

Abstract
Paclitaxel (PTX), one of the key drugs used to treat ovarian cancer, activates the Raf–mitogen‐activated protein kinase kinase (MEK) and phosphatidylinositol 3′‐kinase (PI3K) pathways, both considered to be proliferation and cell‐survival pathways. The present study aimed to clarify whether and how MEK and PI3K inhibitors affect sensitivity to PTX in ovarian cancer cells. We treated five ovarian cancer cell lines using PTX combined with MEK inhibitor (PD98059 [PD]) and PI3K inhibitor (LY294002 [LY]), then assessed cell viability, apoptosis, and expression of phosphorylated (p) MEK and pAkt. We also investigated the effect of combined treatment on survival in a xenograft model. The protein expression levels of MEK, pMEK, Akt, and pAkt were confirmed in all cell lines. pMEK levels increased after PTX treatment in all five ovarian cancer cell lines. Combining PTX with either PD or LY had an additive effect on cell‐growth inhibition. In contrast, we observed a synergistic effect when PTX was combined with both PD and LY. The number of apoptotic cells was significantly higher after treatment with PTX combined with PD and LY, compared with PTX alone or PTX with either PD or LY (P < 0.05). PD with PTX downregulated the protein expression level of pMEK and upregulated pAkt in all five cell lines. Treating nude mice with PTX and PD and LY prolonged survival in an ovarian cancer xenograft model (P < 0.005). These results indicate that further study is warranted for PTX combined with MEK inhibitor and PI3K inhibitor to treat ovarian carcinoma. (Cancer Sci 2007; 98: 2002–2008)
Ovarian cancer is the leading cause of death in women with gynecological cancer. The incidence of ovarian cancer in 2002 was projected to be 205 000 new cases and 125 000 deaths worldwide, representing 4.2% of all cancer deaths in women.( 1 ) More than 75% of patients with ovarian cancer are diagnosed in the advanced stage. Until 1996, standard treatment for advanced ovarian cancer included surgical tumor debulking, followed by adjuvant chemotherapy, consisting of a platinum compound and an alkylating agent.( 2 ) Two pivotal trials, the Gynecologic Oncology Group (GOG) 111 and a European–Canadian study, known as OV‐10, showed that incorporating paclitaxel (PTX) into first‐line therapy improved the survival rate of patients with stage III or IV ovarian cancer.( 3 , 4 ) A subsequent international study, GOG 182‐ICON5, sought to improve the efficacy of standard platinum–taxane therapy by incorporating newer cytotoxic agents in sequential doublet and triplet combinations.( 5 ) Unfortunately, no combination of several agents used in standard therapy has improved overall survival. Although PTX is currently considered an important chemotherapeutic agent, median progression‐free survival is only 18 months and the 5‐year survival rate remains only 30%.( 6 )
Paclitaxel functions by binding to intracellular β‐tubulin, thus stabilizing microtubules. This action blocks normal cell‐cycle progress when the mitotic metaphase and anaphase merge, preventing chromosome segregation and tumor‐cell death. However, the exact signaling cascades responsible are unknown.( 7 ) PTX also has the potential to modulate various signaling pathways, such as the mitogen‐activated protein kinase kinase (MEK)–extracellular signal‐regulated kinase kinase (ERK) signaling cascade and a phosphatidylinositol 3′‐kinase (PI3K)–Akt pathway, which are proliferation and cell‐survival pathways.( 8 , 9 , 10 ) Activating these pathways leads to phosphorylated Bc12 antagonist of cell death (BAD) and B‐cell leukemia/lymphoma (Bcl)‐2, and inhibits apoptosis.( 11 ) Amplified PI3K and activated Akt have been found in 30–40% of ovarian carcinomas and could represent the mechanism of drug resistance in patients with ovarian cancer.( 12 )
Chemotherapy‐induced activation of cell‐survival pathways is observed frequently when conventional anticancer drugs are used, and targeting these survival signals will be invaluable in designing rational and novel combination therapies.( 13 ) Recently, several agents designed to inhibit the kinase components of those signaling pathways, including MEK–ERK and PI3K–Akt, were tested clinically to treat melanoma, lung, and breast cancer.( 14 , 15 ) Our study examines whether MEK or PI3K inhibitors enhance the antitumor effect of PTX in ovarian cancer cells.
