

Abstract
In relation to carcinogenesis, aging and other pathologic conditions, urinary 8‐hydroxydeoxyguanosine (8OHdG) is widely used as a marker for evaluating the effect of oxidative stress on DNA. Because no reports have described how 8OHdG is generated from DNA in vivo or by biological materials, and how it is excreted into urine, the authors investigated the generation of 8OHdG from DNA, using rat liver homogenate. Oxidatively damaged DNA samples containing different levels of 8OHdG were prepared using ultraviolet irradiation with three different concentrations of riboflavin. Following incubation of damaged DNA samples with rat liver homogenates, the generation of 8OHdG from the DNA was determined using high‐performance liquid chromatography with electrochemical detection after ultrafiltration of the incubation mixtures. The generation of 8OHdG was also tested with an anti‐8OHdG antibody. The quantity of 8OHdG generated from the DNA by rat liver homogenates was dependent on the 8OHdG levels in the DNA: almost all 8OHdG in the DNA was released as 8OHdG by rat liver homogenates. Generation of 8OHdG correlated with the degradation of DNA. Interestingly, the generated 8OHdG was stable in the presence of rat liver homogenates, whereas deoxyguanosine (dG) rapidly disappeared in the same conditions. Less than 1/10 000 of dG was converted to 8OHdG when dG was incubated with rat liver homogenate. Incubation of 8‐hydroxyguanine with rat liver homogenates did not generate 8OHdG. These findings suggest that most of the 8OHdG in DNA is released as 8OHdG during DNA degradation and that, because of its stability, 8OHdG is excreted into urine, thus providing a convenient measure of oxidative damage to DNA. (Cancer Sci 2005; 96: 13–19)
Despite the presence of antioxidant defenses and DNA repair systems, oxidative damage to DNA is an inevitable consequence of metabolic activities, of ionizing radiation, and of environmental mutagens. 1 , 2 , 3 Such DNA damage is thought to play an important role in carcinogenesis, in aging and in a number of other pathological conditions. 4 , 5 , 6 Among the many types of oxidative base damage, 8‐hydroxydeoxyguanosine (8OHdG) is the most extensively studied, both because of its mutagenicity, 7 , 8 and because its presence can be determined with high sensitivity. 9 , 10 In reactive oxygen species‐related carcinogenesis, the level of 8OHdG in target tissues appears to play a critical role, 11 , 12 and this has led to 8OHdG being widely used as a marker of oxidative DNA damage. 13 , 14 However, because of the scantness of 8OHdG in DNA, and because of secondary formation during the analysis of 8OHdG in cellular DNA, urinary 8OHdG has been used to evaluate the level of 8OHdG in DNA, and a number of analytical methods have been developed with which to reliably measure 8OHdG in urine. 15 , 16 , 17 , 18 Furthermore, findings show that levels of urinary 8OHdG correlate well with many pathological conditions, particularly with carcinogenesis. 19 , 20 , 21
Even so, although urinary excretion of 8OHdG has been proposed as a candidate biomarker of oxidative stress to DNA, (22) the ultimate source of urinary 8OHdG has not been clarified. In humans, urinary excretion of 8‐hydroxyguanine (8OHG) and 8OHdG is reported to not depend on diet, (23) and may reflect the involvement of different repair mechanisms, namely base excision repair (BER) and nucleotide excision repair (NER). (24) BER is largely responsible for the removal of non‐bulky base adducts, and involves specialized enzymes that recognize a specific repertoire of lesions. In this process, a number of glycosylases have been identified. 25 , 26 These enzymes, however, excise damaged bases, resulting in the excretion of damaged bases, rather than damaged nucleosides, into urine. Another set of human 8OHdG repair enzymes, endonucleases, (27) along with the NER process, which probably acts simply as a back‐up system, (28) are likely to generate 8OHdG from DNA and thus contribute to the presence of 8OHdG in urine. No experimental evidence, however, has been provided to support this conjecture. Findings for several processes other than DNA repair indicate that other channels contribute to the background levels of 8OHdG that are excreted in urine. For example, even though proof of a defined role is still not forthcoming, (18) 8OHdG may derive from sanitation of the nucleotide pool by the action of human MutT homolog (MTH), 29 , 30 or from dead cells. (1) Potential sources of urinary 8OHdG have been collated in a comprehensive review. (31) Thus far, however, there have been neither reports that have described the generation of 8OHdG from DNA through incubation with tissue or cell extracts, nor have any researchers shown any correlation between the amount of 8OHdG generated and the 8OHdG levels in DNA.
