

Abstract
(Cancer Sci 2010; 101: 673–678)
Similar to normal tissue stem cells, cancer stem cells (CSCs) are thought to be quiescent or slow‐cycling and, thereby, insensitive to chemo‐ and radiotherapies. CD44, a cell surface component that interacts with the extracellular matrix, has been found to be highly expressed in CSCs of several solid tumors. However, the relevancy between CD44+ cells and slow‐cycling cells and the underlying mechanisms for the emergence of CD44+ CSCs during tumorigenesis have not been elucidated. Here we show that a gastric gland residing at the squamo‐columnar junction (SCJ) in normal mouse stomach contains CD44+ stem cell‐like slow‐cycling cells and that this characteristic CD44+ gland was expanded by prostaglandin E2 (PGE2) and Wnt signaling in K19‐Wnt1/C2mE mouse, a genetic mouse model for gastric tumorigenesis. The analysis of three transgenic mouse lines, K19‐Wnt1, K19‐C2mE and K19‐Wnt1/C2mE, revealed that the expansion of CD44+ SCJ cells is triggered by PGE2‐mediated signaling and is prominently enhanced by the addition of Wnt activation. Furthermore, each expanded CD44+ gland in gastric tumor of K19‐Wnt1/C2mE mouse contains a few BrdU label‐retaining quiescent or slow‐cycling cells, suggesting that the CD44+ SCJ cells in normal mouse are candidates for the cell‐of‐origin of gastric CSCs. These observations suggest that PGE2‐mediated inflammatory signaling and Wnt signaling cooperatively trigger the expansion of CD44+ slow‐cycling stem‐like cells in SCJ, leading to development of lethal gastric tumors in mice.
CD44, a major cell adhesion molecule, has been implicated in a wide variety of physiological processes and pathological processes, including lymphocyte homing, wound healing, and cell migration, as well as cancer cell growth and metastasis.( 1 , 2 ) CD44 has been recently detected as a cell surface marker of cancer stem cells (CSCs) in several types of cancer.( 3 ) Moreover, a CD44+ subpopulation of cells isolated from several gastric cancer cell lines has been reported to have stem cell‐like properties, which are the capacities to self‐renew and to differentiate.( 4 ) However, the underlying mechanism by which CD44+ CSC‐like subpopulations emerge in the process of tumor development is largely unknown.
Cancer stem cells, also known as cancer‐initiating cells, are responsible for tumor initiation and maintenance. CSCs possess the ability to drive tumor growth and are inherently resistant to chemo‐ and radiotherapies. Recent studies have revealed that melanoma‐initiating cells( 5 ) and leukemia‐initiating cells are a slow‐cycling or quiescent subpopulation.( 6 , 7 ) These quiescent or slow‐cycling properties are thought to be a major reason why CSCs are resistant to cancer therapies targeted at proliferating cells. It has been shown that cell adhesion molecules play an essential role in the quiescence of hematopoietic stem cells by regulating adhesion in the osteoblastic niche.( 8 ) Therefore, we hypothesized that CD44, which is highly expressed in CSCs, is involved in the regulation of their quiescent or slow‐cycling status.
In the present study, we discovered a gastric gland at the squamo‐columnar junction (SCJ) in normal mice, which contains CD44‐expressing stem cell‐like slow‐cycling cells. This unique CD44+ SCJ gastric gland was rapidly expanded in response to the prostaglandin E2 (PGE2)‐mediated signaling in K19‐C2mE mice, a mouse model of gastric hyperplasia. Furthermore, Wnt signal activation significantly enhanced the PGE2‐mediated expansion of the CD44+ gland, resulting in increased numbers of the stem cell‐like slow‐cycling cells, which might contribute to development of lethal gastric tumors in mice.
Materials and Methods
Transgenic mice. K19‐Wnt1, K19‐C2mE, and K19‐Wnt1/C2mE transgenic mice have been described previously.( 9 , 10 ) All animal experiments were carried out according to protocols approved by the Ethics Committee of Keio University (Tokyo, Japan).
BrdU label‐retention assays. Wild‐type mice (8‐ to 10‐weeks‐old) were subjected to sublethal irradiation (8.0 Gy) then injected i.p. with BrdU solution (50 mg/kg body weight) twice a day for 2 days, and killed 12 days later. K19‐Wnt1/C2mE mice were injected i.p. with BrdU solution twice a day for 7 days, and killed 35 days later without irradiation. Tissue samples were processed for immunohistochemical staining with anti‐BrdU (clone Bu20a, 1:50; DakoCytomation, Carpinteria, CA, USA). The mean number of BrdU‐positive cells per 300 tumor (epithelial) cells in four microscopic fields per mouse was calculated. We examined three K19‐Wnt1/C2mE mice.