Materials and Methods
Cell lines and cell cultures. The five human ovarian serous adenocarcinoma cell lines used in the present study (KF, KFTx, SHIN‐3, KOC‐2S, and SK‐OV‐3) were obtained as follows: KF, from Dr Yoshihiro Kikuchi (National Defense Medical College, Tokorozawa, Japan); SHIN‐3, from Dr Yasuhiko Kiyozuka (Nara Medical University, Kashihara, Japan); and KOC‐2S, from Dr Toru Sugiyama (Kurume University, Kurume, Japan). SK‐OV‐3 was obtained from the American Type Culture Collection (Manassas, VA, USA). A PTX‐resistant cell line (KFTx) was established by continuous exposure of KF cells to a stepwise escalating concentration of PTX (Sigma, St Louis, MO, USA) for over 18 months. These cell lines were maintained in RPMI‐1640 medium (Nissui, Tokyo, Japan) with 10% fetal bovine serum, penicillin 100 IU/mL, and streptomycin 50 µm/mL in a humidified atmosphere containing 5% CO2 at 37°C.
Dose–response studies. The sensitivity of the cell lines to PTX was determined by 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐dyphenyltetrazolium bromide (MTT) assay. Briefly, cells were diluted with culture medium to the seeding density of 1–2.5 × 104 cells/mL, plated on 96‐well tissue culture plates at 100 µL/well (Sumitomo Bakelite, Tokyo, Japan), and incubated at 37°C overnight. The next day, the cells were incubated with various concentrations (3–1000 nM) of PTX. After incubation for 72 h, 10 µL MTT solution (5 mg/mL) (Sigma) was added to each well, and the plates were incubated for another 4 h. At the end of that incubation, 100 µL dimethylsulfoxide (Sigma) was added to each well to solubilize the MTT formazan product. Absorbance at 570 nm was measured with a microplate reader (model 550; Bio‐Rad, Richmond, CA, USA). Growth inhibition was calculated as the percentage of viable cells compared with untreated cultures.
Dose–effect analysis. Paclitaxel was combined with the MEK inhibitor PD98059 (PD) (Biomol International, PA, USA) and the PI3K inhibitor LY294002 (LY) (Wako Pure Chemical Industries, Osaka, Japan) at a fixed ratio that spanned the individual IC50 of each drug. The IC50 was determined on the basis of the dose–effect curves, using a standard MTT assay. Median‐effect plot analyses and calculation of the combination index (CI) were analyzed by the method of Chou and Talalay.( 16 ) CalcuSyn software (Biosoft, Freguson, MO, USA) was used to analyze data from the MTT assays in which cells were exposed to agents alone or in combination with anticancer drugs and protein kinase inhibitors. The computer program provides a measure of the combined agents in an additive or synergistic manner. Chou and Talalay defined a parameter, CI, which assesses synergism (CI < 1), additive (CI = 1), or antagonism (CI > 1). The commercial software package was obtained from Calcusyn (Biosoft, Cambridge, UK).
Western blot analysis. Cells were washed twice with phosphate‐buffered saline (PBS) and then lysed in lysis buffer (50 mM Tris‐HCl, 125 mM NaCl, 0.1% Nonidet P (NP)‐40, 5 mM ethylenediaminetetraacetic acid (EDTA), 50 mM NaF, 0.1% phenylmethylsulfonyl fluoride, protease inhibitors [Protease Inhibitor Cocktail Set I; Calbiochem, Germany]). Protein concentrations were measured against a standardized control by using a protein assay kit (Bio‐Rad Laboratories, Hercules, CA, USA). A total of 50 µg protein was separated by electrophoresis on a 10% polyacrylamide gel and transferred to a polyvinylidene difluoride membrane (Millipore, Bedford, MA, USA). The specific antibodies used were rabbit anti‐Akt antibody (1:1000 dilution; Cell Signaling Technology, Beverly, MA, USA), rabbit antiphospho‐Akt (serine 473) antibody (1:1000 dilution; Cell Signaling Technology), rabbit anti‐MEK1 antibody (1:1000 dilution; Cell Signaling Technology), rabbit antiphospho‐MEK1 (threonine 286) antibody (1:500 dilution; Cell Signaling Technology), and mouse anti‐actin antibody (1:1000 dilution; Sigma). These were visualized with secondary antimouse or antirabbit IgG antibody coupled with horseradish peroxidase, using enhanced chemiluminescence according to the manufacturer's recommendation. Immunoblot intensity was quantified using the National Institute of Health (NIH) image program.
Annexin V staining. The annexin V–fluorescein isothiocyanate (FITC) Fluorescence Microscopy Kit I (BD Biosciences Pharmingen, San Diego, CA, USA) was used to assess apoptosis in terms of the externalization of phosphatidylserine residues, according to the specifications of the manufacturer. Briefly, cells were washed twice with cold PBS and once with 1× annexin V binding buffer (10 mmol/L HEPES/NaOH [pH 7.4], 140 mmol/L NaCl, and 2.5 mmol/L CaCl2). The cells were then stained with annexin V–FITC diluted 1:10 in 1× annexin V binding buffer for 15 min at room temperature. Finally, the cells were washed with 1× annexin V binding buffer, then 500 µL 1× annexin V binding buffer was added to each well and the cells were observed under a fluorescence microscope (Olympus, Tokyo, Japan).
