentially functional polymorphisms in DNA repair genes. We selected 48 SNP in 27 DNA repair genes. The SNP selection favored those with a minor allele frequency of at least 5%, those located in the promoter or untranslated region or coding region of the gene, those previously evaluated in relation to cancer, or those with evidence of functional significance. The selected SNP were genotyped using a sequenome mass spectrometry‐based genotyping assay. Of the set of 48 SNP selected, seven SNP that were monomorphic (APEX L104R, E126D, R237A and D283G; and MSH2 rs17224094, rs17217681 and rs17217877), one SNP (RAD52 rs11226) with a genotyping call rate <90.0%, and one SNP (ATR rs2227930) that was deviated from the Hardy–Weinberg equilibrium (HWE; P < 0.05) were excluded from further analysis. The remaining 39 SNP in 26 genes were evaluated for association analysis. The SNP identification numbers, genotype and minor allele frequencies, genotyping call rates and P‐values for HWE of the 39 SNP are shown in Table 1. For quality control, the genotyping analysis was blinded with respect to the patients. In addition, approximately 10% of the samples were randomly selected to be genotyped again by PCR‐RFLP or DNA sequencing.
Table 1.
Genotype frequencies of DNA repair gene polymorphisms analyzed in patients (n = 310) with non‐small‐cell lung cancer
| DNA repair | Gene | Polymorphism† | Genotype‡, n | MAF in healthy populations | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Symbol | Name | ID no. | Base (amino acid) change | 1/1 | 1/2 | 2/2 | CR (%) | MAF | HWE P | Asian† | European† | African† | |
| Base excision repair | PARP1/ADPRT | ADP‐ribosyltransferase | rs1136410 | T>C (V762A) | 97 | 143 | 66 | 98.7 | 0.449 | 0.330 | 0.456 | 0.167 | 0.008 |
| APEX/APE1 | Apurinic endonuclease I | rs1130409 | T>G (D148E) | 103 | 136 | 59 | 96.1 | 0.426 | 0.248 | 0.322 | 0.508 | 0.275 | |
| LIG3 | Ligase III | rs3135967 | IVS3+579A>G | 167 | 106 | 23 | 95.5 | 0.257 | 0.288 | 0.233 | 0.467 | 0.067 | |
| MBD4 | Methyl‐CpG binding domain protein 4 | rs140693 | G>A (E346K) | 129 | 131 | 36 | 95.5 | 0.343 | 0.758 | 0.444 | 0.217 | 0.183 | |
| OGG1 | 8‐Oxoguanine DNA glycosylase | rs1052133 | G>C (C326S) | 84 | 153 | 59 | 95.5 | 0.458 | 0.478 | 0.477 | 0.776 | 0.856 | |
| XRCC1 | X‐ray repair cross‐complementing group 1 | rs3213245 | −77T>C | 234 | 55 | 3 | 94.2 | 0.104 | 0.907 | 0.114 | 0.403 | 0.460 | |
| rs25489 | G>A (R280H) | 231 | 60 | 5 | 94.8 | 0.112 | 0.680 | 0.041 | 0.033 | 0.025 | |||
| rs25487 | G>A (R399Q) | 160 | 96 | 25 | 90.6 | 0.260 | 0.061 | 0.279 | 0.431 | 0.100 | |||
| Direct reversal | MGMT/AGT | O 6 ‐methylguanine‐DNA methyltransferase | rs1625649 | −485G>T | 139 | 142 | 28 | 99.7 | 0.320 | 0.331 | – | – | – |
| rs12917 | C>T (L115F) | 244 | 56 | 7 | 99.0 | 0.114 | 0.089 | 0.178 | 0.125 | 0.175 | |||
| Nucleotide excision repair | XPA | Xeroderma pigmentosum group A | rs3176629 | −501C>T | 252 | 50 | 1 | 97.7 | 0.086 | 0.367 | 0.057 | 0.000 | 0.375 |
| rs1800975 | 114A>G | 83 | 152 | 74 | 99.7 | 0.485 | 0.787 | 0.534 | 0.586 | 0.763 | |||
| XPC | Xeroderma pigmentosum group C | rs2607775 | −27C>G | 272 | 22 | 1 | 95.2 | 0.041 | 0.445 | 0.089 | 0.491 | 0.407 | |
| rs1870134 | G>C (V16L) | 154 | 105 | 27 | 92.3 | 0.278 | 0.149 | 0.182 | 0.000 | 0.000 | |||
| rs2228000 | C>T (A499V) | 157 | 119 | 20 | 95.5 | 0.269 | 0.689 | 0.456 | 0.283 | 0.058 | |||
| rs2228001 | A>C (K939Q) | 119 | 129 | 54 | 97.4 | 0.392 | 0.070 | 0.356 | 0.400 | 0.258 | |||
| ERCC1 | Excision repair cross‐complementing group 1 | rs11615 | C>T (N118N) | 175 | 100 | 19 | 94.8 | 0.235 | 0.362 | 0.289 | 0.650 | 0.017 | |
| XPD/ERCC2 | Xeroderma pigmentosum group D | rs1799793 | G>A (D312N) | 262 | 38 | 3 | 97.7 | 0.073 | 0.231 | 0.102 | 0.314 | 0.068 | |
| rs1052555 | C>T (D711D) | 247 | 38 | 1 | 92.3 | 0.070 | 0.717 | 0.034 | 0.306 | 0.080 | |||
| rs13181 | T>G (K751Q) | 246 | 49 | 2 | 95.8 | 0.089 | 0.795 | 0.081 | 0.333 | 0.183 | |||