Histology and immunohistochemistry. Tissues were fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned at a thickness of 4 μm. Sections were depleted of paraffin then rehydrated in a graded series of ethanol solutions. For histology, sections were stained with H&E. For immunohistochemistry, sections were washed with PBS, subjected to antigen retrieval by heating for 10 min at 100°C in 0.01 M sodium citrate (pH 6.0), and exposed to 3% hydrogen peroxide before incubation with primary antibodies. Immune complexes were detected using a Vectastain Elite Kit (Vector Laboratories, Burlingame, CA, USA) and 3,3′‐diaminobenzidine, and the sections were counterstained with hematoxylin.
Immunofluorescence analysis. Tissues were processed as for immunohistochemistry, with the exception that sections were not exposed to hydrogen peroxide. Sections were incubated for 40 min at room temperature with 10 mm HCl containing pepsin, and incubated for 60 min at room temperature with 2 N HCl then 0.1 M sodium tetraborade (pH 8.5), washed three times with PBS before exposure for 1 h at 30°C to primary antibodies diluted in PBS. The sections were washed three times with PBS, incubated with appropriate Alexa Fluor 488‐conjugated or Texas red‐conjugated secondary antibody (Invitrogen, Tokyo, Japan), diluted in PBS, washed three times with PBS, and mounted in Vectashield mounting medium (Vector Laboratories). Sections were viewed and photographed with a Biorevo BZ‐9000 fluorescence microscope (Keyence, Tokyo, Japan).
Antibodies. The IM7 (diluted 1:100) and CD44v8‐10 (1:100) antibodies were used to detect mouse CD44 and CD44 variant (CD44v), respectively. Proliferating cells were detected with the Ki67 antibody (clone Bu201, 1:50; DakoCytomation).
Preparation of rat monoclonal antibody recognizing mouse CD44v. Female F344 rats were repeatedly immunized with RH7777 cells expressing mouse CD44v8‐10‐GFP. Three days after the final immunization, rat spleen cells were fused to X63 mouse myeloma cells with PEG (Roche Diagnostics, Tokyo, Japan). Selected and established hybridoma clones were inoculated to KSN athymic mice pretreated with tetramethylpentadecane (pristane). Ascitic fluids were precipitated with 50%‐saturated ammonium sulfate. Further purification was done by affinity chromatography with protein G‐conjugated Sepharose (GE Healthcare, Uppsala, Sweden).
Cell preparation and FACS analysis. Mouse gastric tumors from 30‐week‐old K19‐Wnt1/C2mE mice were digested for 3 h at 37°C in Ham’s F12 medium with 5% FCS, 100 U/mL penicillin, 100 μg/mL streptomycin, 5 μg/mL insulin, 300 U/mL collagenase, and 100 U/mL hyaluronidase. This digested solution was vortexed, then the red blood cells were lysed in NH4Cl at room temperature. A single cell suspension was obtained by sequential dissociation of the fragments by gentle pipetting in 0.25% trypsin (1–2 min) and 5 mg/mL dispase with 0.1 mg/mL DNase I (2 min). The solution was then filtered through a 40‐μm nylon mesh. All reagents were purchased from Stemcell Technologies (Vancouver, Canada).
Single suspended cells were incubated with antibodies at 4°C for 30 min. Antibodies for FACS analysis, CD31‐FITC, CD45‐FITC, TER119‐FITC, CD44‐APC, and CD133‐PE were purchased from eBioscience (San Diego, CA, USA). Dissociated Lin− (CD45−, TER119−, CD31−), propidium iodide− (non‐apoptotic) gastric tumor cells were sorted by FACSAria Cell Sorter (BD Biosciences, Tokyo, Japan) using anti‐CD44 antibody.
Quantitative and semiquantitative RT‐PCR analysis. Total RNA was extracted from the sorted tumor cells using Isogen (Nippon Gene, Tokyo, Japan), and was subjected to RT‐PCR with specific primers, and those specific for GAPDH were used as an internal control. Primers for detection of CD44v mRNAs were targeted to exons 5 and 16, as described previously.( 11 ) Quantitative RT‐PCR was carried out using the Thermal Cycler Dice Real Time System (Takara Bio, Tokyo, Japan). The PCR conditions were as follows: 95°C for 2 min, and 40 cycles at 95°C for 30 s, 60°C for 30 s, followed by dissociation‐curve analysis to confirm specificity. Data are presented as means ± SD of triplicates. Primer sets for RT‐PCR were: CD44v forward, 5′‐GGAGATCAGGATGACTCCTTCT‐3′ and reverse, 5′‐AGTCCTTGGATGAGTCTCGATC‐3′; MUC5AC forward, 5′‐CTACCACTCCCTGCTTCTGC‐3′ and reverse, 5′‐TCCTGGCTACACATCGCATA‐3′; CD133 forward, 5′‐GAAAAGTTGCTCTGCGAACC‐3′ and reverse, 5′‐CTCGACCTCTTTTGCAATCC‐3′; and GAPDH forward, 5′‐GTGAAGGTCGGTGTGAACG‐3′ and reverse, 5′‐GACCATGTAGTTGAGGTCAATG‐3′.