DNA laddering. The DNA Laddering Kit (Cayman Chemical Company, Ann Arbor, MI, USA) was used to detect apoptosis according to the specifications of the manufacturer. DNA was isolated from both adherent and suspended cells and subjected to electrophoresis on a 1.8% agarose gel in 89 mM Tris‐HCl, 89 mM boric acid, 2 mM EDTA (pH 8.0) buffer at 50 V for 2 h. A 100‐bp DNA ladder was used as the standard. DNA was visualized by ethidium bromide staining and photographed under ultraviolet illumination.
Ovarian cancer xenograft model. The present study was carried out at the Laboratory Animal Research Center under the control of the animal research committee, in accordance with the Guidelines for Animal Experimentation in the Faculty of Medicine, Tottori University, Yonago, Japan. For these experiments, KOC‐2S or SKOV‐3 cells in log‐phase growth were trypsinized, washed twice with PBS, and centrifuged at 250g. Viable cells were counted, then 2 × 106 viable cells (in 0.5 mL PBS) were injected under aseptic conditions into the peritoneal cavities of female nude mice. Mice were then assigned randomly to one of four groups (10 mice per group) and treatment was started 4 days later as follows. Group 1, intraperitoneal (i.p.) PBS weekly; group 2, i.p. paclitaxel weekly (25 mg/kg per injection); group 3, i.p. PD and LY weekly (7 and 25 mg/kg per injection, respectively) for 4 weeks; and group 4, i.p. PTX with both PD and LY weekly for 4 weeks. Tumors were collected and weighed (five mice per group) on day 28, fixed in 10% neutral buffered formalin (Wako Pure Chemical Industries), and embedded in paraffin for immunohistochemical analysis. Paraffin blocks were sliced in 4‐µm sections and deparaffinized. The expression of phosphorylated (p) Akt and pMEK protein on the tumor tissue sections was detected using the Histofine Simple Stain PO kit (Nichirei, Tokyo, Japan). Slides were counterstained with hematoxylin. The primary antibodies used were anti‐pAKT (dilution 1:100; Cell Signaling Technology) and anti‐pMEK1 (dilution 1:50; Cell Signaling Technology).
Statistical analysis. Statistical analyses were carried out using the GraphPad Prism Version 4 program (GraphPad Software, San Diego, CA, USA). Data are presented as mean ± SD. Means for all data were compared by one‐way analysis of variance with post hoc testing. Survival distributions were calculated using the Kaplan–Meier method, and the significance of apparent differences in survival distribution between groups was tested with log‐rank tests. A P‐value < 0.05 was considered statistically significant.
Results
Paclitaxel activates MEK in ovarian carcinoma cell lines. The effects of PTX on the proliferation of five ovarian cancer cell lines are shown in Fig. 1a. The IC50 to PTX ranged from 22 to 462 nM for those cell lines and, compared with KF cells, the IC50 to PTX was 18‐fold higher for KFTx cells, 5‐fold higher for SK‐OV‐3 cells, 11.2‐fold higher for KOC‐2S cells, and similar to SHIN‐3 cells. The protein expression levels of MEK, pMEK, Akt, and pAkt were confirmed in all cell lines (Fig. 1b). At 3 h after treatment with PTX, pMEK protein expression had increased in all five cell lines (Fig. 2).
Figure 1.
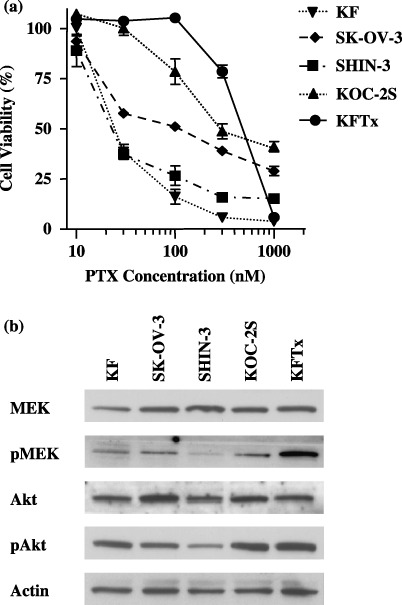
Cytotoxicity of paclitaxel (PTX) in ovarian cancer cell lines and the mitogen‐activated protein kinase kinase (MEK) and phosphatidylinositol 3′‐kinase (PI3K) signaling pathways. (a) Five ovarian cancer cell lines were treated with varying concentrations of PTX, then cell growth inhibition determined using the 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐dyphenyltetrazolium bromide assay. KOC‐2S and KFTx cells are considered to be resistant to PTX. The points represent mean ± SD from six dishes. (b) Protein expression of MEK, phosphorylated (p) MEK, Akt, and pAKT in ovarian cancer cells was determined by western blot analysis. The protein expression levels of MEK, pMEK, Akt, and pAkt were confirmed in all cell lines. The results shown represent duplicate experiments.
