

Abstract
DIXDC1 is the human homolog of Ccd1, a recently identified DIX domain containing protein in zebrafish. It is a positive regulator in the Wnt signaling pathway functioning downstream of Wnt and upstream of Axin. Since Wnt pathway activation is correlated with human colon cancer formation and progression, the biological role of DIXDC1 in human colon cancer was examined. In the current study, up‐regulation of DIXDC1 protein was detected in human colorectal adenocarcinoma tissues and was found to be correlated well with high cell proliferation index. Ectopic over‐expression of DIXDC1 resulted in increased cell proliferation in vitro and accelerated tumorigenesis on nude mice in vivo. We also showed that DIXDC1 promoted G0/G1 to S phase transition concomitantly with up‐regulation of cyclin D1 and down‐regulation of p21 protein. DIXDC1 over‐expression cells showed activation of the PI3K/AKT pathway. Both siRNA knockdown of DIXDC1 and blocking the PI3K pathway using a specific inhibitor caused G1/S phase arrest, as well as down‐regulation of cyclin D1 and up‐regulation of p21 in DIXDC1 over‐expression colon cancer cells. Collectively, this study demonstrates that over‐expression of DIXDC1 might target p21 and cyclin D1 to promote colon cancer cell proliferation and tumorigenesis at least partially through activation of the PI3K/Akt pathway. (Cancer Sci 2009; 100: 1801–1808)
DIXDC1 is the human homolog of Ccd1 (Coiled‐coil‐DIX1), a third type of DIX (Dishevelled‐Axin) domain‐possessing protein and a positive regulator in the Wnt signaling pathway during zebrafish neural patterning.( 1 , 2 , 3 ) DIX domains of Dvl and Axin, two major Wnt downstream mediators, are involved in homomeric or heteromeric protein interactions and are essential for the signal transduction.( 4 ) Human DIXDC1 was recently isolated as an Axin2 C‐terminus binding protein.( 5 ) Since frame‐shift mutations in the Axin2 gene resulting in complete deletion of the C‐terminal DIX domain of the Axin2 protein were detected in more than 25% of colorectal cancers with microsatellite instability,( 6 ) DIXDC1 was postulated to be associated with colorectal cancer tumorigenesis.
In the current study, we aimed to investigate the biological role of DIXDC1 in human colon cancer formation and progression. We provide evidence that up‐regulation of DIXDC1 was observed in colorectal adenocarcinoma tissues and was correlated with high proliferation index. Over‐expression of DIXDC1 was found to enhance cell proliferation by promoting G0/G1 to S phase transition in colorectal cancer cells through the activation of the PI3K pathway.
Materials and Methods
Cells and reagents. DLD1, LS174T, and 293T cell lines were obtained from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). Rabbit anti‐DIXDC1( 5 ) antibody was a kind gift from Dr Wan‐Guo Liu (Louisiana State University, Baton Rouge, New Orleans, LA, USA). Mouse anti‐Ki‐67 antibody and supervision antirabbit or antimouse detection reagent (HRP) were purchased from Changdao Biotech (Shanghai, China). Rabbit anti‐Akt, anti‐phospho‐Akt antibodies, and PI3K inhibitor LY294002 were purchased from Cell Signaling Technology (Danvers, MA, USA). Mouse anti‐cyclin D1 and anti‐β‐catenin antibodies were purchased from BD Biotechnology (BD Biotechnology, Franklin Lakes, NJ, USA). Mouse anti‐p21 antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Mouse anti‐β‐actin antibody was from Sigma (St. Louis, MO, USA). Lipofectamine 2000 was purchased from Invitrogen (Carlsbad, CA, USA). ECL Western Blotting Substrate System was purchased from Pierce (Rockford, IL, USA). HiPerFect Transfection Reagent was purchased from Qiagen (Duesseldorf, Germany).
Tissue samples. All cases in this study were retrieved from the files of Department of Pathology, Hua Shan Hospital (Shanghai, China). The study was approved by the institutional review board at Shanghai Hua Shan Hospital and all patients provided consent. Cases included were normal colon tissue (n = 20), colon adenoma (n = 20), and colon adenocarcinoma (n = 84). None of these cancer cases were previously treated with chemotherapy. All cases were diagnosed by two experienced pathologists without discrepancy.
Immunohistochemical staining. Serial sections of formalin‐fixed and paraffin‐embedded tissues were subjected to antigen retrieval by microwaving in 0.1 M citrate solution (pH 6.0) for 10 min, then incubated with the rabbit anti‐DIXDC1 antibody or mouse anti‐Ki67 antibody at 4°C overnight. After incubation in supervision antirabbit or antimouse detection reagent for 30 min at room temperature, the sections were developed in 0.05% diaminobenzidine containing 0.01% hydrogen peroxidase.
Stable transfection. DLD1 cells were transfected with pcDNA3.0‐DIXDC1 containing full length DIXDC1 cDNA( 5 ) (kindly provided by Dr Wan‐Guo Liu of Louisiana State University) or pcDNA 3.0 empty vector with Lipofectamine 2000 according to the manufacturer's instructions. Cells were 1:10 passaged at 48 h post‐transfection and selected in growth medium containing G418 (900 µg/mL) for 30 days. A single surviving clone was isolated by ring cloning and expanded into a stable cell line.
Co‐immunoprecipitation and western blotting. Cells were lysed in lysis buffer (50 mM Tris‐HCl [pH 8.0], 150 mM NaCl, 15 mM MgCl2, 5 mM EDTA, and 1% NP‐40 with Protease Inhibitor Cocktail [Roche, Mennheim, Switzerland]). Cell extracts were clarified by centrifugation. For immunoprecipitation, cell lysates were precleared with protein G‐agarose beads (Roche) for 2 h at 4°C, then incubated with 2‐µg DIXDC1 antibody and beads for 4 h at 4°C. The immunocomplexes were pelleted and washed three times with the lysis buffer. The immunocomplexes and cell lysate protein were separated on 10% SDS‐PAGE gel and blotted on polyvinylidene fluoride membrane. The expression of DIXDC1, β‐catenin, p21, cyclin D1, Akt, phosphor‐Akt, and β‐actin protein were detected using antibodies mentioned in ‘Cells and reagents’. The protein band intensities were measured using a FluroChem8800 imaging system (Alpha Innotech, San Leandro, CA, USA).