Statistical analysis. Data are presented as means ± SD and were analyzed using the unpaired Student’s t‐test in Excel 2007 (Microsoft, Redmond, WA, USA). A value of P < 0.05 was considered statistically significant.
Results
CD44 differentially expressed in undifferentiated cells located at center of gastric tumors in K19‐Wnt1/C2mE mice. K19‐Wnt1/C2mE mouse, which overexpress Wnt1, COX2 and microsomal prostaglandin E synthase 1 (mPGES‐1) in gastric epithelial cells, is a genetic model for gastric carcinogenesis.( 9 ) Simultaneous activation of both Wnt and PGE2 pathways causes large dysplastic gastric tumors, and all of these transgenic animals became moribund at approximately 30–50 weeks of age. One hundred percent of the gastric tumors in K19‐Wnt1/C2mE mice arose from the region of the SCJ (Fig. 1A–C).( 9 ) Although keratin 19 promoter is active in pancreas and intestines as well as stomach, we did not find any tumor formation in the tissues other than stomach because the expression of transgenes, COX2 and mPGES‐1, was silenced in those tissues by an unknown mechanisms, as we previously described.( 12 )
Figure 1.
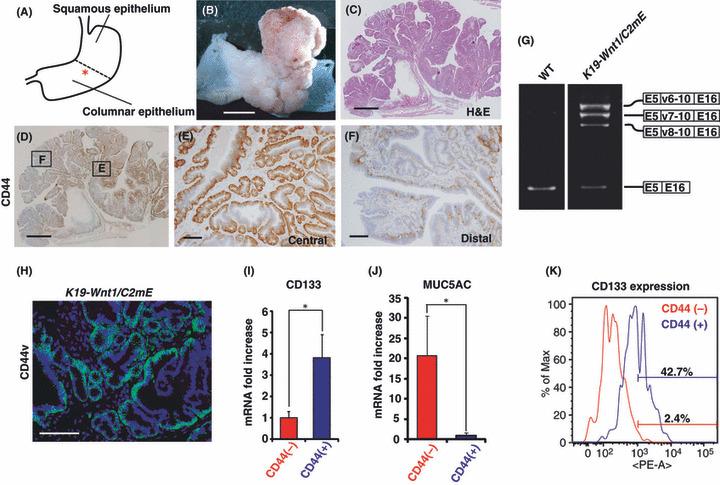
Pattern of CD44 expression in gastric tumors of K19‐Wnt1/C2mE mice. (A–C) Left panel shows a diagram of the wild‐type mouse stomach (A). The asterisk indicates the squamo‐columnar junction region. Macroscopic image (B) and H&E staining (C) of the stomach from a 30‐week‐old K19‐Wnt1/C2mE mouse. (D–F) Immunohistochemical staining for CD44 (D) in the stomach of a 30‐week‐old K19‐Wnt1/C2mE mouse. The central (E) and distal (F) region of the gastric tumor is shown at higher magnification in the middle and right panels, respectively. Scale lines = 5 mm (B), 1 mm (C,D), and 100 μm (E,F). (G) Total RNA isolated from gastric mucosa of 30‐week‐old wild‐type and gastric tumors of 30‐week‐old K19‐Wnt1/C2mE mice, was subjected to RT‐PCR analysis with specific primers targeted to exon 5 and 16 of CD44 mRNA. Direct sequencing of the PCR products revealed that CD44 variant (CD44v) (v6‐10, v7‐10, v8‐10) was predominantly expressed in the gastric tumors of K19‐Wnt1/C2mE mice. (H) Immunofluorescence analysis of CD44v in gastric tumors of 30‐week‐old K19‐Wnt1/C2mE mice. Scale line = 100 μm. (I) Expression of CD133 in CD44+ cells relative to that in CD44− cells. Lin− (CD45−, TER119−, CD31−), propidium iodide− (non‐apoptotic) gastric tumor cells were separated by FACS using anti‐CD44 antibody and subjected to quantitative RT‐PCR analysis. *P < 0.01. (J) Expression of MUC5AC in CD44− cells relative to that in CD44+ cells were determined by quantitative RT‐PCR analysis using a similar method to that outlined in (G). *P < 0.01. (K) Flow cytometric analysis of CD133 expression in CD44+ or CD44− cells. A control was included in the negative fraction (0–103).