Figure 2.

Paclitaxel (PTX) activated the mitogen‐activated protein kinase kinase (MEK) signaling pathway. Phosphorylated (p) MEK was clearly upregulated 3 h after treatment with PTX (the KF, SK‐OV‐3, and SHIN‐3 cell lines at 100 nM, and the KOC2S and KFTx cell lines at 1 µM) in all ovarian cancer cell lines tested. Immunoblot intensity was quantified using the National Institute of Health (NIH) Image program. The results shown represent duplicate experiments. C, control.
Paclitaxel combined with both the MEK and PI3K inhibitors enhances PTX‐induced cell death. We next examined cell proliferation after treating with PTX with or without both PD and LY or each individually. At 3 days after exposure, growth inhibition was measured using the MTT assay. A negligible increase in PTX‐induced cell‐growth inhibition was detected in the presence of either PD or LY in all cell lines (Fig. 3a). Surprisingly, PTX combined with PD and LY substantially suppressed cell growth (P < 0.01) and increased annexin V‐positive cells in all cell lines (P < 0.05) (Fig. 3b). Moreover, 36 h after treating with PTX combined with both PD and LY, the DNA had fragmented in all cells.
Figure 3.

The effects of paclitaxel (PTX) combined with mitogen‐activated protein kinase kinase (MEK) and phosphatidylinositol 3′‐kinase (PI3K) inhibitors on ovarian cancer cells. (a) Five ovarian cancer cell lines were treated with varying concentrations of PTX along with phosphate‐buffered saline, or 25 µM PD98059 (PD) and/or 5 µM LY294002 (LY) for 72 h compared with the control. Cell‐growth inhibition was determined using the 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐dyphenyltetrazolium bromide assay. Cell proliferation was significantly suppressed by PTX combined with MEK and PI3K inhibitors compared with other treatment conditions (P < 0.01). Points represent mean ± SD from six dishes. (b) Cells were treated with 100 nM PTX (the KF, SK‐OV‐3, and SHIN‐3 cell lines) or 300 nM PTX (the KOC‐2S and KFTx cell lines) in the presence or absence of 50 µM PD and/or 10 µM LY for 36 h, then stained with annexin V–fluorescein isothiocyanate. The number of apoptotic cells increased significantly after treating with PTX combined with both the MEK and PI3K inhibitors, compared with the other treatments (P < 0.05). The points represent mean ± SD from three dishes. (Inset) DNA fragmentation was also observed 48 h after treatment with a combination of PTX with both PD and LY in all cell lines. Lane 1, no treatment; lane 2, LY+PD+PTX; and lane 3, 100‐bp DNA ladder as standard. The results shown represent duplicate experiments. (c) The combination effects of PD and LY were synergistic with those of PTX in ovarian cancer cells. PTX was combined with PD and LY at a fixed ratio that spanned the individual IC50 of each drug. Data were analyzed using the method of Chou and Talalay.( 16 ) The results shown represent duplicate experiments. p, phosphorylated.
To confirm the synergistic activity of combining PTX with both PD and LY, CI values were analyzed using the method of Chou and Talalay.( 16 ) All of the CI values at an effective dose of 50, 75, and 90 (effective dose means the percentage inhibition of cell growth using the drug combinations in the actual experiment) were less than 0.9 for the five cell lines (Fig. 3c). The CI values were more than 0.9 for all of the five cell lines when PTX was combined with PD or LY (data not shown). Thus, when PTX was combined with both PD and LY, synergistic effects were found in both PTX‐sensitive (KF, SK‐OV‐3, and SHIN‐3) and PTX‐resistant (KOC‐2S and KFTx) cell lines.
Mitogen‐activated protein kinase kinase inhibitor combined with PTX downregulates the MEK‐signaling pathway and upregulates the Akt pathway. PD98059 combined with PTX decreased the PTX‐upregulated protein expression levels of pMEK (Fig. 4). The protein expression level of pAkt was upregulated after treating with PD with or without PTX in KF and KOC‐2S cells. PTX combined with PD and LY downregulated the protein expression levels of both pMEK and pAkt. Similar results were obtained in the other three cell lines.
Figure 4.