| XPF/ERCC4 | Xeroderma pigmentosum group F | rs1799801 | T>C (S835S) | 187 | 100 | 18 | 98.4 | 0.223 | 0.348 | 0.318 | 0.250 | 0.110 | |
| XPG/ERCC5 | Xeroderma pigmentosum group G | rs17655 | G>C (D1104H) | 94 | 141 | 57 | 94.2 | 0.437 | 0.752 | 0.478 | 0.733 | 0.467 | |
| Mismatch repair | MLH1 | MutL (E. coli) homolog 1 | rs1800734 | −93A>G | 87 | 143 | 68 | 96.1 | 0.468 | 0.530 | 0.489 | 0.800 | 0.917 |
| rs1799977 | A>G (I219V) | 280 | 18 | 0 | 96.1 | 0.030 | 0.591 | 0.078 | 0.333 | 0.042 | |||
| MSH2 | MutS (E. coli) homolog 2 | rs1863332 | −434A>C | 182 | 91 | 16 | 93.2 | 0.213 | 0.306 | 0.178 | 0.058 | 0.183 | |
| rs3732183 | IVS10+12A>G | 110 | 142 | 54 | 98.7 | 0.408 | 0.487 | 0.400 | 0.658 | 0.283 | |||
| Double strand break repair | BRCA1 | Breast cancer 1 | rs799917 | C>T (P871L) | 138 | 135 | 26 | 96.5 | 0.313 | 0.384 | – | – | – |
| BRCA2 | Breast cancer 2 | rs144848 | T>G (N372H) | 159 | 124 | 15 | 96.1 | 0.258 | 0.139 | 0.307 | 0.292 | 0.125 | |
| LIG4 | Ligase 4 | rs1805388 | C>T (T9I) | 228 | 66 | 7 | 97.1 | 0.133 | 0.399 | 0.056 | 0.167 | 0.158 | |
| NBS1 | Nijmegen breakage syndrome | rs1063045 | C>T (L34L) | 98 | 134 | 61 | 94.5 | 0.437 | 0.228 | 0.455 | 0.283 | 0.292 | |
| rs1805794 | C>G (Q185E) | 101 | 137 | 63 | 97.1 | 0.437 | 0.193 | 0.400 | 0.292 | 0.150 | |||
| RAD51 | Rad51 (S. cerevisiae) homolog | rs1801320 | 170G>C | 233 | 55 | 5 | 94.5 | 0.111 | 0.409 | 0.146 | 0.067 | 0.250 | |
| XRCC3 | X‐ray repair cross‐complementing group 3 | rs1799796 | IVS6‐14A>G | 143 | 120 | 40 | 97.7 | 0.330 | 0.069 | 0.289 | 0.317 | 0.125 | |
| DNA damage response | ATM | Ataxia‐telangiectasia mutated | rs664143 | IVS61+60C>T | 74 | 152 | 72 | 96.1 | 0.497 | 0.728 | 0.556 | 0.508 | 0.325 |
| ATR | Ataxia‐telangiectasia and Rad3‐related | rs2227928 | T>C (M211T) | 102 | 144 | 53 | 96.5 | 0.418 | 0.860 | 0.422 | 0.583 | 0.833 | |
| rs2229032 | G>A (R2425Q) | 236 | 61 | 5 | 97.4 | 0.118 | 0.646 | 0.122 | 0.125 | 0.033 | |||
| CHEK1/CHK1 | Check point kinase 1 | rs521102 | IVS11+35C>T | 115 | 133 | 56 | 98.1 | 0.403 | 0.114 | 0.413 | 0.500 | 0.214 | |
| CHEK2/CHK2 | Check point kinase 2 | rs2267130 | IVS8‐200T>C | 164 | 111 | 30 | 98.4 | 0.280 | 0.087 | 0.271 | 0.396 | 0.239 | |
| TP53 | Tumor protein p53 | rs1042522 | G>C (R72P) | 106 | 136 | 60 | 97.4 | 0.424 | 0.176 | 0.409 | 0.233 | 0.669 | |
†Information about single nucleotide polymorphisms (SNP) and SNP identification and MAF in other ethnic populations (Asian, European and African) were obtained from the NCBI database (http://ncbi.nih.gov). The transcription start site was counted as +1 in the reference sequences. ‡Wild‐type allele is denoted by 1 and the polymorphic allele by 2. CR, call rate; HWE, Hardy–Weinberg equilibrium; MAF, minor allele frequency.
Statistical analysis. The demographic and clinical information was compared across genotype and stage using chi square tests for categorical variables. The HWE was tested using a goodness‐of‐fit chi square test with one degree of freedom. The degree of linkage disequilibrium between polymorphisms was measured using Haploview.( 11 ) The haplotypes and haplotype frequencies were estimated based on a Bayesian algorithm using the Phase program.( 12 ) Overall survival (OS) was measured from the day of surgery until the date of death or the date of the last follow up. Disease‐free survival (DFS) was calculated from the day of surgery until recurrence or death from any cause. The survival estimates were calculated using the Kaplan–Meier method. The differences in OS or DFS across different genotypes were compared using the log‐rank test. Hazard ratios (HR) and 95% confidence intervals (CI) were estimated using multivariate Cox proportional hazards models, with adjustment for age (≤64 versus >64 years), gender (male versus female), smoking status (never‐ versus ever‐smoker), pathological stage (I versus II–IIIA) and adjuvant therapy. All analyses were performed using the Statistical Analysis System for Windows, version 9.1 (SAS Institute, Cary, NC, USA).