Serum starvation and flow cytometry. Cells at 80–90% confluence were incubated for 36 h in the RPMI‐1640 medium containing 0.5% FBS, then cultured back in RPMI‐1640 medium with 10% FBS for 12 h. Cells were harvested and fixed in citric acid buffer for 30 min. After centrifugation, the cell pellets were re‐suspended and treated in phosphate buffer solution (PBS) with 100 mg/L RNase and 10 mg/L propidium iodide for 20 min. Apoptotic cell fraction and cell cycle distribution were analyzed by FACScan cytometry (Becton‐Dickinson, San Jose, CA, USA).
In vitro cell proliferation assay. All proliferation assays were carried out in sextuplet wells in parallel 96‐well microtiter plates. Cells were seeded at 5 × 103 per well. MTT (3‐[4,5‐dimethylthiazol‐2‐yl]‐2,5‐diphenyltetrazolium bromide) assay was used to evaluate cell proliferation viability as previously described.( 7 )
Tumorigenicity assay in nude mice. Male BALB/C nude mice (6 weeks old) were obtained from the animal facilities of the Chinese Academy of Sciences. The animal experiments were performed in accordance with the institutional animal welfare guidelines. The mice were randomized into two groups (six mice per group) for inoculation with DLD1‐pcDNA3.0 or DLD1‐DIXDC1‐1 cells. Mice were injected subcutaneously with 1 × 107 cells in 200 µL PBS into the right foreleg. One week after injection, the size of tumor was determined by measurement of the subcutaneous tumor mass with a caliper every 4 days. Five weeks after injection, mice were sacrificed. Tumors were weighed and tumor volumes were calculated according to the formula length × width × height × 0.5236.( 8 , 9 )
Reporter assays. All constructs used in the luciferase assay were kind gifts from Dr Wan‐Guo Liu (Louisiana State University). 293T cells were transfected using LipofectAMINE 2000 in 24‐well plates with 0.1 µg per well of TOPFlash or FOPFlash luciferaser reporter together with 0.1 µg pcDNA3.0 or pcDNA3.0‐DIXDC1 plasmid. Cell extracts were prepared at 24 h after transfection by a detergent lysis method (Promega, Madison, WI, USA). Transfection efficiencies were normalized by pRL‐TK reporter activity. Reporter activities (mean ± SD) are presented as the fold increases of TOPFlash activity from the cells transfected with empty vector.
Short‐interference RNA. AllStars Negative Control siRNA (cat. no. 1027280) and DIXDC1 siRNA were obtained from Qiagen (cat. no. 1027020). The sequences of siRNA duplex targeting DIXDC1 are: sense, 5′‐r (AUGCCUUGCAGCAGAGAU)dTdT‐3′; antisense, 5′‐r (AUCUCUGCUGCAAGGCAU)dCdC‐3′. LS174T cells were transfected at ~30% confluence. Transfection was performed with HiPerFect Transfection Reagent (Qiagen). Cells transfected with DIXDC1 siRNA or control siRNA were subjected to western blot analysis and proliferation assay.
Statistical analysis. All values are expressed as mean ± SD. The Student's t‐ test, χ2 ‐test, and Kendall's rank correlation test were used to evaluate the experimental data. Differences were considered to be significant at P < 0.05.
Results
Overexpression of DIXDC1 enhances human colon cell proliferation and tumorigenesis. To evaluate the biological role of DIXDC1 in the proliferation of human colon cancer cells, stable DIXDC1 transfected clones were made. We chose DLD1 for the study because this cell line was detected to have relatively low endogenous DIXDC1 protein expression (data not shown; available upon request). As shown in Figure 1(a), two stable DIXDC1 transfectants, namely DLD1‐DIXDC1‐1 and DLD1‐DIXDC1‐3, were detected to have increased levels of DIXDC1 protein compared with the pcDNA3.0 transfectant. These two clones were selected to be used for the following experiments.
Figure 1.
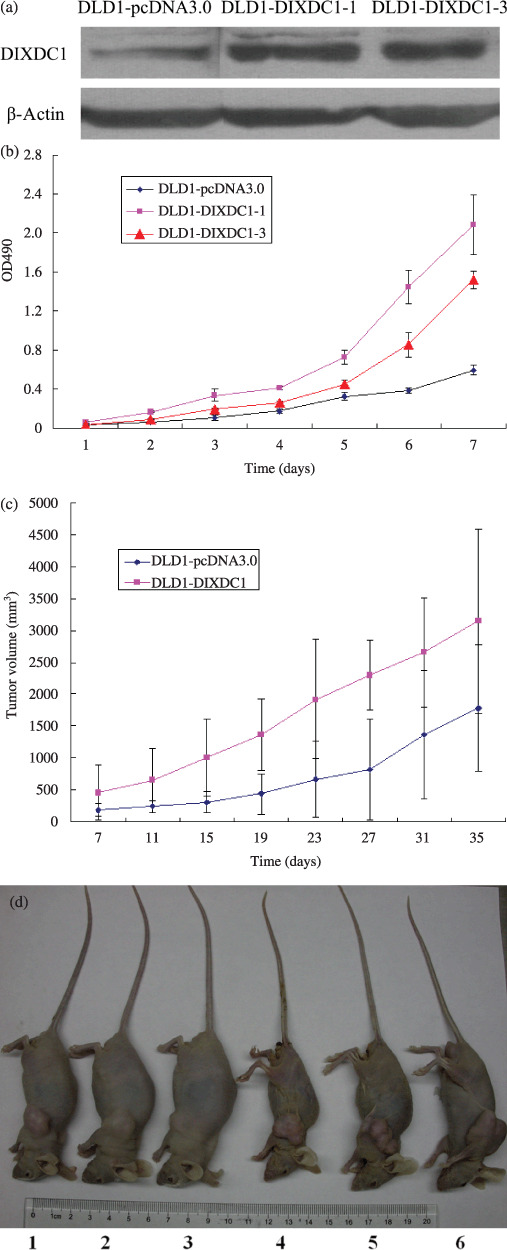
Effects of DIXDC1 on cell proliferation and tumorigenesis of DLD1 cells. (a) Photograph of a representative western blot analysis of DIXDC1 protein expression in pcDNA3.0 and pcDNA3.0‐DIXDC1‐transfected DLD1 cells. Protein levels were normalized to β‐actin. (b) Detection of cell proliferation by MTT assay as described in ‘Materials and Methods’. Columns, mean of the six experiments; bars, SD. (c) DIXDC1 promoted tumorigenecity in nude mice. (d) Photograph of representative mice injected with DLD1‐pcDNA3.0 cells (1–3) and DLD1‐DIXDC1‐1 cells (4–6).