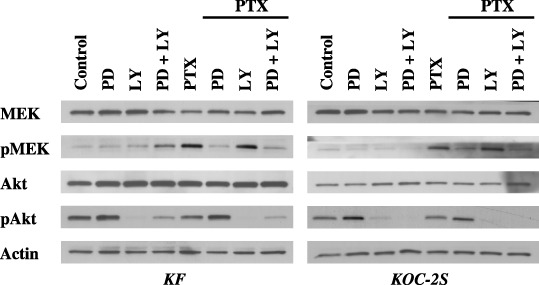
The mitogen‐activated protein kinase kinase (MEK) inhibitor combined with paclitaxel (PTX) upregulated the Akt‐signaling pathway. Each of the cell lines was treated with 100 nM or 1 µM PTX as well as with phosphate‐buffered saline or 25 µM PD98059 (PD) and/or 5 µM LY294002 (LY) for 3 h. The cells were then collected and the protein expression levels of MEK, phosphorylated (p) MEK, Akt, and pAKT were tested by western blotting. PD prevents the activation of MEK by Raf or MEK kinase by binding to the inactive form of MEK, and does not inhibit the phosphorylation of other Raf or MEK kinase substrates. After treatment with PD combined with PTX, the protein expression levels of pMEK were downregulated; however, those of pAkt were upregulated. The results shown represent duplicate experiments.
Paclitaxel combined with PD and LY prolongs survival in mice injected with ovarian cancer cells. Immunohistochemical and western blot analysis of tumor tissues verified that PD and LY downregulated the MEK and Akt signaling pathways in tumor cells. As expected, pMEK and pAkt proteins were effectively downregulated only in tumors from mice treated with both PD and LY (Fig. 5a,b). The results of PTX combined with PD and LY on tumor growth are shown in Figure 5c. In nude mice bearing KOC‐2S, the mean tumor weight of peritoneally disseminated tumors in the group treated with PTX combined with PD and LY (0.53 ± 0.22 g) was significantly lower than that of the group treated with PBS (3.71 ± 0.56 g), PD and LY (3.32 ± 0.52 g), or PTX alone (2.62 ± 0.56 g) (P < 0.05). Similarly, in nude mice bearing SK‐OV‐3, the mean tumor weight of peritoneally disseminated tumors in the group treated with PTX combined with PD and LY (0.32 ± 0.25 g) was significantly lower than that treated with PBS (2.61 ± 0.82 g), PD and LY (2.47 ± 0.53 g), or PTX alone (1.47 ± 0.45 g) (P < 0.05). Mice treated with PTX combined with PD and LY survived significantly longer than those treated with PBS, PD and LY, or PTX alone (P < 0.005) (Fig. 5d). The median survival times were 38 days for PTX treatment, 52 days for PTX with PD and LY treatment, and 30 days for PBS treatment. These findings indicate that PTX combined with both PD and LY resulted in prolonged survival in nude mice bearing KOC‐2S ovarian cancer cells.
Figure 5.
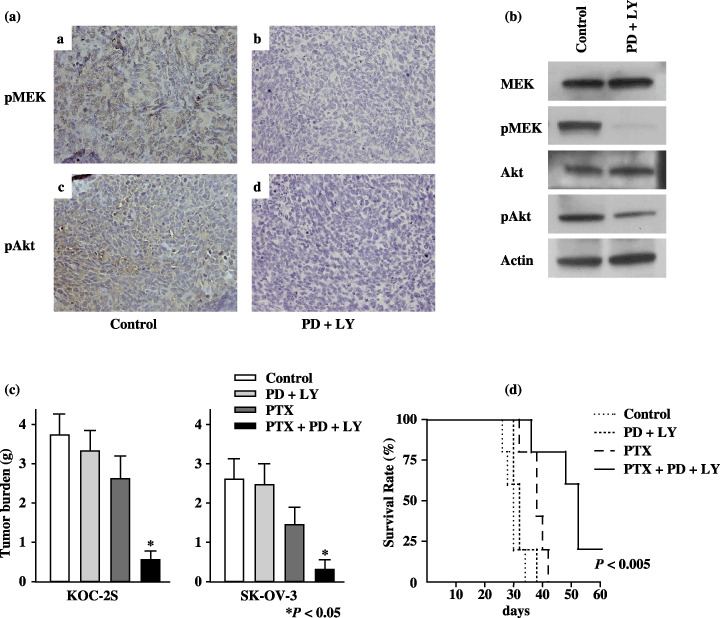
Treatment with paclitaxel (PTX) combined with both mitogen‐activated protein kinase kinase (MEK) and phosphatidylinositol 3′‐kinase (PI3K) inhibitors prolongs survival in mice with implanted KOC‐2S cells. (a) Immunohistochemical stains of representative tumor tissue samples from mice implanted with KOC‐2S cells and treated with 7 mg/kg PD98059 (PD) and 25 mg/kg LY294002 (LY). The brown staining at the upper and lower left indicates the phosphorylated (p) MEK and pAkt proteins, respectively. The results shown represent duplicate experiments. (b) The levels of pMEK and pAkt protein were determined 24 h after intravenous treatment with phosphate‐buffered saline (PBS), or PD and LY by western blotting. Those proteins were effectively downregulated only in tumors from mice treated with PD and LY. The results shown represent duplicate experiments. (c) Female nude mice (five per group) were given an intraperitoneal (i.p.) injection of 2 × 106 KOC‐2S cells or SK‐OV‐3 cells followed by weekly i.p. injections of 25 mg/kg PTX, and/or 7 mg/kg PD and 25 mg/kg LY for 4 weeks. Tumors were collected and weighed on day 28. In KOC‐2S cell‐ and SK‐OV‐3 cell‐inoculated mice, the weight of the peritoneally disseminated tumors was significantly lower in the mice treated with PTX combined with PD and LY than with the other treatments (P < 0.05). (d) Female nude mice (5 per group) were given an intraperitoneal (i.p.) injection of 2 × 106 KOC‐2S cells followed by weekly i.p. injections of 25 mg/kg PTX, and/or 7 mg/kg PD and 25 mg/kg LY for 4 weeks. Treatment with PTX, PD, and LY prolonged survival relative to treatment with PBS, PD and LY, or PTX (P < 0.005).