Results
Patient characteristics and clinical predictors. The clinical and pathological characteristics of patients and the association with OS and DFS are shown in Table 2. There were 114 deaths (36.8%). The estimated 5‐year OS and DFS for all the patients was 53.3% (95% CI = 45.8–60.2%) and 44.0% (95% CI = 36.9–50.9%). Upon univariate analysis, the pathological stage was significantly associated with OS and DFS (P < 0.0001 for OS and DFS).
Table 2.
Univariate analysis for overall survival by demographics, smoking status, histological type and pathological stage
| Variables | No. cases | Overall survival | Disease‐free survival | ||||
|---|---|---|---|---|---|---|---|
| No. deaths (%)† | 5Y‐OSR (%)‡ | Log‐rank P | No. events (%)† | 5Y‐DFSR (%)‡ | Log‐rank P | ||
| Overall | 310 | 114 (36.8) | 53 | – | 148 (47.7) | 44 | – |
| Age (years) | |||||||
| ≤64 | 147 | 50 (34.0) | 55 | 0.44 | 68 (46.3) | 45 | 0.79 |
| >64 | 163 | 64 (39.3) | 52 | 80 (48.1) | 43 | ||
| Gender | |||||||
| Male | 245 | 98 (40.0) | 51 | 0.17 | 121 (49.4) | 43 | 0.74 |
| Female | 65 | 16 (24.6) | 67 | 27 (41.5) | 51 | ||
| Smoking status | |||||||
| Never | 61 | 16 (26.2) | 67 | 0.24 | 27 (44.3) | 47 | 0.97 |
| Ever | 249 | 98 (39.4) | 51 | 121 (48.6) | 44 | ||
| Pack‐years§ | |||||||
| ≤39 | 150 | 60 (40.0) | 48 | 0.99 | 74 (49.3) | 41 | 0.58 |
| >39 | 99 | 38 (39.4) | 56 | 47 (47.5) | 48 | ||
| Histological type¶ | |||||||
| Squamous cell carcinoma | 189 | 70 (37.0) | 54 | 0.50 | 88 (46.6) | 46 | 0.50 |
| Adenocarcinoma | 116 | 42 (36.2) | 52 | 58 (50.0) | 40 | ||
| Pathological stage | |||||||
| I | 177 | 45 (25.4) | 61 | <0.0001 | 59 (33.3) | 56 | <0.0001 |
| II | 46 | 23 (50.0) | 44 | 29 (63.0) | 31 | ||
| IIIA | 87 | 46 (52.9) | 43 | 60 (69.0) | 28 | ||
| Adjuvant therapy†† | |||||||
| No | 62 | 36 (58.1) | 42 | 0.57 | 42 (67.7) | 27 | 0.75 |
| Yes | 71 | 33 (46.5) | 46 | 47 (66.2) | 31 | ||
†Row percentage. ‡Five‐year overall survival rate (5Y‐OSR) and 5‐year disease free survival rate (5Y‐DFSR): proportion of survival derived from Kaplan–Meier analysis. §In ever‐smokers. ¶Five large‐cell carcinomas were excluded from this analysis. ††In pathological stage II + IIIA: 55 cases received chemotherapy; five cases received radiotherapy; and two cases received chemotherapy and radiotherapy.
Genotype frequencies and effect on OS and DFS. Thirty‐three SNP and their haplotypes were not significantly associated with survival outcome. Six SNP (PARP1 rs1136410T>C [V762A], XRCC1 rs25489G>A [R280H], MGMT rs12917C>T [L115F], XPC rs2228000C>T [A499V], XPD rs1799793G>A [D312N] and MSH2 rs3732183A>G [IVS10+12]) were significantly associated with survival outcome in multivariate analysis. Patients with the PARP1 rs1136410 CC genotype had a significantly better OS and DFS compared with those carrying the TT or TC genotype (adjusted HR [aHR] for OS = 0.50, 95% CI = 0.28–0.88, P = 0.02; and aHR for DFS = 0.48, 95% CI = 0.23–0.78, P = 0.003). The XRCC1 rs25489, XPC rs2228000 and MSH2 rs3732183 SNP exhibited a better survival outcome under a dominant model for the variant allele for each SNP. The MGMT rs12917C>T exhibited a worse OS under a dominant model for the variant T allele, and the XPD rs1799793G>A exhibited a worse OS under a recessive model for the variant allele (Table 3). The association of MGMT rs12917C>T genotypes with the survival of patients was further examined after categorizing the patients by smoking status. The rs12917 CT or TT genotype exhibited a significantly worse OS in ever‐smokers (aHR for OS = 1.91, 95% CI = 0.11–3.29, P = 0.02), but not in never‐smokers (aHR = 1.10, 95% CI = 0.47–2.58, P = 0.82). The genotype and polymorphic allele frequencies of the six polymorphisms were not significantly different according to patient‐ or tumor‐related factors, such as age, gender, smoking status, histological subtype, pathological stage or adjuvant therapy (data not shown).
Table 3.