Compared with the DLD1‐pcDNA3.0 cells, both DLD1‐DIXDC1‐1 and DLD1‐DIXDC1‐3 cells showed a significantly increased rate of cell proliferation (Fig. 1b), indicating that DIXDC1 can promote DLD1 cell proliferation in vitro. To confirm this effect in vivo, we performed tumorigenesis assays. Five weeks after injection, all mice had subcutaneous tumors. As shown in Figure 1(c), tumors in the DLD1‐DIXDC1‐1 inoculating group grew much faster than those in the DLD1‐pcDNA3.0 inoculating group. The average final tumor volume in DLD1‐pcDNA3.0 group was 1.78 ± 0.99 cm3, compared with 3.15 ± 1.44 cm3 in the DLD1‐DIXDC1‐1 inoculating group. In addition to the difference of tumor volume, we also noticed that the mice injected with DLD1‐DIXDC1‐1 cells displayed obvious cachexia compared with the mice receiving DLD1‐pcDNA3.0 (Fig. 1d). The final mice weight in the DLD1‐pcDNA3.0 injecting group was 22.6 ± 4.4 g, whereas that in the DLD1‐DIXDC1‐1 injecting group was 18.7 ± 3.5 g. Difference in the final tumor volume between these two groups was significant (t‐test, P < 0.05). These results indicated that DIXDC1 can promote cell proliferation in vitro and tumorigenesis in vivo.
Up‐regulation of DIXDC1 protein in human colorectal cancer tissues and its correlation with high proliferation index. To confirm the cell proliferating effect of DIXDC1 in human colon cancer cells in vivo, we performed immunohistochemical assays to analyze DIXDC1 and Ki67 expression on human colon cancer and matched normal tissues. DIXDC1 protein was detected mainly in the cytoplasm of cancer cells. Positive DIXDC1 staining was observed in tumor cells in 69 of 84 (82.1%) cases of colon adenocarcinoma, and four of 20 (20%) cases of adenoma. None of the normal colon epithelial cells showed positive DIXDC1 staining (Fig. 2a,b). Statistical analysis revealed there was a significant correlation of DIXDC1 activity with the human colon cancer formation (χ2 ‐test, P < 0.001).
Figure 2.
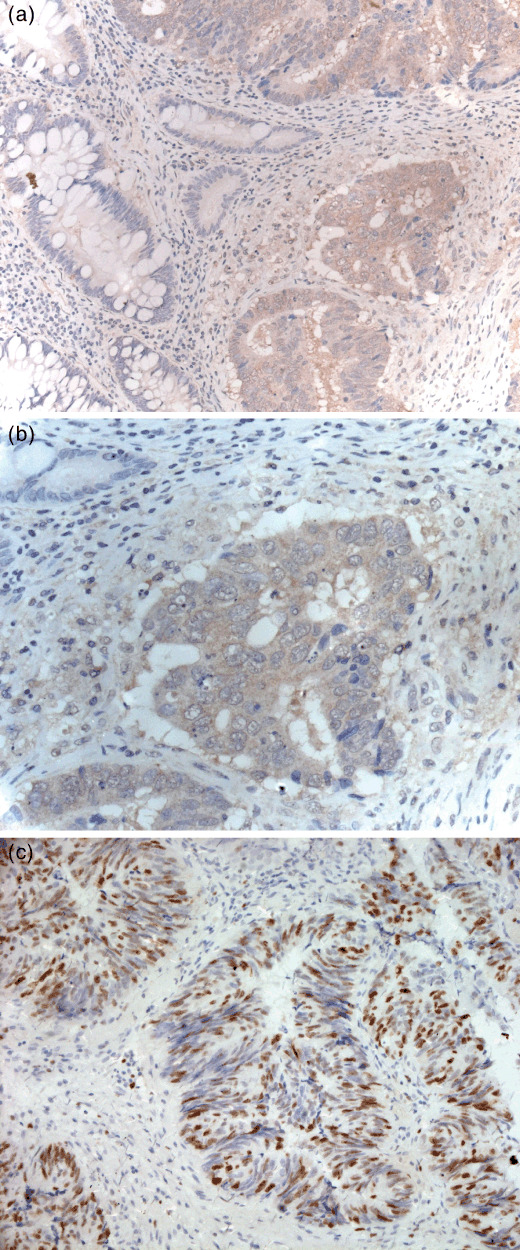
Up‐regulation of DIXDC1 in human colon cancer tissues. Immunostaining with anti‐DIXDC1 antibody was performed on human colon cancer and matched normal tissues. Representative photograph of positive DIXDC1 immunostaining (a) and positive Ki‐67 immunostaining (c) in colon cancer cells. (b) High power view of (a).
In determining the correlation of DIXDC1 expression with cell proliferating index,( 10 , 11 ) the Ki‐67 immunoreactivity of the cases were semi‐quantitatively scored as negative or occasional positive cells, 0; <25% cells positive, 1+; 26–50% cells positive, 2+; 51–75% cells positive, 3+; 76–100% cells positive, 4+ (Fig. 2c), and the intensity of DIXDC1 staining was graded as negative (0–), mild (1+), moderate (2+), and intense (3+). Statistical analysis revealed that there was a correlation of DIXDC1 expression with Ki67 index (Kendall's rank correlation test, P < 0.05; Table 1).