Discussion
Our study of the cytotoxicity of PTX in ovarian cancer cells revealed that the protein expression levels of pMEK increased in all five cell lines after PTX treatment. Several other studies have shown that the MEK–ERK pathway was activated after treatment with PTX and other microtubule‐interacting agents in different cell models.( 17 , 18 )
The MEK–ERK signaling cascade has a role in many cellular programs, such as proliferation, differentiation, and movement, and is activated by a variety of receptor tyrosine kinases.( 19 ) Activation of the MEK–ERK pathway during G1 phase of the cell cycle has been well documented.( 20 ) This pathway is also thought necessary for mitosis, and activated ERK localizes to kinetochores, asters, and the midbody during mitosis.( 21 ) Approximately 40% of ERK also colocalizes to microtubules.( 22 ) As a result, microtubule damage by PTX may alter protein expression associated with microtubules, such as MEK and ERK. Moreover, the effects of combining MEK–ERK pathway inhibitors and PTX treatment have been evaluated in several types of human cancer cell lines.( 23 , 24 , 25 , 26 ) McDaid and Horwitz reported that the degree of activation of the MEK–ERK pathway governed whether the combined effects of PTX and MEK inhibition were additive, synergistic, or antagonistic.( 18 ) However, in the present study, we found that the additive effect of MEK inhibitor combined with PTX in all of the ovarian cancer cell lines tested expressed a range of pMEK protein expression levels after PTX treatment. Enhanced PTX‐induced cell death in the presence of MEK inhibitor may relate to suppression of the survival‐signaling function of the MEK–ERK pathway, although the exact mechanism is unclear.( 27 )
In human cancers, activation of the P13K and Akt pathways is thought to modulate sensitivity to PTX.( 10 , 28 , 29 ) Besides its role in metabolic regulation, the PI3K pathway helps regulate tumor progression and cell death by multiple mechanisms, including phosphorylation of murine double minute (MDM)2, mammalian target of rapamycin (mTOR), glycogen synthase kinase 3β, nuclear factor‐κB, forkhead in human rhabdomyosarcoma (FKHR), and BAD.( 28 ) In ovarian cancer, gene amplification of AKT2, the PI3K catalytic subunit p110, or the regulatory p85 subunit has been reported,( 29 ) and overexpression of activated Akt was shown to decrease PTX‐induced apoptosis.( 9 , 30 ) Several reports also showed that inhibiting activated PI3K enhances PTX‐induced apoptosis in human cancer cells, including ovarian cancer.( 31 ) In our study, we observed the additive effect of PI3K inhibitor and PTX in all ovarian cancer cell lines studied, regardless of the pAkt expression levels. Interestingly, upregulation of pAkt protein expression levels was seen after treating the cells with MEK inhibitor and PTX. Overexpression of activated Akt may decrease the sensitivity to the combination of MEK inhibitor and PTX. Furthermore, our study is the first to show clearly that PTX combined with both MEK and Akt inhibitors delivers a synergistic effect on PTX‐induced cytotoxicity in both PTX‐sensitive and ‐resistant cell lines. A recent study showed that inhibiting MEK and ERK upregulates Akt kinase activity.( 32 ) Inhibiting MEK pharmacologically not only activates Akt, but also prolongs cell survival in normal cells.( 32 ) These findings suggest that simultaneously suppressing the MEK–ERK and PI3K–Akt pathways could dramatically intensify PTX‐induced apoptosis in ovarian cancer cells. Targeting these two pathways may be important in treating ovarian cancer.