Overall survival and disease‐free survival according to the polymorphisms in DNA repair genes
| Polymorphism/Genotype | No. cases (%)† | Overall survival | Disease‐free survival | ||||||
|---|---|---|---|---|---|---|---|---|---|
| No. deaths (%)‡ | 5YSR (%)§ | HR (95% CI)¶ | P¶ | No. events (%)‡ | 5YSR(%)§ | HR (95% CI)¶ | P¶ | ||
| PARP1/ADPRT rs1136410†† | |||||||||
| TT | 97 (31.7) | 40 (41.2) | 48 | 1.00 | – | 52 (53.6) | 37 | 1.00 | 0.97 |
| TC | 143 (46.7) | 56 (39.2) | 52 | 0.94 (0.62–1.42) | 0.76 | 73 (51.1) | 41 | 0.99 (0.69–1.42) | 0.005 |
| CC | 66 (21.2) | 14 (21.2) | 68 | 0.47 (0.26–0.87) | 0.02 | 19 (28.8) | 66 | 0.47 (0.28–0.80) | – |
| TT + TC | 240 (78.4) | 96 (40.0) | 51 | 1.00 | – | 115 (52.1) | 40 | 1.00 | 0.003 |
| CC | 66 (21.2) | 14 (21.2) | 68 | 0.50 (0.28–0.88) | 0.02 | 19 (28.8) | 66 | 0.48 (0.23–0.78) | – |
| XRCC1 rs25489†† | |||||||||
| GG | 231 (78.6) | 87 (37.7) | 51 | 1.00 | – | 115 (48.8) | 41 | 1.00 | – |
| GA | 60 (20.4) | 16 (26.7) | 68 | 0.57 (0.33–0.97) | 0.04 | 21 (35.0) | 61 | 0.58 (0.36–0.92) | 0.02 |
| AA | 3 (1.0) | 1 (33.3) | 67 | 0.78 (0.10–6.19) | 0.82 | 1 (33.3) | 67 | 0.54 (0.07–4.12) | 0.55 |
| GG | 231 (78.6) | 87 (37.7) | 51 | 1.00 | – | 115 (48.8) | 41 | 1.00 | – |
| GA + AA | 63 (21.4) | 17 (27.0) | 68 | 0.61 (0.36–1.03) | 0.06 | 22 (34.9) | 61 | 0.59 (0.37–0.94) | 0.03 |
| MGMT rs12917†† | |||||||||
| CC | 244 (79.5) | 84 (34.4) | 57 | 1.00 | – | 113 (46.3) | 47 | 1.00 | – |
| CT | 56 (18.2) | 24 (42.9) | 43 | 1.63 (1.02–2.58) | 0.04 | 29 (51.8) | 35 | 1.38 (0.91–2.09) | 0.13 |
| TT | 7 (2.3) | 3 (42.9) | 30 | 1.75 (0.54–5.88) | 0.35 | 3 (42.9) | 51 | 1.41 (0.44–4.59) | 0.59 |
| CC | 244 (79.5) | 84 (34.4) | 57 | 1.00 | – | 113 (46.3) | 47 | 1.00 | – |
| CT + TT | 63 (20.5) | 27 (42.9) | 42 | 1.67 (1.07–2.61) | 0.02 | 32 (50.8) | 35 | 1.39 (0.93–2.08) | 0.11 |
| XPC rs2228000†† | |||||||||
| CC | 157 (53.0) | 63 (40.1) | 49 | 1.00 | – | 79 (50.3) | 42 | 1.00 | – |
| CT | 119 (40.2) | 37 (31.1) | 60 | 0.62 (0.41–0.93) | 0.02 | 50 (42.0) | 50 | 0.68 (0.48–0.96) | 0.03 |
| TT | 20 (6.8) | 5 (25.0) | 60 | 0.71 (0.28–1.79) | 0.47 | 9 (45.0) | 43 | 1.02 (0.51–2.04) | 0.97 |
| CC | 157 (53.0) | 63 (40.1) | 49 | 1.00 | – | 79 (50.3) | 42 | 1.00 | – |
| CT + TT | 139 (47.0) | 42 (30.2) | 61 | 0.66 (0.44–0.99) | 0.04 | 59 (42.5) | 49 | 0.74 (0.53–1.05) | 0.09 |
| XPD rs1799793†† | |||||||||
| GG | 262 (86.5) | 99 (37.8) | 51 | 1.00 | – | 128 (48.9) | 42 | 1.00 | – |
| GA | 38 (12.5) | 10 (26.3) | 76 | 0.52 (0.26–1.05) | 0.07 | 15 (38.5) | 61 | 0.64 (0.38–1.14) | 0.13 |
| AA | 3 (1.0) | 2 (66.7) | 0 | 5.79 (1.33–25.26) | 0.02 | 2 (66.7) | 0 | 3.15 (0.74–13.38) | 0.12 |
| GG + GA | 300 (99.0) | 153 (39.5) | 54 | 1.00 | – | 143 (47.7) | 44 | 1.00 | – |
| AA | 3 (1.0) | 2 (66.7) | 0 | 5.95 (1.36–26.00) | 0.02 | 2 (66.7) | 0 | 3.10 (0.73–13.19) | 0.13 |
| MSH2 rs3732183†† | |||||||||
| AA | 110 (36.0) | 45 (40.8) | 47 | 1.00 | – | 56 (50.9) | 44 | 1.00 | – |
| AG | 142 (46.4) | 45 (31.7) | 57 | 0.64 (0.42–0.96) | 0.03 | 61 (43.0) | 47 | 0.73 (0.51–1.05) | 0.08 |
| GG | 54 (17.6) | 22 (40.7) | 50 | 0.75 (0.45–1.29) | 0.28 | 29 (53.7) | 35 | 0.96 (0.81–1.51) | 0.85 |
| AA | 110 (36.0) | 45 (40.8) | 47 | 1.00 | – | 56 (50.9) | 44 | 1.00 | – |
| AG + GG | 196 (64.0) | 67 (34.2) | 55 | 0.68 (0.48–0.99) | 0.05 | 90 (45.9) | 44 | 0.79 (0.59–1.11) | 0.18 |
†Column percentage. ‡Row percentage. §Five‐year survival rate (5YSR): proportion of survival derived from Kaplan–Meier analysis. ¶Hazard ratio (HR), 95% confidence interval (CI) and their corresponding P‐values were calculated using multivariate Cox proportional hazard models, adjusted for age, gender, smoking status, tumor histology, pathological stage and adjuvant therapy. ††Genotype failure: four cases for rs1136410; 16 cases for rs25489; three cases for 12917; 14 cases for 2228000, seven cases for rs1799793; and six cases for rs3732183.