In the present report, to more clearly elucidate the source of urinary 8OHdG, the authors investigated whether 8OHdG is generated from DNA by rat liver homogenate, and whether the amounts of generated 8OHdG correspond with the levels of oxidative damage in DNA.
Materials and Methods
Materials. 8OHdG and 8OHG were obtained from Cayman Chemical (Ann Arbor, MI, USA). Calf thymus DNA, bovine serum albumin, alkaline phosphatase, control mouse IgG1, protease inhibitor cocktail, deoxyguanosine (dG) and ethidium bromide were obtained from Sigma Chemical (St Louis, MO, USA). Nuclease P1 came from Seikagaku Corporation (Tokyo, Japan). IgG1 class mouse monoclonal anti‐8OHdG antibody (Clone N45.1) was purchased from the Japanese Aging Control Institute (Shizuoka, Japan). DNA marker and loading buffer were from BEXEL Biotechnology (Union City, CA, USA). All other reagents were reagent grade and purchased from Nacalai Tesque (Kyoto, Japan).
Preparation of rat liver homogenate. Male Wistar rats aged 11 weeks were killed under deep ether anesthesia and the livers were promptly removed, frozen in liquid nitrogen, and stored at −80°C until needed. Using a Teflon‐glass homogenizer, livers were homogenized in five volumes of ice‐cold homogenization buffer (20 mmol/L Tris‐HCl pH 7.4, containing 0.25 mol/L sucrose and 1% v/v protease inhibitor cocktail). The homogenates were filtered through nylon mesh to remove clumps of connective tissue attached to unbroken cells and then were stored at −80°C. The contaminated DNA concentration in the homogenates was 0.04 mg/mL.
Preparation of oxidatively damaged DNA. Calf thymus DNA was dissolved with Dulbecco's phosphate‐buffered saline (DPBS) at 2.0 mg/mL and incubated with three different concentrations of riboflavin (50, 10, 2 µg/mL). The mixtures were then irradiated with ultraviolet (UV) at 365 nm (UVGL‐58; UVP, Upland, CA, USA) for 10 min at room temperature. The dose of irradiated UV was calculated to be 0.6 J/cm2. After irradiation, DNA was precipitated and washed with ethanol. The DNA was then dissolved with DPBS and precipitated again with ethanol to remove residual riboflavin. Finally the DNA was dissolved in DPBS at 1.0 mg/mL. The levels of 8OHdG in the damaged DNA were determined as described below.
8OHdG release from oxidatively damaged DNA. For the indicated durations, 10‐µL samples of oxidatively damaged DNA were incubated with 15 µL of rat liver homogenate at 37°C. After this, the incubation mixtures were diluted with 75 µL of double distilled water and ultrafiltered with YM10 (Millipore; Billerica, MA, USA) at 13 400 g for 30 min. The quantities of 8OHdG in the ultrafiltrates were determined as described below. In the degradation experiments, dG, 8OHG, or 8OHdG were incubated with the rat liver homogenates, and the mixtures were ultrafiltered as described above.
Determination of 8OHdG and dG. The level of 8OHdG in the damaged DNA was determined as described previously. (32) Briefly, DNA was heat denatured and then digested sequentially with nuclease P1 and alkaline phosphatase. The generated 8OHdG was determined using an electrochemical detector (ECD, Coulochem II; ESA, Chelmsford, MA, USA) and dG with a UV detector: both methods were combined with previously described (33) high‐performance liquid chromatography (HPLC). As described above, the dG levels in the dG‐degradation experiment and the quantities of 8OHdG that were generated from damaged DNA after incubation with rat liver homogenates were determined. The authors also detected some 8OHG under the same conditions as for the 8OHdG determination; however, because of the close proximity of other peaks close to the 8OHG peak, an accurate determination of the small amounts of 8OHG was not possible.