We first examined the expression and distribution of the CSC marker CD44 in gastric tumors of 30‐week‐old K19‐Wnt1/C2mE mice (Fig. 1D). Immunohistochemical analysis revealed that CD44 expression is predominantly present in the center of the gastric tumors (Fig. 1E) and weakly present toward the periphery of the tumor (Fig. 1F). These results indicate that the majority of CD44+ tumor cells are preferentially located in the central regions of gastric tumors. Given that the increased expression of CD44 splicing variant isoforms (CD44v) is associated with tumor progression in various human adenocarcinomas,( 13 , 14 ) we next investigated CD44v expression in tumors of K19‐Wnt1/C2mE mice. Using RT‐PCR analysis we detected the expression of three variant isoforms in addition to the standard form, and the DNA sequence analysis revealed that those variant isoforms included exons v6‐10, v7‐10, and v8‐10 (Fig. 1G). To further investigate the expression of CD44v in K19‐Wnt1/C2mE mice, we generated an antibody against the mouse CD44 v8‐10 epitope and found that CD44v is expressed predominantly in tumor cells of K19‐Wnt1/C2mE mice (Fig. 1H).These results are consistent with previous observations in human gastric adenocarcinoma.( 14 , 15 )
To characterize CD44+ tumor cells, we isolated CD44+ or CD44− tumor cells from gastric tumors of K19‐Wnt1/C2mE mice by FACS and examined the mRNA expression of CD133, a potential marker of undifferentiated cells in the gastrointestinal tract,( 16 ) and the gastric epithelial differentiation marker MUC5AC.( 17 ) CD44+ tumor cells show a high level of CD133 expression (Fig. 1I) and a low level of MUC5AC expression compared to CD44− cells (Fig. 1J). Furthermore, FACS analysis revealed that a large population of CD44+ cells (42.7%) express high levels of CD133 compared to CD44− cells (Fig. 1K). Based on these findings, we assumed that CD44, which is expressed in undifferentiated gastric tumor cells that are located in the center of the tumor, plays a role in the maintenance and development of gastric tumors.
CD44+ gland at SCJ region of normal mouse stomach contains some slow‐cycling cells. The SCJ region corresponds to the esophagus–columnar junction (EC junction) in humans and is a frequent site of tumor development.( 18 , 19 ) Therefore, we speculated that the normal SCJ region would contain cells that are susceptible to gastric carcinogenesis. In the corpus of the normal mouse stomach, CD44+ gastric epithelial cells were rarely detected. However, we found one gland in the vicinity of the SCJ that contains cells expressing high levels of CD44 (Fig. 2A). CD44+ cells tended to be localized along the basal side of the gland. We then immunohistochemically investigated whether these CD44+ cells express the standard form or variant isoform of CD44. We found that CD44+ cells located at SCJ gland express CD44 variant isoform like tumor cells (Fig. 2B). This result suggests that gastric tumor in K19‐Wnt1/C2mE mice might be a result of neoplastic expansion of CD44v+ cells at the SCJ gland. To further characterize CD44+ cells located in the SCJ gland, we examined the proliferation status in these cells. Such CD44+ cells in the SCJ gland were mostly negative for Ki67, a marker of cell proliferation (Fig. 2B), suggesting that they are quiescent or slow‐cycling stem‐like cells located near proliferative progenitor cells.
Figure 2.