Table 1.
Correlation of DIXDC1 expression with Ki67 index in 84 cases of colon cancer
| DIXDC1 | Ki67 | ||||
|---|---|---|---|---|---|
| – | + | + + | + + + | + + + + | |
| – | 0 | 10 | 5 | 0 | 0 |
| + | 0 | 9 | 16 | 0 | 0 |
| + + | 0 | 0 | 11 | 12 | 4 |
| + + + | 0 | 1 | 2 | 8 | 6 |
Kendall's rank correlation, P < 0.05.
DIXDC1 promoted G0/G1 to S phase transition: up‐regulation of cyclin D1 and decrease of p21 were involved. To explore the possible roles of DIXDC1 in controlling colon cancer cell proliferation, we measured the cell cycle distribution and apoptotic cell fraction in DIXDC1‐transfected cells by flow cytometry. The percentage of cells re‐entrying into S‐phase after serum starvation in DLD1‐DIXDC1‐1 was 43.32% ± 1.06, which was significantly higher than that in DLD1‐pcDNA3.0 cells (23.98% ± 1.51) (Fig. 3a,b, P < 0.01). However, no significant difference was found in the apoptotic cell fraction between DLD1‐pcDNA3.0 and DLD1‐DIXDC1‐1 cells (Fig. 3c). These data indicated that DIXDC1 might promote colon cancer cell growth through accelerating the G0/G1 to S phase transition in the cell cycle.
Figure 3.
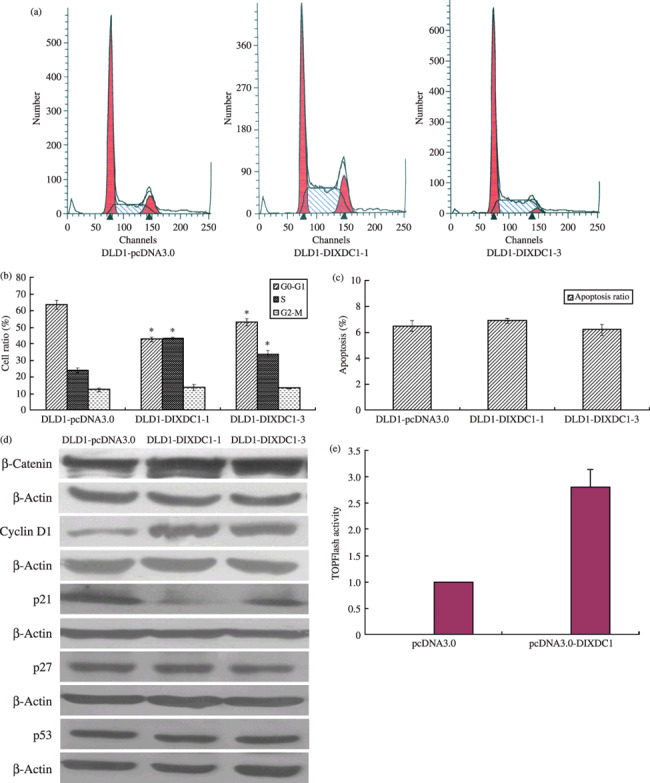
Effects of DIXDC1 on cell cycle progression and cell cycle regulators expression in DLD1 cells. (a) Representative photograph of FACS analysis of cell cycle distribution of DLD1‐pcDNA3.0, DLD1‐DIXDC1‐1, and DLD1‐DIXDC1‐3 cells. (b) Percentage of cells in the G0/G1, S, and G2/M phases were counted. (c) Overexpression of DIXDC1 has no effect on apoptosis of DLD1 cells. Columns, mean of three independent experiments; bars, SD; *P < 0.01. (d) Overexpression of DIXDC1 up‐regulated cyclin D1 and β‐catenin, but down‐regulated p21 protein in DLD1 cells. (e) DIXDC1 increased the expression of the Wnt target gene. 293T cells were transfected with TOPFlash together with DIXDC1 or empty pcDNA3.0 vector. Luciferase activity was assayed as described in ‘Materials and Methods’. Columns, mean of three independent experiments; bars, SD.
To identify molecules that are regulated by DIXDC1 and are responsible for the cell proliferative effect caused by DIXDC1 in DLD1 cells, the expressions of cyclin‐dependent kinase or inhibitors were detected using western blotting. Immunoblotting analysis demonstrated that DLD1‐DIXDC1 cells contained a greater amount of cyclin D1 and lower amount of p21 protein, compared with DLD1‐pcDNA3.0 cells (Fig. 3d). No obvious difference was detected in the amount of p27 and p53 protein between DLD1‐pcDNA3.0 and DLD1‐DIXDC1‐1 cells (Fig. 3d). These data demonstrated that up‐regulation of cyclin D1 and down‐regulation of p21 might contribute to the DIXDC1‐induced G0/G1 to S phase cell cycle progression.
β‐Catenin, a key binding partner of the T‐cell factor/lymphoid‐enhancer factor (TCF/LEF) family,( 12 , 13 ) was also found to be up‐regulated in DIXDC1‐transfected cells (Fig. 3d). We then performed luciferase assay. As shown in Figure 3(e), the TCF binding site reporter gene activity (TPOFlash) was higher in DIXDC1 transfection cells compared to the empty vector transfection cells, indicating DIXDC1 might up‐regulate β‐catenin and the Wnt pathway target gene cyclin D1.
PI3K signaling pathway was involved in DIXDC1‐induced colon cancer cell proliferation. To investigate the possible cell signaling pathways involved in the regulation of cyclin D1 and p21 to promote cell cycle progression, we performed immunoblotting analysis to determine expression of various components of the cell‐signaling pathway. The Akt activity, which was indirectly measured through western blot analysis of the phosphorylated Akt (p‐Akt) level, was increased in DLD1‐DIXDC1 cells compared with the DLD1‐pcDNA3.0 control cells; however, the total Akt protein level remained similar in DIXDC1 and pcDNA3.0‐transfected cells (Fig. 4a). To confirm this in vivo, DIXDC1 and p‐Akt protein expressions in the xenograph tumors from nude mice were determined by immunohistochemistry. As shown in Figure 4(b), the tumor cells from the DLD1‐DIXDC1‐1 injected mice (Fig. 4b‐1, b‐3) displayed stronger staining of cytoplasm DIXDC1 and p‐Akt compared to the tumors from DLD1‐pcDNA3.0‐injected mice (Fig. 4b‐2, b‐4). These results indicate that DIXDC1 could activate the PI3K/Akt pathway.