Absorption of 8OHdG with anti‐8OHdG antibody. After incubation of oxidatively damaged DNA with rat liver homogenates at 37°C for 18 h, the incubation mixture was ultrafiltered through YM10, then the ultrafiltrate was incubated with either anti‐8OHdG antibody, control IgG1 or DPBS at 37°C for 60 min. The molar ratio of 8OHdG to antibody or control IgG1 was 1:4. The mixtures were then ultrafiltered again with YM10, and the ultrafiltrates (final ultrafiltrates) were applied to the HPLC–ECD system to determine the quantity of 8OHdG.
DNA degradation determined using electrophoresis. After DNA was incubated with rat liver homogenates as described above, the incubation mixtures were loaded onto 2% agarose gels containing 0.5 × TBE (45 mM Tris‐boric acid with 1 mM ethylenediamine tetra‐acetic acid, pH 8.0) and ethidium bromide, and then electrophoresed with 0.5 × TBE buffer. The separated fragments were made visible on the agarose gel using a UV transilluminator and DNA profiles were taken using a camera.
Protein assay. The protein concentrations of rat liver homogenates were determined using a Bio‐Rad protein assay solution (Bio‐Rad, Hercules, CA, USA). Bovine serum albumin was used as a standard.
Statistical analysis. Data are presented as means ± standard error. Statistical analyses were carried out using one‐way anova; P‐values of <0.05 were considered to be statistically significant.
Results
Generation of 8OHdG from oxidatively damaged DNA by rat liver homogenate. After subjecting the DNA to UV irradiation with different concentrations of riboflavin, 8OHdG in DNA was concentration‐dependently induced. For 10 µg of DNA, the amount of induced 8OHdG was: 26.7 ng with riboflavin 50 µg/mL, 12.5 ng with 10 µg/mL, or 3.7 ng with 2 µg/mL (Table 1).
Table 1.
Different levels of 8‐hydroxydeoxyguanosine (8OHdG) in damaged DNA and generation by rat liver homogenates of 8OHdG from the DNA
| Quantity of 8OHdG in 10 µg DNA (ng) | Quantity of 8OHdG released (ng) | Quantity of 8OHdG released(%) | |
|---|---|---|---|
| A | 26.7 ± 0.59 | 18.58 ± 0.55 | 69.7 ± 3.5 |
| B | 12.5 ± 0.10 | 8.99 ± 0.39 | 72.1 ± 3.2 |
| C | 3.7 ± 0.02 | 2.78 ± 0.08 | 75.1 ± 2.6 |
Ultraviolet irradiation with (A) 50 µg/mL, (B) 10 µg/mL, or (C) 2 µg/mL riboflavin induced different levels of 8OHdG. The percentage released indicates the ratio of the amount of released 8OHdG to the total amount of 8OHdG in the DNA. Data from a typical experiment conducted in triplicate are presented as mean ± SE.
When oxidatively damaged DNA (prepared using UV irradiation with 50 µg/mL riboflavin) was incubated with varying concentrations of rat liver homogenates, concentration‐dependent generation of 8OHdG was observed up to an 8 mg/mL concentration (Fig. 1). The authors found no further increase in 8OHdG generation from DNA at homogenate concentrations of >8 mg/mL. In the following experiments, to maximally generate 8OHdG, 12 mg/mL of rat liver homogenate was thus used. In contrast, no 8OHdG was generated when, for 24 h, rat liver homogenates were incubated alone without damaged DNA, or when the damaged DNA was incubated with homogenization buffer but without homogenate.
Figure 1.
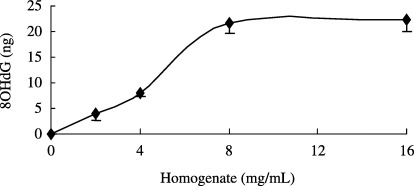
Release of 8‐hydroxydeoxyguanosine (8OHdG) from oxidatively damaged DNA. After ultraviolet irradiation in the presence of 50 µg/mL riboflavin, consequently damaged DNA was incubated with the indicated concentrations of rat liver homogenates at 37°C for 24 h. After incubation, the reaction mixtures were ultrafiltered and the quantity of 8OHdG in the ultrafiltrate was determined as described in Materials and Methods. Results from two independent experiments conducted in duplicate are presented as mean ± SE.