Combined effect of DNA repair gene SNP on OS and DFS. Because the rs1136410 TT or TC, rs25489 GG, rs12917 CT or TT, rs2228000 CC, rs1799793 AA and rs3732183 AA genotypes were associated with worse survival outcomes, we considered these six genotypes as bad genotypes and then evaluated their combined effects by grouping the patients based on the number of bad genotypes. As shown in Table 4 and Figure 1, OS and DFS were decreased as the number of bad genotypes increased (P trend < 0.0001 for OS and DFS). Based on multivariate analysis, patients carrying three, and four or five bad genotypes had significantly worse OS and DFS compared with those carrying zero or one bad genotype (aHR for OS = 3.53, 95% CI = 1.25–9.97, P = 0.02, and aHR for DFS = 3.31, 95% CI = 1.41–7.76, P = 0.006; and aHR for OS = 5.47, 95% CI = 1.87–16.00, P = 0.002, and aHR for DFS = 4.42, 95% CI = 1.82–10.74, P = 0.001, respectively).
Table 4.
Combined effects of the six polymorphisms in the DNA repair genes on overall survival and disease‐free survival
| No. bad genotypes | No. cases (%)† | Overall survival | Disease‐free survival | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 5YSR (%)‡ | Log‐rank P | HR (95% CI)§ | P§ | 5YSR (%)‡ | Log‐rank P | HR (95% CI)§ | P§ | ||
| 0 or 1¶ | 28 (10.0) | 81 | 0.001 | 1 | – | 73 | 0.001 | 1 | – |
| 2 | 94 (33.4) | 62 | – | 1.99 (0.69–5.78) | 0.21 | 50 | – | 2.11 (0.88–5.03) | 0.09 |
| 3 | 111 (39.5) | 50 | – | 3.53 (1.25–9.97) | 0.02 | 41 | – | 3.31 (1.41–7.76) | 0.006 |
| 4 or 5 | 48 (17.1) | 38 | – | 5.47 (1.87–16.00) | 0.002 | 30 | – | 4.42 (1.82–10.74) | 0.001 |
| P trend | – | – | – | <0.0001 | – | <0.0001 | – | ||
†Twenty‐nine cases, in which genotyping failed for any of the six single nucleotide polymorphisms, were excluded from this analysis. The number in parenthesis is the column percentage. ‡Five‐year survival rate (5YSR): proportion of survival derived from Kaplan–Meier analysis. §Hazard ratio (HR), 95% confidence interval (CI) and their corresponding P‐values were calculated using multivariate Cox proportional hazard models, adjusted for age, gender, smoking status, tumor histology, pathological stage and adjuvant therapy. ¶The number of patients with zero bad genotypes was six.
Figure 1.
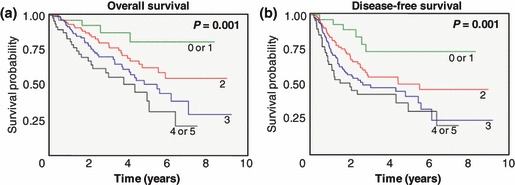
Kaplan–Meier survival estimates of overall survival (a) and disease‐free survival (b) according to the number of bad genotypes of the six single nucleotide polymorphisms (rs1136410, rs25489, rs12917, rs2228000, rs1799793 and rs3732183). P‐values are from the log‐rank test.
Discussion
Given that the prognosis of cancer patients likely involves multistep, multigenic pathways, it is unlikely that any single polymorphism would have a dramatic effect on survival outcome, and it is important to undertake a pathway‐based analysis that assesses the combined effects of polymorphisms that interact in the same pathway. Therefore, we used a multigenic approach to evaluate the associations of a panel of SNP in DNA repair genes with the survival outcome of patients with surgically‐resected NSCLC. In the present study, six SNP (rs1136410, rs25489, rs12917, rs2228000, rs1799793 and rs3732183) and their combined genotypes were associated with the survival outcome of patients. These findings suggest that the SNP in DNA repair genes, particularly their combined genotypes, can be used as prognostic factors for patients with surgically resected NSCLC.
The most significant finding of this study was that combined analysis of multiple SNP in the same or relevant pathways had a much higher potential value in terms of unraveling polymorphisms of prognostic importance. Although six SNP were associated with survival outcome in individual SNP analysis, considering the borderline CI and multiple comparisons, the impact of any individual SNP on survival outcomes would be minimal. However, when these six SNP were combined, OS and DFS decreased as the number of bad genotypes increased (P trend < 0.0001 for OS and DFS). Matullo et al. ( 13 ) reported a dose‐dependent relationship between the number of adverse alleles in DNA repair gene SNP and the level of DNA adducts in peripheral blood cells, suggesting a stepwise decrease in DNA repair capacity (DRC) as the number of adverse alleles increases, and providing biological plausibility and validity to our approach.