Figure 4.
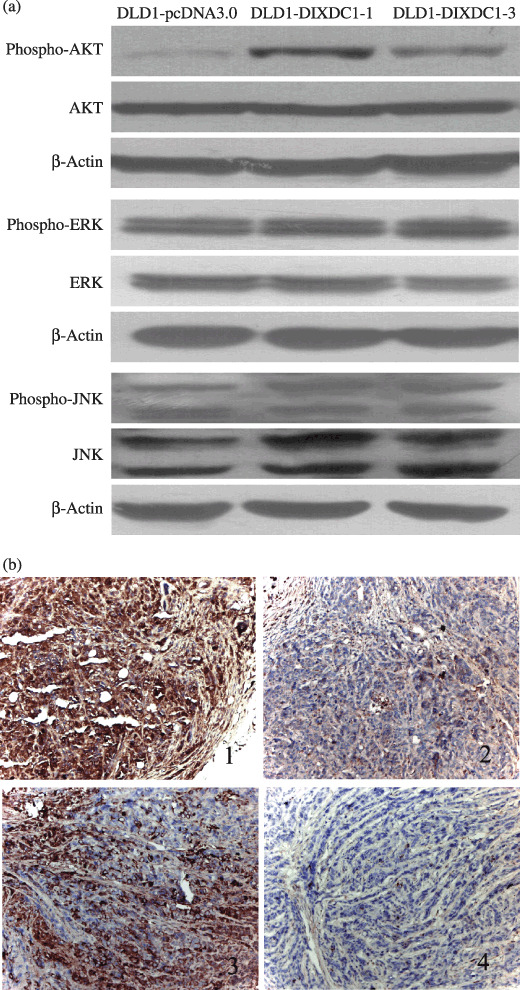
Overexpression of DIXDC1 activated the PI3K/Akt pathway in DLD1 cells. (a) Cell lysates were subjected to immunoblotting using antibodies against Akt, p‐Akt, ERK, p‐ERK, JNK, and p‐JNK. (b) Immunohistochemical staining on DLD1 xenograph tumors. Tumors from DLD1‐DIXDC1‐1‐injected mice showed strong cytoplasmic DIXDC1 (Fig. 4b‐1) and (p)‐Akt staining (Fig. 4b‐2), compared with tumors from DLD1‐pcDNA3.0‐injected mice (Figs 4b‐3 and 4b‐4).
To examine whether the PI3K/Akt pathway was involved in the DIXDC1‐related proliferation of DLD colon cancer cells, DLD1‐DIXDC1‐1 cells were treated with LY294002 (10 µM), a specific inhibitor of the PI3K pathway,( 14 ) followed by cell cycle distribution and western blot analysis. Treatment of DLD1‐DIXDC1 cells with LY294002 at a concentration of 10 µM for 12 h and 24 h almost completely inhibited the p‐Akt activity (Fig. 5a). Compared to the DMSO treated cells, the S‐phase cell percentage of DLD1‐DIXDC1 cells was decreased by 34.7% at 12 h and 56% at 24 h after LY204002 treatment, while that of DLD1‐pcDNA 3.0 cells was decreased by 8% and 12.5% at 12 h and 24 h after LY294002 treatment, respectively (Fig. 5b). Western blot analysis showed that the amount of p21 increased 1.67‐fold and 1.96‐fold at 12 h and 24 h after incubation with LY294002, respectively (Fig. 5a). Cyclin D1 and β‐catenin protein expression in the DLD1‐DIXDC1‐1 cells was decreased by 30.2% and 20.3%, respectively, at 12 h and 55.6%, and 28.5% at 24 h after LY294002 treatment. We then performed co‐immunoprecipitation to detect if there was any association between DIXDC1 and activated Akt. As shown in Figure 5(c), phosphorylated Akt was detected to be co‐immunoprecipitated with DIXDC1 in DIXDC1 overexpression cells but not in control cells. All these data indicate that the PI3K/Akt pathway was involved in DIXDC1‐induced cell proliferation promotion.
Figure 5.