Finally, we confirmed the importance of the MEK–ERK and PI3K–Akt pathways in PTX therapy in vivo in an ovarian cancer xenograft model. Combining MEK inhibitor, PI3K inhibitor, and PTX prolonged the survival of these mice compared with those treated with MEK and PI3K inhibitors or PTX alone. The present study provides clear evidence of the downregulation of both the MEK and PI3K pathways, which enhances the effects of PTX treatment in vitro and in vivo.
In summary, our study shows that MEK inhibitor combined with PTX upregulates protein expression of pAkt, a kinase critical to cell survival. Furthermore, simultaneously inhibiting MEK and PI3K dramatically enhances PTX‐induced apoptosis in vitro, and this combined treatment prolongs the survival of nude mice with ovarian cancer cells. We hope that this combination therapy will improve the survival of patients with advanced ovarian cancer.
Acknowledgments
This work was supported by Grant‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (17244120, H. Itamochi).
References
- 1. Sankaranarayanan R, Ferlay J. Worldwide burden of gynaecological cancer: the size of the problem. Best Pract Res Clin Obstet Gynaecol 2006; 20: 207–25. [DOI] [PubMed] [Google Scholar]
- 2. Bristow RE, Tomacruz RS, Armstrong DK et al . Survival effect of maximal cytoreductive surgery for advanced ovarian carcinoma during the platinum era: a meta‐analysis. J Clin Oncol 2002; 20: 1248–59. [DOI] [PubMed] [Google Scholar]
- 3. McGuire WP, Hoskins WJ, Brady MF et al . Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage III and stage IV ovarian cancer. N Engl J Med 1996; 334: 1–6. [DOI] [PubMed] [Google Scholar]
- 4. Piccart MJ, Bertelsen K, James K et al . Randomized intergroup trial of cisplatin‐paclitaxel versus cisplatin‐cyclophosphamide in women with advanced epithelial ovarian cancer: 3 years results. J Nat Cancer Inst 2000; 92: 699–708. [DOI] [PubMed] [Google Scholar]
- 5. Ahmedin J, Rebecca S, Elizabeth W et al . Cancer statistics, 2007. CA Cancer J Clin 2007; 57: 43–66. 17237035 [Google Scholar]
- 6. Du Bois A, Pfisterer J. Future options for first‐line therapy of advanced ovarian cancer. Int J Gynecol Cancer 2005; 15: 42–50. [DOI] [PubMed] [Google Scholar]
- 7. Rowinsky EK, Donehower RC, Jones RJ, Tucker RW. Microtubule changes and cytotoxicity in leukemic cell lines treated with taxol. Cancer Res 1988; 48: 4093–100. [PubMed] [Google Scholar]
- 8. Sunters A, Madureira PA, Pomeranz KM et al . Paclitaxel‐induced nuclear translocation of FOXO3a in breast cancer cells is mediated by c‐Jun NH2‐terminal kinase and Akt. Cancer Res 2006; 66: 212–20. [DOI] [PubMed] [Google Scholar]
- 9. Qiu LW, Jiang Q et al . Targeted inhibition of transient activation of the EGFR‐mediated cell survival pathway enhances paclitaxel‐induced ovarian cancer cell death. Int J Oncol 2005; 27: 1441–8. [PubMed] [Google Scholar]
- 10. Mabuchi S, Ohmichi M, Kimura A et al . Inhibition of phosphorylation of BAD and Raf‐1 by Akt sensitizes human ovarian cancer cells to paclitaxel. J Biol Chem 2002; 277: 33 490–500. [DOI] [PubMed] [Google Scholar]
- 11. Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell 2002; 108: 153–64. [DOI] [PubMed] [Google Scholar]
- 12. Bellacosa A, De Feo D, Godwin AK et al . Molecular alterations of the AKT2 oncogene in ovarian and breast carcinomas. Int J Cancer 1995; 64: 280–5. [DOI] [PubMed] [Google Scholar]
- 13. Liu SQ, Yu JP, Yu HG, Lv P, Chen HL. Activation of Akt and ERK signaling pathways induced by etoposide confer chemoresistance in gastric cancer cells. Dig Liver Dis 2006; 38: 310–18. [DOI] [PubMed] [Google Scholar]
- 14. Downward J. Cancer biology: signatures guide drug choice. Nature 2006; 439: 274–5. [DOI] [PubMed] [Google Scholar]