This study suggests that DNA repair gene polymorphisms may influence malignant phenotypes of NSCLC. There is a growing realization that genetic polymorphisms strongly influence not only the development of cancer, but also cancer progression and prognosis.( 14 ) DNA repair gene polymorphisms have been shown to be associated with DRC, and genetic and epi‐genetic alterations lead to clonal expansion of abnormal cells through the acquisition of a proliferative advantage.( 13 , 15 , 16 , 17 ) Therefore, it is possible that functional polymorphisms in DNA repair genes may affect the natural history of lung cancer, such as tumor histopathology including the stage or grade of disease, the rate of disease progression, or the propensity for metastasis, thereby influencing the survival outcome.
In the present study, six SNP were associated with survival outcome. The XPC rs2228000C>T (A499V) SNP exhibited a better survival outcome under a dominant model for the variant T allele. The XPC protein plays a key role in nucleotide excision repair (NER) that removes bulky DNA adducts that are typically generated by polycyclic aromatic hydrocarbons in tobacco smoke. The XPC protein binds with HR23B to form a complex that recognizes damaged DNA and initiates NER through the recruitment of other components of the repair apparatus, such as XPA, transcription factor IIH (TFIIH) and XPG. Because the A499V SNP is located in the region of the HR23B interaction domain of XPC, the Ala‐to‐Val change may affect the XPC‐HR23B interaction, thereby influencing NER capacity.( 18 ) Consistent with our finding, it has been reported that subjects with the rs2228000 CT or TT genotype had a higher DRC compared with those with the rs2228000 CC genotype.( 19 , 20 ) For the XPD rs1799793G>A SNP, patients with homozygous variant A alleles exhibited worse survival outcome compared with those carrying the GG or GA genotype. The XPD protein is an ATP‐dependent helicase, a subunit of TFIIH that is essential for NER. Because the rs1799793G>A [D312N] SNP is located in the domain of interaction between the XPD protein and its helicase activator, p44 protein, inside the transcription factor IIH complex, it has been suggested that this SNP may thwart XPD‐p44 protein interaction, thereby leading to reduced NER capacity.( 21 ) In agreement with this suggestion, the rs1799793 AA genotype has been reported to be associated with reduced DRC( 15 ) and an increased risk of lung cancer in a meta‐analysis.( 22 ) Therefore, our finding of an association of the XPD rs1799793 AA genotype with a worse survival outcome is biologically plausible. In the present study, MGMT rs12917C>T was associated with a worse survival outcome under a dominant model of the variant T allele. MGMT removes mutagenic O 6 ‐alkylguanine DNA adducts originating from exposure to alkylating agents, such as nitrosamines present in tobacco smoke.( 23 ) It has been suggested that the rs12917C>T [L115F] SNP may reduce the stability of the MGMT protein, thereby reducing MGMT‐mediated repair of DNA alkyl adducts.( 23 , 24 ) Hill et al. ( 25 ) reported that individuals with the rs12917 CT or TT genotype had a significantly higher level of tobacco‐specific nitrosamine‐induced chromosome aberrations compared with those with the CC genotype. Therefore, our finding of an association of the rs12917 CT or TT genotype with a worse survival outcome is also biologically plausible. In addition, it is conceivable that the MGMT rs12917C>T SNP may play an important role in determining the prognosis of lung cancer in ever‐smokers. For the MSH2 rs3732183A>G (IVS10+12A>G) SNP, we found that the AG or GG genotype was associated with a better survival outcome compared with the AA genotype. The hMSH2 gene is one of the mismatch repair genes. It has been reported that multiple alternatively spliced isoforms of hMSH2 mRNA are present in normal human tissues. Some of the isoforms would produce truncated proteins that lack exon 2–8 or exon 13, and the others are in‐frame deletions lacking exon 5 or exon 2–7. The relative level of expression of each splicing variant has shown considerable interindividual variation.( 26 ) Although the functional relevance of this SNP is still unknown, this splicing site polymorphism may influence the alternative splicing of hMSH2, resulting in interindividual variation in the levels of expression of the hMSH2 splicing variants.( 27 ) Our finding is in agreement with the results of previous studies, in which this SNP was related to a significantly decreased risk of lung cancer and a better survival outcome in patients with colorectal cancer in a dominant model for the variant G allele.( 27 , 28 )
PARP1 plays a critical role in base excision repair (BER), which is a major DNA repair pathway for small base adducts produced by oxidation, methylation and environmental carcinogens.( 29 ) PARP1 acts as a sensor of DNA streak breaks during BER. In addition to its role as a nick sensor, PARP1 also interacts with the scaffold protein, XRCC1, and may accelerate the recruitment of DNA repair proteins to strand interruption. The PARP1 rs1136410T>C [V762A] SNP, located within the carboxy‐terminal catalytic domain, has been reported to influence PARP activity, thereby affecting BER capacity.( 30 ) A Chinese study group reported that the rs1136410T>C SNP was associated with a significantly increased risk of esophageal SCC and lung cancer.( 31 , 32 ) Lockett et al. ( 30 ) also reported that this SNP was associated with a significantly increased risk of prostate cancer in American Caucasians. However, in contrast to these studies, Li et al. ( 33 ) reported that this SNP was associated with a significantly decreased risk of SCC of the head and neck in American Caucasians. In addition, the rs1136410T>C SNP has been reported to be associated with a non‐significantly reduced risk of melanoma and breast cancer in American Caucasians and Chinese women, respectively.( 34 , 35 ) Our finding that the rs1136410T>C SNP was associated with better survival outcome is in agreement with some reports,( 33 , 34 , 35 ) but in disagreement with others.( 30 , 31 , 32 ) XRCC1 acts as a facilitator or coordinator in BER through its interaction with PARP1, DNA polymerase β and DNA ligase III. Provided that the rs25489G>A [R280H] SNP is located in the vicinity of the PARP1 interaction domain of XRCC1, this SNP may alter XRCC1‐PARP1 interaction and thereby influence BER capacity.( 36 ) Discrepancy exists with respect to the association between the rs25489G>A SNP and lung cancer; indeed, both an increased and decreased risk have been reported.( 37 ) In the present study, the XRCC1 rs25489G>A SNP was associated with a better survival outcome in the dominant model for the variant A allele. Although it is difficult to decipher the mechanism(s) for the discrepant finding from different studies, including an oppositely directed (flip‐flop) association, the different genetic backgrounds in the study populations or interactive effects of multiple polymorphisms might contribute to the discrepancy.( 38 ) Another possible explanation for the discrepancy between different studies might be that different cell types of cancer have different etiologies and different carcinogenesis pathways.