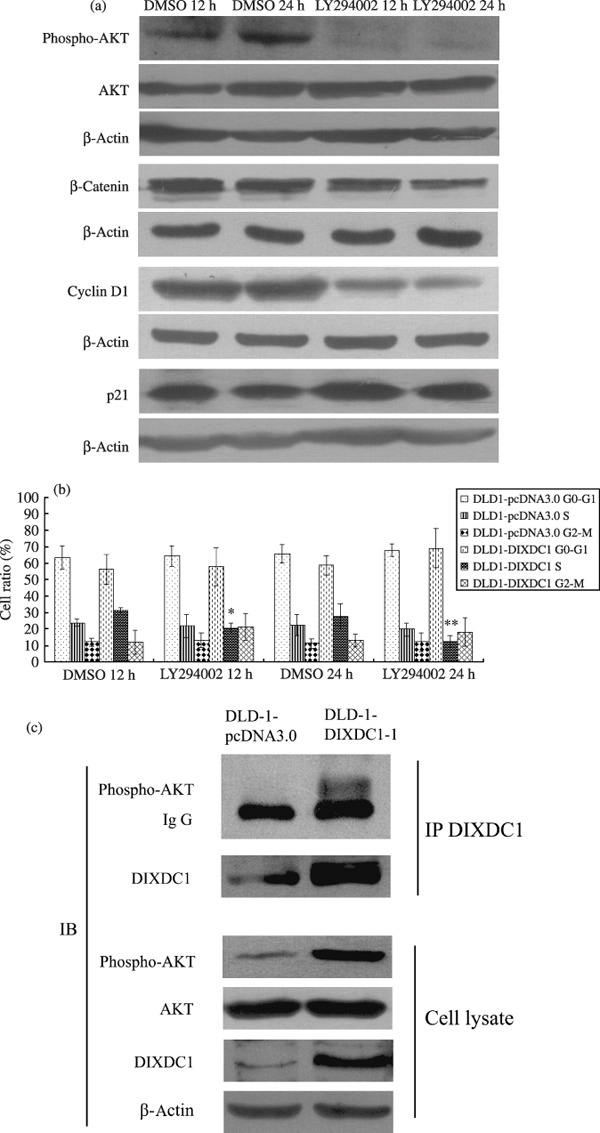
The PI3K pathway was involved in DIXDC1‐induced p21, cyclin D1 alteration. (a) DLD1‐DIXDC1‐1 cells were treated with 10 µM LY294002 or DMSO vehicle control for 12 h and 24 h, then were harvested and subjected to immunoblotting analysis using Akt, p‐Akt, p21, cyclin D1, and β‐catenin antibodies. (b) Blocking the PI3K pathway with LY294002 decreased the S‐phase cell fraction in DLD1‐DIXDC1‐1 cells. Cells were treated with LY292002 (10 µM) or DMSO for 12 h or 24 h, and were then collected for flow cytometry analysis. The percentages of cells in the G0/G1, S, and G2/M phases were counted. Columns represent the mean of three independent experiments; bars, SD. *P < 0.01, compared with DMSO 12 h; **P < 0.01, compared with DMSO 24 h. (c) DIXDC1 interacted with p‐Akt in DIXDC1 overexpression cells. Cell lysates were immunoprecipitated with anti‐DIXDC1 antibody. The immunoprecipitates and cell lysates were probed with anti‐phospho‐Akt, anti‐Akt, and anti‐DIXDC1 antibodies. IP, immnunoprecipitation; IB, immunoblot.
Knockdown of DIXDC1 inhibited G1/S transition and PI3K/Akt activity in human colon cancer cells. LS174T was selected for knockdown of DIXDC1 to clarify the role of endogenous DIXDC1 in cell proliferative activity because of its relatively high DIXDC1 protein level (data not shown; available upon request). As shown in Figure 6(a), DIXDC1 protein level in LS174T cells was dramatically knocked down after transfection with DIXDC1 siRNA for 60 h. Knockdown DIXDC1 in LS174T cells markedly inhibited the cell proliferation rate (Fig. 6b), increased G1‐phase cell populations (41.25% ± 0.007 vs 51.64% ± 0.007), and decreased S‐phase populations (34.45% ± 0.02 vs 21.99% ± 0.006), compared to the control siRNA‐transfected cells (Fig. 6c). DIXDC1 siRNA‐transfected LS174T cells were detected to have lower p‐Akt activity, compared with the control siRNA‐transfected cells. Knockdown of DIXDC1 also reduced the amount of cyclin D1 and β‐catenin, while it increased the amount of p21 protein in LS174T cells (Fig. 6d). Similar phenomena were observed when DLD1‐DIXDC1‐1 cells were transfected with DIXDC1 siRNA (data not shown; available upon request). These findings implicated a direct role of DIXDC1 in cell cycle progression through activation of the PI3K/Akt pathway in colon cancer.
Figure 6.
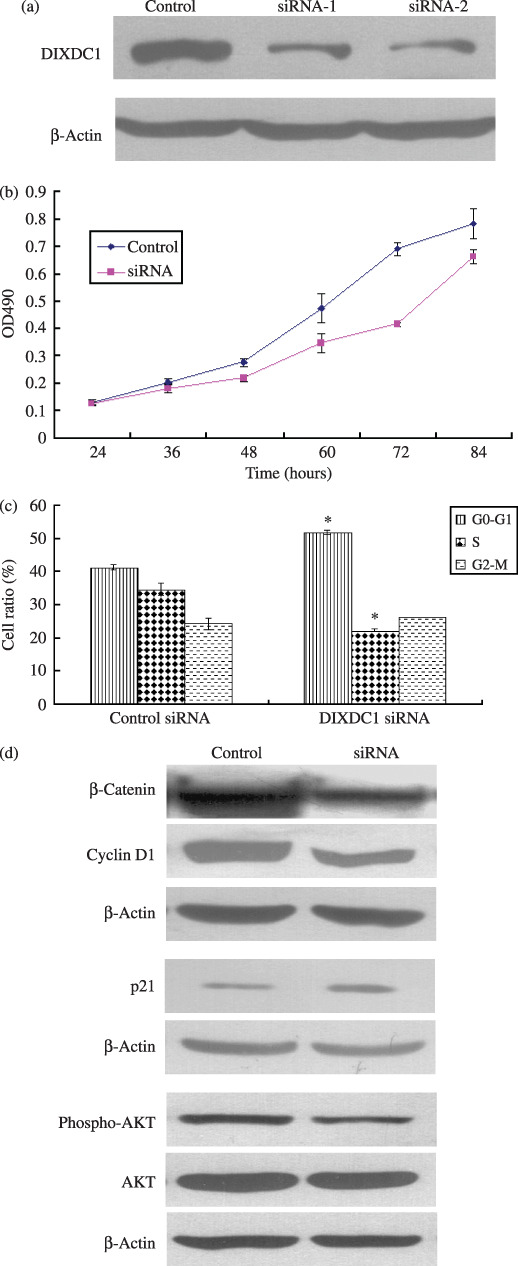
Knockdown DIXDC1 on the proliferative activity of LS174T colon cancer cells. (a) Knockdown of DIXDC1 protein at 60 h after by DIXDC1 siRNA transfection as shown by the immunoblotting analysis. (b) Cells were plated in 96‐well plates at 24 h after DIXDC1 siRNA transfection and detected for OD490 at the indicated time interval with MTT assay. (c) The G1/S cell cycle transition of LS174T cells was inhibited by knockdown of DIXDC1. Columns, mean of three independent experiments; bars, SD. *P < 0.01, compared with control group. (d) LS174T cells were lysed at 60 h after DIXDC1 siRNA transfection. Cell lysates were subjected to immunoblotting with β‐catenin, cyclin D1, p21, Akt, and p‐Akt antibodies as described in ‘Materials and Methods’. Protein levels were normalized to β‐actin.