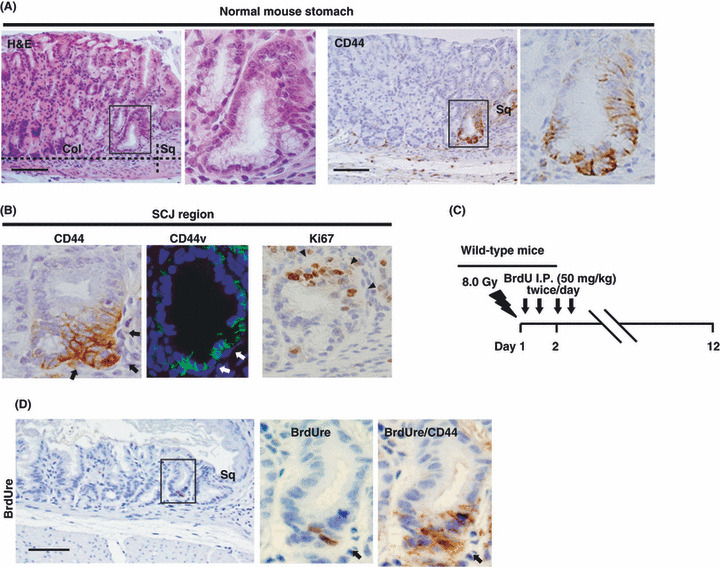
CD44+ gland cells at the squamo‐columnar junction (SCJ) region in mouse stomach include label‐retaining cells. (A) H&E staining and immunohistochemical staining for CD44 in the SCJ region in wild‐type mouse stomach. The boxed regions in the left panels are shown at higher magnification in the right panels. Col, columnar epithelium; Sq, squamous epithelium. (B) Immunohistochemical staining for CD44 and Ki67 and immunofluorescence analysis of CD44 variant (CD44v) of the SCJ region in wild‐type mouse stomach. Black arrows (white arrows) and arrowheads indicate mutually exclusive expression of CD44 (CD44v) and Ki67, respectively. (C) The protocol for the BrdU label‐retention assay using wild‐type mice. (D) Immunohistochemical staining for BrdU in the SCJ region 10 days after BrdU injection. The boxed region in the left panel is shown at a higher magnification in the middle panel and the right panel shows double staining for BrdU and CD44 in the same section. Arrows indicate BrdU label‐retaining cells in the SCJ region. Scale bars = 100 μm.
We carried out BrdU labeling assays (Fig. 2C) to investigate whether CD44+ gastric cells in the SCJ gland showed a characteristic peculiar to tissue stem cells, that is, slow cycling and retention of the BrdU label in the nucleus over several weeks.( 20 ) Immunohistochemical analysis done by sequentially staining with antibodies against BrdU and CD44 revealed that a few label‐retaining cells (LRCs) were located in the base of the SCJ gland and that those LRCs expressed high levels of CD44 (Fig. 2D). These results suggested that these rare CD44+ SCJ cells have stem‐like quiescent or slow‐cycling characteristics.
PGE2‐mediated signaling is essential for the expansion of CD44+ SCJ gland cells. Given that duplication of gastric glands is thought to be mediated by stem cell division,( 21 ) we hypothesized that CD44+ SCJ cells would be cells‐of‐origin of the gastric tumors that develop in K19‐Wnt1/C2mE mice. Our hypothesis was supported by immunohistochemical analysis of the SCJ region in K19‐Wnt1/C2mE mice at 12, 20, and 30 weeks of age, which showed that the CD44+ gland cells expanded concomitantly with gastric tumor development (Fig. 3A,B).
Figure 3.
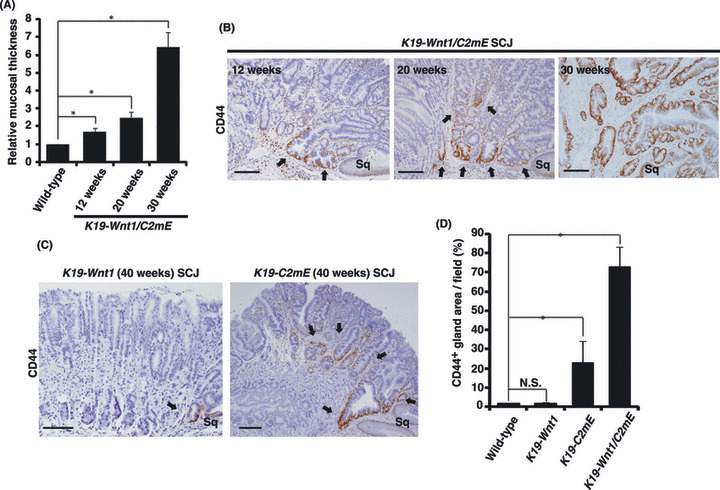
CD44+ gland cells are expanded in K19‐C2mE mice but not in K19‐Wnt1 mice. (A) The gastric mucosal thickness, which reflects the size of the tumor, in K19‐Wnt1/C2mE mice at the indicated ages relative to that in age‐matched wild‐type mice. Data are means ± SD from three mice. *P < 0.05 versus wild‐type mice. (B) Immunohistochemical staining for CD44 at the squamo‐columnar junction (SCJ) region in the stomach of K19‐Wnt1/C2mE mice at 12, 20, and 30 weeks of age. CD44+ gland cells (arrows) are expanded concomitant with tumor development. Scale bars = 100 μm. (C) Immunohistochemical staining for CD44 at the SCJ region of K19‐Wnt1 or K19‐C2mE mice at 40 weeks of age. Arrows indicate CD44+ gastric glands at the SCJ region. Scale bars = 100 μm. (D) Percentage of CD44+ gland cell area per field of wild‐type, K19‐Wnt1, and K19‐C2mE mice at 40 weeks of age, and K19‐Wnt1/C2mE mice at 30 weeks of age. Data are means ± SD from three mice of each genotype. *P < 0.01.