As shown in Figure 2, 8OHdG was time‐dependently generated from oxidatively damaged DNA after incubation with rat liver homogenates. When DNA samples with different degrees of damage were treated for the same incubation time, the quantities of 8OHdG that were generated correlated with the 8OHdG levels in DNA. The greater the presence of 8OHdG in DNA, the greater its release by rat liver homogenates. After a 24‐h incubation period, approximately 70% of the 8OHdG in DNA was released as 8OHdG (Table 1), and no significant differences in the percentage of 8OHdG released from the original levels of 8OHdG in the different samples were found.
Figure 2.
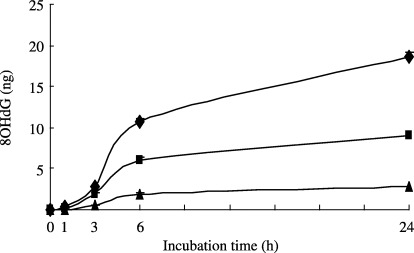
Time course of 8‐hydroxydeoxyguanosine (8OHdG) generation from DNA with different levels of 8OHdG. Ultraviolet irradiation with (◆) 50 µg/mL, (▪) 10 µg/mL or (▴) 2 µg/mL riboflavin‐damaged DNA samples were incubated with rat liver homogenates at 37°C for 1, 3, 6, or 24 h. Then the incubation mixtures were ultrafiltered through YM10 and the quantities of 8OHdG in the ultrafiltrates were determined as described in Materials and Methods. Results from a typical experiment conducted in triplicate is presented as mean ± SE.
Absorption of 8OHdG with anti‐8OHdG antibody. To confirm the generation of 8OHdG from DNA by rat liver homogenates, the absorption of 8OHdG by anti‐8OHdG antibody was tested. As Figure 3 shows, the final ultrafiltrate of the reaction mixture of rat liver homogenates and DNA peaked at the same elution time as authentic 8OHdG. Control IgG1 did not absorb the peak in the ultrafiltrate; however, anti‐8OHdG antibody at the same concentration as control IgG1 absorbed the peak almost completely.
Figure 3.

High‐performance liquid chromatography–electrochemical detector chromatogram of the final ultrafiltrates incubated with anti‐8‐hydroxydeoxyguanosine (8OHdG) antibody or control IgG1. After incubation of DNA (damaged by ultraviolet irradiation with 50 µg/mL riboflavin) with rat liver homogenates at 37°C for 18 h, the incubation mixture was ultrafiltered. The ultrafiltrate was incubated with Dulbecco's phosphate‐buffered saline (B), control IgG1 (C), or anti‐8OHdG antibody (D), then the mixtures were ultrafiltered again with YM10. Quantities of 8OHdG in the final ultrafiltrates were determined as described in Materials and Methods. Peak A indicates authentic 8OHdG, and ‘I’ indicates the injection points of samples.
Degradation of DNA by rat liver homogenates. As Figure 4 shows, rat liver homogenates degraded DNA that had been damaged by UV irradiation in the presence of 50 µg/mL riboflavin. Oxidatively damaged DNA was considerably degraded even after 1 h of incubation, and the DNA degradation by rat liver homogenates was dependent on the incubation time. In contrast, when damaged DNA was incubated for 24 h without rat liver homogenates, it was not degraded (C24 vs C0). Although the DNA was extensively damaged by UV irradiation in the presence of riboflavin, its size was larger than 1000 bp (C0).
Figure 4.