Discussion
Colon cancer is one of the most common causes of cancer incidence worldwide.( 15 ) It has been well documented that aberrant activation of the Wnt signaling pathway is associated with a variety of human cancers, particular colorectal cancer.( 16 , 17 , 18 ) DIXDC1 is the human homolog of Ccd1, a recently identified novel DIX domain–containing protein, and a positive regulator in the Wnt pathway functioning downstream of Wnt and upstream of Axin.( 1 , 2 , 5 ) The biological role of DIXDC1 in the formation and progression of human colon cancer remains largely unknown.
In this study, we provide evidence that DIXDC1 overexpression could promote colon cancer cell proliferation both in vitro and in vivo through facilitated G1/S phase transition, which is a very common phenotype in cancer cells. Precise coordination of the S and M phases of the eukaryotic cell cycle is critical not only for normal cell division, but also for effective growth arrest under conditions of stress.( 19 ) Many components of the cell cycle kinase or kinase inhibitors are involved in mediating G1/S cell transition.( 20 , 21 ) To better understand the mechanisms that facilitate the G1/S cell cycle transition mediated by DIXDC1, the protein levels of some of the cell cycle kinase inhibitors were determined using immunoblotting. Our results indicated that promoted G1/S cell cycle progression in DIXDC1 overexpression colon cancer cells might be facilitated by DIXDC1‐mediated p21 down‐regulation and cyclin D1 up‐regulation.
In the current study, β‐catenin was found to be accumulated in DIXDC1 overexpression cells. Cyclin D1, a downstream target of Wnt, can be transactivated upon Wnt/β‐catenin activation. Ccd1 was previously reported to be a positive regulator in the Wnt pathway. The luciferase assay results showing that DIXDC1 could increase TOPFlash activity further indicated that the up‐regulation of cyclin D1 by DIXDC1 might be through a Wnt/βcatenin dependent mechanism.
p21 is a widely accepted tumor suppressor protein and a negative regulator in the G1/S transition.( 22 ) It can interact with cyclin‐dependent kinase 4/6 complexes; thereby inhibiting their activity and cell proliferation.( 23 , 24 ) Down‐regulation or loss of p21 expression has been reported to be found in a variety of human cancers, and is correlated with uncontrolled cell growth.( 25 , 26 ) p21 can be up‐regulated through p53‐dependent trans‐activation or another mechanism.( 27 ) We showed in the present study that DIXDC1 could down‐regulate p21 through a p53‐independent mechanism.
Previous studies have revealed the roles of the cell‐signaling pathway in the regulation of cell proliferation. Our data suggest that DIXDC1 overexpression could target p21 and cyclin D1 to promote colon cancer cell proliferation through PI3K pathway activation. First, the association of endogenous phosphorylated Akt with DIXDC1 could be detected in DIXDC1 overexpression cells. Second, DIXDC1 could increase p‐Akt and cyclin D1 expression, but decrease p21 expression in colon cancer cells. Third, blocking the PI3K pathway using LY294002 or down‐regulating DIXDC1 using short‐interference RNA decreased the p‐Akt protein level as well as the S‐phase cell fraction, and up‐regulated p21 and down‐regulated cyclin D1 in colon cancer cells. Moreover, overexpression of the DIXDC1 protein was observed in human colon cancer tissues and was well correlated with high cell proliferation index. Thus, the cell proliferation promoting effect of DIXDC1 by the PI3K pathway was confirmed both in vitro and in vivo.
The PI3K/Akt pathway has been demonstrated to be an important mediator in modulating cell proliferation or apoptosis.( 28 ) PI3K pathway activation could promote G1/S cell cycle promotion through transactivation of cyclin D1.( 29 ) Participation of the PI3K/Akt pathway in Wnt‐induced cell proliferation has been reported in previous research.( 30 , 31 ) We showed in the present study that DIXDC1 could up‐regulate cyclin D1 through a Wnt/β‐catenin mechanism. Mechanisms other than PI3K/Akt activation and p21, cyclin D1 alteration could also exist which are responsible for the proliferative activity of DIXDC1. Nevertheless, our research strongly suggests the coupling of the PI3K pathway with DIXDC1 activation in modulating colon cancer cell proliferation.
Taken together, the results of this study demonstrated that overexpression of DIXDC1 could promote colon cancer cell proliferation through PI3K pathway activation, and p21 and cyclin D1 were targeted in this process. Modulation of the tumor proliferation effect through inhibiting PI3K activation mediated by DIXDC1 overexpression might be used as a potential target for colon cancer prevention and therapy.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (no. 30700363) and research funding from the Shanghai Health Bureau in China (no. 2007087) for Dr Li Zheng.