In the present study, six SNP in DNA repair genes were found to be associated with the survival outcome of patients with early stage NSCLC, and the combined genotypes of these six SNP to be independent prognostic markers for those patients. Consequently, in addition to the pathological stage, testing for the presence of these six SNP may help identify patient subgroups at high risk of a poor disease outcome, thereby helping to refine therapeutic decisions in the treatment of NSCLC. However, larger studies are required to confirm our findings in other ethnic populations. In addition, further studies are needed to determine the effects of these SNP on the survival outcome of other types of cancer because different cell types of cancer have different etiologies and different carcinogenesis pathways.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
This study was supported by the National R&D Program for Cancer Control Ministry of Health & Welfare (0720550‐2) and the Regional Technology Innovation Program of the Ministry of Commerce, Industry and Energy (RTI04‐01‐01) of Republic of Korea.
References
- 1. Jemal A, Siegel R, Ward E et al. Cancer statistics, 2008. CA Cancer J Clin 2008; 58: 71–96. [DOI] [PubMed] [Google Scholar]
- 2. Miller YE. Pathogenesis of lung cancer: 100 year report. Am J Respir Cell Mol Biol 2005; 33: 216–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang X, Miao X, Sun T et al. Functional polymorphisms in cell death pathway genes FAS and FASL contribute to risk of lung cancer. J Med Genet 2005; 42: 479–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Park JY, Park JM, Jang JS et al. Caspase 9 promoter polymorphisms and risk of primary lung cancer. Human Mol Genet 2006; 15: 1963–71. [DOI] [PubMed] [Google Scholar]
- 5. Heist RS, Zhou W, Chirieac LR et al. MDM2 polymorphism, survival, and histology in early‐stage non‐small cell lung cancer. J Clin Oncol 2007; 25: 2243–7. [DOI] [PubMed] [Google Scholar]
- 6. Heist RS, Zhai R, Liu G et al. VEGF polymorphisms and survival in early‐stage non‐small cell lung cancer. J Clin Oncol 2008; 26: 856–62. [DOI] [PubMed] [Google Scholar]
- 7. Yoo SS, Choi JE, Lee WK et al. Polymorphisms in the CASPASE genes and survival in patients with early stage non‐small cell lung cancer. J Clin Oncol 2009; 27: 5823–9. [DOI] [PubMed] [Google Scholar]
- 8. Hoeijmakers JHJ. Genome maintenance mechanisms for preventing cancer. Nature 2001; 411: 366–74. [DOI] [PubMed] [Google Scholar]
- 9. Ronen A, Glickman BW. Human DNA repair genes. Environ Mol Mutagen 2001; 37: 241–283. [DOI] [PubMed] [Google Scholar]
- 10. Mountain CF. Revisions in the international system for staging lung cancer. Chest 1997; 111: 1710–7. [DOI] [PubMed] [Google Scholar]
- 11. Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 2005; 21: 263–5. [DOI] [PubMed] [Google Scholar]
- 12. Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 2001; 68: 978–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Matullo G, Dunning AM, Guarrera S et al. DNA repair polymorphisms and cancer risk in non‐smokers in a cohort study. Carcinogenesis 2006; 27: 997–1007. [DOI] [PubMed] [Google Scholar]
- 14. Lotkionov A. Common gene polymorphisms, cancer progression and prognosis. Cancer Lett 2004; 208: 1–33. [DOI] [PubMed] [Google Scholar]
- 15. Spitz MR, Wu X, Wang Y et al. Modulation of nucleotide excision repair capacity by XPD polymorphisms in lung cancer patients. Cancer Res 2001; 61: 1354–7. [PubMed] [Google Scholar]
- 16. Sakano S, Matsumoto H, Yamamoto Y et al. Association between DNA repair gene polymorphisms and p53 alterations in Japanese patients with muscle‐invasive bladder cancer. Pathobiology 2006; 73: 295–303. [DOI] [PubMed] [Google Scholar]
- 17. Candiloro LM, Dobrobic A. Detection of MGMT methylation in normal individuals is strongly associated with the T allele of the rs16906252 MGMT promoter single nucleotide polymorphism. Cancer Prev Res 2009; 2: 862–7. [DOI] [PubMed] [Google Scholar]