Wnt/β‐catenin signaling is known to promote the proliferation and self‐renewal of stem cells or progenitors during the development of a variety of tissues.( 22 ) PGE2 signaling has also been found to enhance the number of stem cells and multipotent progenitors in the hematopoietic system.( 23 ) Therefore, we next investigated whether the expansion of CD44+ SCJ cells in the transgenic mice is mediated by PGE2 or Wnt1 signaling. CD44+ SCJ cells were significantly expanded in 40‐week‐old K19‐C2mE mice, which overexpress COX2 and mPGES‐1 and develop preneoplastic hyperplastic legions. In contrast, CD44+ SCJ cells were not expanded in K19‐Wnt1 mice, which overexpress Wnt1 (Fig. 3C,D). These data indicate that PGE2‐mediated signaling plays a major role in the expansion of CD44+ SCJ cells. Importantly, Wnt1 expression strongly enhanced the expansion of CD44+ SCJ cells in the presence of PGE2 signaling (Fig. 3D), leading to gastric tumorigenesis. PGE2 signaling has been shown to induce cyclic AMP/protein kinase A‐mediated phosphorylation of β‐catenin, leading to the stabilization of β‐catenin and activation of Wnt signaling in hematopoietic stem/progenitor cells.( 24 ) Taken together, these results suggest that the effects of PGE2 signaling are prominently enhanced by Wnt signaling in CD44+ stem‐like cells.
Majority of expanding slow‐cycling tumor cells express high levels of CD44. If the increased number of CD44+ gland cells in K19‐Wnt1/C2mE mice tumors is due to expansion of the SCJ gland, then slow‐cycling LRCs might exist among the CD44+ gland cells. To test this hypothesis, we carried out assays to measure long‐term BrdU label retention using K19‐Wnt1/C2mE mice (Fig. 4A). Immunohistochemical analysis revealed that LRCs are mainly detected in CD44+ gland cells located in the central region of tumors, whereas only a few LRCs were found in the CD44− cells or cells expressing low levels of CD44 in the periphery of the tumors (Fig. 4B,C). These findings suggest that gastric tumors in the transgenic mouse model are initiated by expansion of specific SCJ gland cells consisting of CD44+ quiescent or slow‐cycling stem‐like cells.
Figure 4.
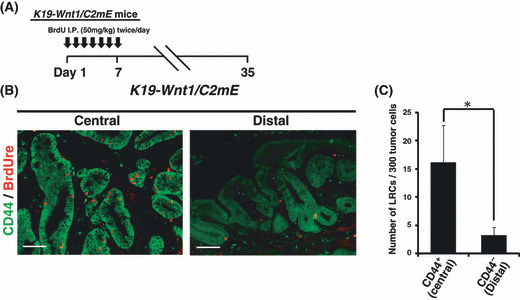
Slow‐cycling CD44+ cells in K19‐Wnt1/C2mE mice. (A) The protocol of long‐term BrdU label‐retaining assay using K19‐Wnt1/C2mE mice. (B) Immunofluorescence analysis of double staining for BrdU label‐retaining/CD44 in gastric tumors of 30‐week‐old K19‐Wnt1/C2mE mice. Scale bars = 50 μm. (C) Number of label‐retaining cells in CD44+ tumor cells at the central region or CD44− tumor cells in the periphery of gastric tumors, respectively. Data are means ± SD from four microscopic fields. *P < 0.01.
Discussion
In this study, we found that the SCJ region of the normal mouse stomach contains a gland consisting of CD44+ cells, of which a few are quiescent or slow‐cycling stem cell‐like gastric cells. Furthermore, we showed that this characteristic gland containing the stem‐like CD44+ SCJ cells expands in response to PGE2‐mediated signaling and constructs preneoplastic lesions. The cell fate in the SCJ region is determined by conflicting extrinsic signals from the two adjacent epithelial tissues, namely squamous and glandular tissues, and this region of the mouse stomach is thus considered to be highly susceptible to oncogenesis.( 25 ) Therefore, we assumed that the CD44+ slow‐cycling cells in the normal stomach SCJ would be candidates for the cell‐of‐origin of gastric CSCs in K19‐Wnt1/C2mE mice.