References
- 1. Shiomi K, Uchida H, Keino‐Masu K, Masu M. Ccd1, a novel protein with a DIX domain, is a positive regulator in the Wnt signaling during zebrafish neural patterning. Curr Biol 2003; 13: 73–7. [DOI] [PubMed] [Google Scholar]
- 2. Soma K, Shiomi K, Keino‐Masu K, Masu M. Expression of mouse Coiled‐coil‐DIX1 (Ccd1), a positive regulator of Wnt signaling, during embryonic development. Gene Expr Patterns 2006; 6: 325–30. [DOI] [PubMed] [Google Scholar]
- 3. Shiomi K, Kanemoto M, Keino‐Masu K, Yoshida S, Soma K, Masu M. Identification and differential expression of multiple isoforms of mouse Coiled‐coil‐DIX1 (Ccd1), a positive regulator of Wnt signaling. Brain Res Mol Brain Res 2005; 135: 169–80. [DOI] [PubMed] [Google Scholar]
- 4. Kishida S, Yamamoto H, Hino S, Ikeda S, Kishida M, Kikuchi A. DIX domains of Dvl and axin are necessary for protein interactions and their ability to regulate beta‐catenin stability. Mol Cell Biol 1999; 19: 4414–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang X, Zheng L, Zeng Z et al . DIXDC1 isoform, 1‐DIXDC1, is a novel filamentous actin‐binding protein. Biochem Biophys Res Commun 2006; 347: 22–30. [DOI] [PubMed] [Google Scholar]
- 6. Liu W, Dong X, Mai M et al . Mutations in AXIN2 cause colorectal cancer with defective mismatch repair by activating beta‐catenin/TCF signalling. Nat Genet 2000; 26: 146–7. [DOI] [PubMed] [Google Scholar]
- 7. Zheng L, Ren JQ, Li H, Kong ZL, Zhu HG. Downregulation of wild‐type p53 protein by HER‐2/neu mediated PI3K pathway activation in human breast cancer cells: its effect on cell proliferation and implication for therapy. Cell Res 2004; 14: 497–506. [DOI] [PubMed] [Google Scholar]
- 8. Plonowski A, Schally AV, Nagy A, Kiaris H, Hebert F, Halmos G. Inhibition of metastatic renal cell carcinomas expressing somatostatin receptors by a targeted cytotoxic analogue of somatostatin AN‐238. Cancer Res 2000; 60: 2996–3001. [PubMed] [Google Scholar]
- 9. Massfelder T, Lang H, Schordan E et al . Parathyroid hormone‐related protein is an essential growth factor for human clear cell renal carcinoma and a target for the von Hippel‐Lindau tumor suppressor gene. Cancer Res 2004; 64: 180–8. [DOI] [PubMed] [Google Scholar]
- 10. Kim HJ, Andersson LC, Bouton D, Warner M, Gustafsson JA. Stromal growth and epithelial cell proliferation in ventral prostates of liver X receptor knockout mice. Proc Natl Acad Sci U S A 2009; 106: 558–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Millar EK, Dean JL, McNeil CM et al . Cyclin D1b protein expression in breast cancer is independent of cyclin D1a and associated with poor disease outcome. Oncogene 2009; 28: 1812–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. He TC, Sparks AB, Rago C et al . Identification of c‐MYC as a target of the APC pathway. Science 1998; 281: 1509–12. [DOI] [PubMed] [Google Scholar]
- 13. Shtutman M, Zhurinsky J, Simcha I et al . The cyclin D1 gene is a target of the beta‐catenin/LEF‐1 pathway. Proc Natl Acad Sci U S A 1999; 96: 5522–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Samuels Y, Diaz LA Jr, Schmidt‐Kittler O et al . Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell 2005; 7: 561–73. [DOI] [PubMed] [Google Scholar]
- 15. Midgley R, Kerr D. Colorectal cancer. Lancet 1999; 353: 391–9. [DOI] [PubMed] [Google Scholar]
- 16. Bienz M, Clevers H. Linking colorectal cancer to Wnt signaling. Cell 2000; 103: 311–20. [DOI] [PubMed] [Google Scholar]
- 17. Peifer M, Polakis P. Wnt signaling in oncogenesis and embryogenesis – a look outside the nucleus. Science 2000; 287: 1606–9. [DOI] [PubMed] [Google Scholar]
- 18. Fodde R, Smits R, Clevers H. APC, signal transduction and genetic instability in colorectal cancer. Nat Rev Cancer 2001; 1: 55–67. [DOI] [PubMed] [Google Scholar]
- 19. Rizki A, Lundblad V. Defects in mismatch repair promote telomerase‐independent proliferation. Nature 2001; 411: 713–6. [DOI] [PubMed] [Google Scholar]
- 20. Motokura T, Arnold A. Cyclin D and oncogenesis. Curr Opin Genet Dev 1993; 3: 5–10. [DOI] [PubMed] [Google Scholar]
- 21. Pines J. Cyclins: wheels within wheels. Cell Growth Differ 1991; 2: 305–10. [PubMed] [Google Scholar]
- 22. Niculescu AB 3rd, Chen X, Smeets M, Hengst L, Prives C, Reed SI. Effects of p21 (Cip1/Waf1) at both the G1/S and the G2/M cell cycle transitions: pRb is a critical determinant in blocking DNA replication and in preventing endoreduplication. Mol Cell Biol 1998; 18: 629–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cai K, Dynlacht BD. Activity and nature of p21 (WAF1) complexes during the cell cycle. Proc Natl Acad Sci U S A 1998; 95: 12254–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ogryzko VV, Wong P, Howard BH. WAF1 retards S‐phase progression primarily by inhibition of cyclin‐dependent kinases. Mol Cell Biol 1997; 17: 4877–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Egilmez R, Elagoz S, Kanik EA. Cdk1/P34Cdc2 and P21waf expression in colorectal adenomas and carcinomas. J Exp Clin Cancer Res 2001; 20: 549–52. [PubMed] [Google Scholar]
- 26. Stein JP, Ginsberg DA, Grossfeld GD et al . Effect of p21WAF1/CIP1 expression on tumor progression in bladder cancer. J Natl Cancer Inst 1998; 90: 1072–9. [DOI] [PubMed] [Google Scholar]
- 27. Parker SB, Eichele G, Zhang P et al . p53‐independent expression of p21Cip1 in muscle and other terminally differentiating cells. Science 1995; 267: 1024–7. [DOI] [PubMed] [Google Scholar]
- 28. Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev 1999; 13: 2905–27. [DOI] [PubMed] [Google Scholar]
- 29. Liang J, Pan Y, Zhang D et al . Cellular prion protein promotes proliferation and G1/S transition of human gastric cancer cells SGC7901 and AGS. Faseb J 2007; 21: 2247–56. [DOI] [PubMed] [Google Scholar]
- 30. Kim SE, Lee WJ, Choi KY. The PI3 kinase‐Akt pathway mediates Wnt3a‐induced proliferation. Cell Signal 2007; 19: 511–8. [DOI] [PubMed] [Google Scholar]
- 31. Fukumoto S, Hsieh CM, Maemura K et al . Akt participation in the Wnt signaling pathway through Dishevelled. J Biol Chem 2001; 276: 17479–83. [DOI] [PubMed] [Google Scholar]