- 15. Powis G, Ihle N, Kirkpatrick DL. Practicalities of drugging the phosphatidylinositol‐3‐kinase/Akt cell survival signaling pathway. Clin Cancer Res 2006; 12: 2964–6. [DOI] [PubMed] [Google Scholar]
- 16. Chou TC, Talalay P. Quantitative analysis of dose–effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 1984; 22: 27–55. [DOI] [PubMed] [Google Scholar]
- 17. Shtil AA, Mandlekar S, Yu R et al . Differential regulation of mitogen‐activated protein kinases by microtubule‐binding agents in human breast cancer cells. Oncogene 1999; 18: 377–84. [DOI] [PubMed] [Google Scholar]
- 18. McDaid HM, Horwitz SB. Selective potentiation of paclitaxel (taxol)‐induced cell death by mitogen‐activated protein kinase kinase inhibition in human cancer cell lines. Mol Pharmacol 2001; 60: 290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McCubrey JA, Steelman LS, Abrams SL et al . Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Adv Enzyme Regul 2006; 46: 249–79. [DOI] [PubMed] [Google Scholar]
- 20. Ussar S, Voss T. MEK1 and MEK2, different regulators of the G1/S transition. J Biol Chem 2004; 279: 43 861–9. [DOI] [PubMed] [Google Scholar]
- 21. Zecevic M, Catling AD, Eblen ST et al . Active MAP kinase in mitosis: localization at kinetochores and association with the motor protein CENP‐E. J Cell Biol 1998; 142: 1547–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reszka AA, Seger R, Diltz CD, Krebs EG, Fischer EH. Association of mitogen‐activated protein kinase with the microtubule cytoskeleton. Proc Natl Acad Sci USA 1995; 92: 8881–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brognard J, Dennis PA. Variable apoptotic response of NSCLC cells to inhibition of the MEK/ERK pathway by small molecules or dominant negative mutants. Cell Deaths Differ 2002; 9: 893–904. [DOI] [PubMed] [Google Scholar]
- 24. Hu Y, Bally M, Dragowska WH, Mayer L. Inhibition of mitogen‐activated protein kinase/extracellular signal‐regulated kinase kinase enhances chemotherapeutic effects on H460 human non‐small cell lung cancer cells through activation of apoptosis. Mol Cancer Ther 2003; 2: 641–9. [PubMed] [Google Scholar]
- 25. Mackeigan JP, Collins TS, Ting JPY. MEK inhibition enhances paclitaxel‐induced tumor apoptosis. J Biol Chem 2000; 275: 38 953–6. [DOI] [PubMed] [Google Scholar]
- 26. McDaid HM, Lopez‐Barcons L, Grossman A et al . Enhancement of the therapeutic efficacy of taxol by the mitogen‐activated protein kinase kinase inhibitor CI‐1040 in nude mice bearing human heterotransplants. Cancer Res 2005; 65: 2854–60. [DOI] [PubMed] [Google Scholar]
- 27. Orr GA, Verdier‐Pinard P, McDaid H, Horwitz SB. Mechanisms of taxol resistance related to microtubules. Oncogene 2003; 22: 7280–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vivanco I, Sawyers CL. The phosphatidylinositol 3‐kinase AKT pathway in human cancer. Nat Rev Cancer 2002; 2: 489–501. [DOI] [PubMed] [Google Scholar]
- 29. Lee JT Jr, Steelman LS, McCubrey JA. Phosphatidylinositol 3′‐kinase activation leads to multidrug resistance protein‐1 expression and subsequent chemoresistance in advanced prostate cancer cells. Cancer Res 2004; 64: 8397–404. [DOI] [PubMed] [Google Scholar]
- 30. Del Bufalo D, Di Castro V, Biroccio A et al . Endothelin‐1 protects ovarian carcinoma cells against paclitaxel‐induced apoptosis: requirement for Akt activation. Mol Pharmacol 2002; 61: 524–32. [DOI] [PubMed] [Google Scholar]
- 31. Hu L, Hofmann J, Lu Y, Mills GB, Jaffe RB. Inhibition of phosphatidylinositol 3′‐kinase increases efficacy of paclitaxel in in vitro and in vivo ovarian cancer models. Cancer Res 2002; 62: 1087–92. [PubMed] [Google Scholar]
- 32. Sinha D, Bannergee S, Schwartz JH, Lieberthal W, Levine JS. Inhibition of ligand‐independent ERK1/2 activity in kidney proximal tubular cells deprived of soluble survival factors up‐regulates Akt and prevents apoptosis. J Biol Chem 2004; 279: 10 962–72. [DOI] [PubMed] [Google Scholar]