The normal gastric stem/progenitor cells are thought to be located in the isthmus/neck region of gastric glands.( 21 ) It is not known whether the SCJ region of mice stomach, which corresponds to human EC junction, contains gastric stem/progenitor cells. However, both the human EC junction and mouse SCJ region are common sites of tumor development.( 18 , 25 ) Based on this evidence and our present study, we presume that these junctional regions in human and mouse might contain stem/progenitor cells that expand in response to the activation of PGE2‐mediated signaling and give rise to gastric tumor, although further investigations are needed to support this concept. Furthermore, we found large numbers of CD44+ gastric glands in human adenocarcinomas and adjacent metaplasias, but not in the normal gastric epithelium (data not shown). Thus, it is possible that a few CD44+ stem‐like cells, similar to the mouse SCJ cells, reside in human normal gastric epithelium and that metaplastic changes, often triggered by chronic inflammation, are induced by the expansion of CD44+ stem‐like cells through the activation of PGE2 signaling.
PGE2, an inflammatory mediator released at the site of tissue inflammation, promotes colon adenoma growth in APC mutant mice,( 26 ) and triggers the expansion of hematopoietic stem cells.( 23 ) These observations suggest that PGE2 and its downstream signaling effectors potentiate the self‐renewal ability of stem/progenitor cells in vivo. Importantly, we previously reported that tumor necrosis factor‐α, a mediator of PGE2‐induced inflammatory signaling, triggers sustained proliferation of progenitor cells in gastric tumors of K19‐C2mE mice.( 10 ) Therefore, PGE2 and its downstream targets might play key roles in the regulation of CD44+ slow‐cycling cell expansion.
Aberrant activation of Wnt signaling, which regulates the self‐renewal of stem/progenitor cells, has been shown to trigger colon tumorigenesis.( 22 ) In the present study, we found that the activation of Wnt signaling alone is not sufficient for CD44+ SCJ cell expansion in the absence of PGE2 signaling, as seen in K19‐Wnt1 mice. In contrast, Wnt signaling cooperatively promotes PGE2‐induced CD44+ gland cell expansion in K19‐Wnt1/C2mE mice. These results indicate that Wnt signaling stimulates the proliferation of CD44+ stem‐like cells during inflammation, leading to the enhancement of PGE2‐induced expansion of CD44+ quiescent or slow‐cycling cells in gastric tumorigenesis.
It is important to note that CD44+ cells in the normal SCJ gland were LRCs and/or Ki67− cells (Fig. 2B), suggesting that CD44 expression is related to quiescent or slow‐cycling characteristics in normal stem cells. However, in the glands expanded by the cooperative activation of PGE2 and Wnt, CD44 expression was found not only in LRCs and Ki67− cells but also in some Ki67+ cycling cells (data not shown). These findings indicate that CD44 expression does not strictly correlate with the quiescent or slow‐cycling status in the context of cancer cells. Further investigations on the functional association of CD44 with various cell cycle regulators in both normal and cancer cells are warranted.
Taken together, our present findings suggest that the eradication of the CD44+ subpopulation consisting of quiescent or slow‐cycling cells and the suppression of Wnt/PGE2‐mediated CD44+ cell expansion would be an interesting strategy for the development of efficient therapies against gastric cancers.
Acknowledgments
We thank I. Ishimatsu, N. Suzuki, and Y. Ito for technical assistance, K. Arai for help in preparation of the manuscript, and M. Miyasaka for providing the anti‐pan‐CD44 antibody (IM7). This work was supported by grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (to O.N. and H.S.).