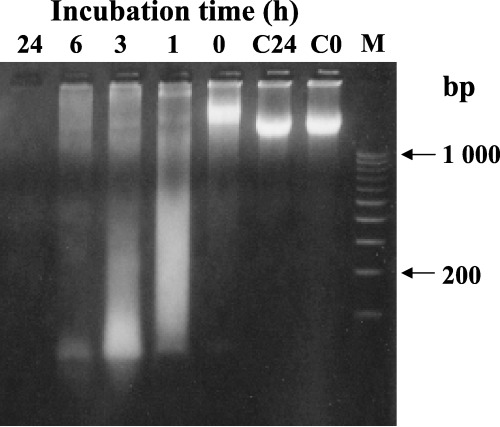
Electrophoretic determination of DNA degradation by rat liver homogenates. DNA (damaged by ultraviolet irradiation with 50 µg/mL riboflavin) was incubated with rat liver homogenates at 37°C for the indicated durations. Then incubation mixtures containing 1.0 µg DNA were subjected to electrophoresis as described in Materials and Methods. C0, C24: samples of damaged DNA were incubated with homogenization buffer at 37°C for 0 h (C0) or 24 h (C24). M, marker DNA fragments, sizes of marker fragments are indicated on the right.
Changes in the amount of dG, 8OHG or 8OHdG in the presence of rat liver homogenates. As Figure 5A shows, when dG was incubated with rat liver homogenates, a small amount of 8OHdG was detected at 0 h of incubation, and this amount increased slightly after 6 h of incubation. At 6 h, however, the amount of 8OHdG generated from dG was <1/10 000 of dG (Fig. 5A). In the presence of rat liver homogenates, dG disappeared rapidly. More than 70% of dG had disappeared even after 0.5 h of incubation: after 3 h none could be detected. By contrast, in the presence of rat liver homogenates, 8OHG and 8OHdG did not breakdown significantly after 6 h, and more than 75% of 8OHG and 8OHdG were detected unchanged even after 24 h of incubation (Fig. 5B,C). 8OHdG was not generated from 8OHG by rat liver homogenates.
Figure 5.
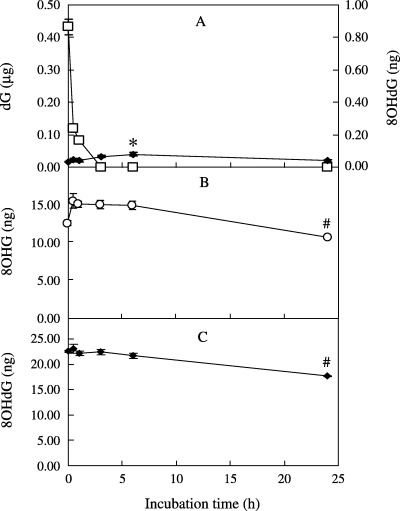
Effect of incubation time on (□) quantity of deoxyguanosine (dG), (○) 8‐hydroxyguanine (8OHG), or (◆) 8‐hydroxydeoxyguanosine (8OHdG) in the presence of rat liver homogenates. (A) dG (0.5 µg). (B) 8OHG (16 ng). (C) 8OHdG (25 ng). Amounts were calculated from samples of 10 µg of DNA that were damaged by ultraviolet irradiation with 50 µg/mL riboflavin. These were incubated at 37°C with rat liver homogenates for 0, 0.5, 1, 3, 6 or 24 h. After incubation, the mixtures were ultrafiltered and the quantities of dG, 8OHG, or 8OHdG in the ultrafiltrates were determined as described in Materials and Methods. Data from a typical experiment conducted in triplicate are presented as mean ± SE. *P < 0.05, significantly increased when compared with 0 h; #P < 0.05, significantly decreased when compared with 0 h.
Discussion
Urinary 8OHdG is widely used as a biological marker with which to evaluate oxidative stress in the body. 22 , 34 , 35 Its usefulness, however, has so far been limited because we do not know enough about how 8OHdG comes to be present in urine. 23 , 31 No evidence has been presented that 8OHdG is released from DNA by tissues, cells, or by their extracts. Here, the authors clearly show that 8OHdG is generated from DNA by rat liver homogenates (Fig. 1). Because other compounds that are present in rat liver homogenates might have, in HPLC analysis, produced peaks at the same position as 8OHdG, the authors tested with an anti‐8OHdG antibody. The antibody absorbed the peak almost completely, indicating that 8OHdG was generated from DNA (Fig. 3). Our data also show that the quantity of 8OHdG generated from the DNA corresponds with the level of oxidative damage in the DNA (Fig. 2). These findings indicate that, in vivo, 8OHdG is generated from DNA, and that the amounts of generated 8OHdG are useful for evaluating oxidative damage in DNA. However, attention should be paid when determining the 8OHdG quantity (see Fig. 2).