- 18. Bernardes de Jesus BM, Bjoras M, Coin F et al. Dissection of the molecular defects caused by pathogenic mutations in the DNA repair factor XPC. Mol Cell Biol 2008; 28: 7225–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shen J, Desai M, Agrawal DO et al. Polymorphisms in nucleotide excision repair genes and DNA repair capacity phenotype in sisters discordant for breast cancer. Cancer Epidemiol Biomarkers Prev 2006; 15: 1614–9. [DOI] [PubMed] [Google Scholar]
- 20. Zhu Y, Yang H, Chen Q et al. Modulation of DNA damage/DNA repair capacity by XPC polymorphisms. DNA Repair 2008; 7: 141–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Benhamou S, Sarasin A. ERCC2/XPD gene polymorphisms and cancer risk. Mutagenesis 2002; 17: 463–9. [DOI] [PubMed] [Google Scholar]
- 22. Benhamou S, Sarasin A. ERCC2/XPD gene polymorphisms and lung cancer: a huge review. Am J Epidemiol 2005; 161: 1–14. [DOI] [PubMed] [Google Scholar]
- 23. Tubbs JL, Pegg AE, Tainer JA. DNA binding, nucleotide flipping, and helix‐turn‐helix motif in base repair by O‐alkylguanine‐DNA alkyltransferase and its implication for cancer chemotherapy. DNA Repair 2007; 6: 1100–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Inoue R, Abe M, Nakabeppu Y et al. Characterization of human polymorphic DNA repair methyltransferase. Pharmacogenetics 2000; 10: 59–66. [DOI] [PubMed] [Google Scholar]
- 25. Hill CE, Wickliffe JK, Wolfe KJ et al. The L84F and the I143V polymorphisms in the O 6 ‐methylguanine‐DNA‐methlytransferase (MGMT) gene increase human sensitivity to the genotoxic effects of the tobacco‐specific nitrosamine carcinogen NNK. Pharmacogenet Genomics 2005; 15: 571–8. [DOI] [PubMed] [Google Scholar]
- 26. Mori Y, Shiwaku H, Fukushige S et al. Alternative splicing of hMSH2 in normal human tissues. Hum Genet 1997; 99: 590–5. [DOI] [PubMed] [Google Scholar]
- 27. Jung CY, Choi JE, Park JM et al. Polymorphisms in the hMSH2 gene and risk of primary lung cancer. Cancer Epidemiol Biomarkers Prev 2006; 15: 762–8. [DOI] [PubMed] [Google Scholar]
- 28. Kim JG, Chae YS, Sohn SK et al. IVS10+12A>G polymorphism in hMSH2 gene associated with prognosis for patients with colorectal cancer. Ann Oncol 2010; 21: 525–9. [DOI] [PubMed] [Google Scholar]
- 29. Masutani M, Nakagama H, Sugimura T. Poly(ADP‐ribosyl)ation in relation to cancer and autoimmune disease. Cell Mol Life Sci 2005; 62: 769–83. [DOI] [PubMed] [Google Scholar]
- 30. Lockett KL, Hall MC, Xu J et al. The ADPRT V762A genetic variant contributes to prostate cancer susceptibility and deficient enzyme function. Cancer Res 2004; 64: 6344–8. [DOI] [PubMed] [Google Scholar]
- 31. Hao B, Wang H, Zhou K et al. Identification of genetic variants in base excision repair pathways and their associations with risk of esophageal squamous cell carcinoma. Cancer Res 2004; 64: 4378–84. [DOI] [PubMed] [Google Scholar]
- 32. Zhang X, Miao X, Liang G et al. Polymorphisms in DNA base excision repair genes ADPRT and XRCC1 and risk of lung cancer. Cancer Res 2005; 65: 722–26. [PubMed] [Google Scholar]
- 33. Li C, Hu Z, Lu J et al. Genetic polymorphisms in DNA base‐excision repair genes ADPRT, XRCC1, and APE1 and the risk of squamous cell carcinoma of the head and neck. Cancer 2007; 110: 867–75. [DOI] [PubMed] [Google Scholar]
- 34. Li C, Liu Z, Wang LE et al. Genetic variants of the ADPRT, XRCC1, and APE1 genes and risk of cutaneous melanoma. Carcinogenesis 2006; 27: 1894–1901. [DOI] [PubMed] [Google Scholar]
- 35. Zhai X, Liu J, Hu Z et al. Polymorphisms of ADPRT Val762Ala and XRCC1 Arg399Glu and risk of breast cancer in Chinese women: a case‐control study. Oncol Rep 2006; 15: 247–52. [PubMed] [Google Scholar]
- 36. Tuimala J, Szekely G, Gundy S et al. Genetic polymorphisms of DNA repair and xenobiotics‐metabolizing enzymes: role in mutagen sensitivity. Carcinogenesis 2002; 23: 1003–8. [DOI] [PubMed] [Google Scholar]
- 37. Schneider J, Classen V, Helmig S. XRCC1 polymorphism and lung cancer risk. Expert Rev Mol Diagn 2008; 8: 761–780. [DOI] [PubMed] [Google Scholar]
- 38. Lin PI, Vance JM, Pericak‐Vance MA et al. No gene is an island: the flip‐flop phenomenon. Am J Hum Genet 2007; 80: 531–8. [DOI] [PMC free article] [PubMed] [Google Scholar]