References
- 1. Ponta H, Sherman L, Herrlich PA. CD44: from adhesion molecules to signalling regulators. Nat Rev 2003; 4: 33–45. [DOI] [PubMed] [Google Scholar]
- 2. Aruffo A, Stamenkovic I, Melnick M, Underhill CB, Seed B. CD44 is the principal cell surface receptor for hyaluronate. Cell 1990; 61: 1303–13. [DOI] [PubMed] [Google Scholar]
- 3. Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer 2008; 8: 755–68. [DOI] [PubMed] [Google Scholar]
- 4. Takaishi S, Okumura T, Tu S et al. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells 2009; 27: 1006–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zabierowski SE, Herlyn M. Melanoma stem cells: the dark seed of melanoma. J Clin Oncol 2008; 26: 2890–4. [DOI] [PubMed] [Google Scholar]
- 6. Ito K, Bernardi R, Morotti A et al. PML targeting eradicates quiescent leukaemia‐initiating cells. Nature 2008; 453: 1072–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Holtz M, Forman SJ, Bhatia R. Growth factor stimulation reduces residual quiescent chronic myelogenous leukemia progenitors remaining after imatinib treatment. Cancer Res 2007; 67: 1113–20. [DOI] [PubMed] [Google Scholar]
- 8. Arai F, Suda T. Maintenance of quiescent hematopoietic stem cells in the osteoblastic niche. Ann N Y Acad Sci 2007; 1106: 41–53. [DOI] [PubMed] [Google Scholar]
- 9. Oshima H, Matsunaga A, Fujimura T, Tsukamoto T, Taketo MM, Oshima M. Carcinogenesis in mouse stomach by simultaneous activation of the Wnt signaling and prostaglandin E2 pathway. Gastroenterology 2006; 131: 1086–95. [DOI] [PubMed] [Google Scholar]
- 10. Oshima M, Oshima H, Matsunaga A, Taketo MM. Hyperplastic gastric tumors with spasmolytic polypeptide‐expressing metaplasia caused by tumor necrosis factor‐alpha‐dependent inflammation in cyclooxygenase‐2/microsomal prostaglandin E synthase‐1 transgenic mice. Cancer Res 2005; 65: 9147–51. [DOI] [PubMed] [Google Scholar]
- 11. Okamoto I, Morisaki T, Sasaki J et al. Molecular detection of cancer cells by competitive reverse transcription‐polymerase chain reaction analysis of specific CD44 variant RNAs. J Natl Cancer Inst 1998; 90: 307–15. [DOI] [PubMed] [Google Scholar]
- 12. Oshima H, Oshima M, Inaba K, Taketo MM. Hyperplastic gastric tumors induced by activated macrophages in COX‐2/mPGES‐1 transgenic mice. EMBO J 2004; 23: 1669–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tanabe KK, Ellis LM, Saya H. Expression of CD44R1 adhesion molecule in colon carcinomas and metastases. Lancet 1993; 341: 725–6. [DOI] [PubMed] [Google Scholar]
- 14. Mayer B, Jauch KW, Gunthert U et al. De‐novo expression of CD44 and survival in gastric cancer. Lancet 1993; 342: 1019–22. [DOI] [PubMed] [Google Scholar]
- 15. Miwa T, Watanabe A, Yamada Y et al. Progression in gastric carcinoma relative to the ratio of CD44 epithelial variant transcript to CD44 hematopoietic variant transcript. Cancer 1996; 77: 25–9. [DOI] [PubMed] [Google Scholar]
- 16. Zou GM. Cancer initiating cells or cancer stem cells in the gastrointestinal tract and liver. J Cell Physiol 2008; 217: 598–604. [DOI] [PubMed] [Google Scholar]
- 17. Hollingsworth MA, Swanson BJ. Mucins in cancer: protection and control of the cell surface. Nat Rev Cancer 2004; 4: 45–60. [DOI] [PubMed] [Google Scholar]
- 18. Odze RD. Unraveling the mystery of the gastroesophageal junction: a pathologist’s perspective. Am J Gastroenterol 2005; 100: 1853–67. [DOI] [PubMed] [Google Scholar]
- 19. Qiao XT, Ziel JW, McKimpson W et al. Prospective identification of a multilineage progenitor in murine stomach epithelium. Gastroenterology 2007; 133: 1989–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Potten CS, Owen G, Booth D. Intestinal stem cells protect their genome by selective segregation of template DNA strands. J Cell Sci 2002; 115: 2381–8. [DOI] [PubMed] [Google Scholar]
- 21. McDonald SA, Greaves LC, Gutierrez‐Gonzalez L et al. Mechanisms of field cancerization in the human stomach: the expansion and spread of mutated gastric stem cells. Gastroenterology 2008; 134: 500–10. [DOI] [PubMed] [Google Scholar]
- 22. Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature 2005; 434: 843–50. [DOI] [PubMed] [Google Scholar]
- 23. North TE, Goessling W, Walkley CR et al. Prostaglandin E2 regulates vertebrate haematopoietic stem cell homeostasis. Nature 2007; 447: 1007–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Goessling W, North TE, Loewer S et al. Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell 2009; 136: 1136–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bleuming SA, He XC, Kodach LL et al. Bone morphogenetic protein signaling suppresses tumorigenesis at gastric epithelial transition zones in mice. Cancer Res 2007; 67: 8149–55. [DOI] [PubMed] [Google Scholar]
- 26. Wang D, Wang H, Shi Q et al. Prostaglandin E(2) promotes colorectal adenoma growth via transactivation of the nuclear peroxisome proliferator‐activated receptor delta. Cancer Cell 2004; 6: 285–95. [DOI] [PubMed] [Google Scholar]