A semiquantitative assay of 8OHG using the 8OHdG detection system showed that the quantities of 8OHG produced from DNA were approximately 1/25 of the quantities of 8OHdG after incubation of DNA with rat liver homogenates, and that the proportion did not vary with the 8OHdG levels in DNA (data not shown). It might be thought that just 70% of 8OHdG was released from DNA after a 24‐h incubation period (Table 1). However, Figure 5C shows that 75% of 8OHdG could be recovered after the same incubation time in the presence of rat liver homogenates. Thus, in this system, the authors considered that most of the 8OHdG in damaged DNA was released as 8OHdG.
The authors were surprised that most of the 8OHdG was released from DNA by the rat liver homogenates. At first, it was considered that 8OHdG was generated during the DNA repair process, because dG, a DNA degradation product, (1) was barely detectable in the ultrafiltrates. The result of electrophoresis (Fig. 4), however, indicated that the generation of 8OHdG co‐occurred during DNA degradation. In the presence of rat liver homogenates, it is possible that dG rapidly disappeared, which was confirmed as shown in Figure 5A. When DNA was incubated with rat liver homogenates for 1 h, the electrophoretic mobility of oxidatively damaged DNA was decreased, probably due to the interaction between the DNA and the proteins in the homogenate.
It is also possible that 8OHdG is generated from dG or 8OHG. In particular, generation from dG has been reported in the co‐presence of oxidants. 9 , 36 , 37 Commercially available dG preparations usually contain 1–5 molecules of 8OHdG per 100 000 of dG (data not shown). When dG was incubated with rat liver homogenates, however, <1/10 000 of the dG was converted to 8OHdG during 6 h of incubation (Fig. 5A). Meanwhile, 8OHdG was not generated when 8OHG was incubated with rat liver homogenates (Fig. 5B). These findings indicate that, during DNA degradation, 8OHdG was generated directly from DNA. The authors suggest that 8OHdG is generated when oxidative stress causes tissue or cell destruction: in such conditions, both oxidative DNA damage and tissue or cell homogenates could be produced. Because oxidative stress induces apoptosis, 38 , 39 and DNA is extensively degraded during apoptosis, (40) apoptotic cells might also be sources of 8OHdG. The authors are now investigating whether living cells could also generate 8OHdG from DNA using a cell culture system.
Liver contains many types of nuclease 41 , 42 , 43 , 44 that degrade DNA to nucleotides. In turn, these can be dephosphorylated to nucleosides by the phosphatases that are also present in the liver. 45 , 46 Some nucleases in the liver are reported to be sensitive to NaCl, 47 , 48 and when NaCl was added to the incubation mixtures, NaCl at concentrations of more than 150 mmol/L inhibited the generation of 8OHdG (data not shown). The finding further supports our conclusion that 8OHdG generation is coupled with DNA degradation. Thus it seems plausible that, in the present experiment, the nucleases and phosphatases present in the liver were responsible for the generation of 8OHdG from DNA. Additionally, in support of this conclusion, the technique for determining 8OHdG in DNA uses nuclease P1, an exonuclease, and alkaline phosphatase. 11 , 33 , 49 Further study, however, is required to identify which enzyme or enzymes are responsible for the generation of 8OHdG from DNA. Furthermore, investigation as to which organ most efficiently generates 8OHdG may eventually make it possible to use urinary 8OHdG to evaluate organ‐specific oxidative stress. In contrast to rat liver homogenates, Fpg protein, a bacterial homolog of oxoguanine glycosylase that acts as a DNA BER enzyme, 50 , 51 generated 8OHG from DNA, but not 8OHdG (data not shown).
It is interesting that, while dG rapidly disappeared under the same conditions, in the presence of rat liver homogenates more than 75% of 8OHG and 8OHdG remained unchanged up to 24 h of incubation (Fig. 5). These findings suggest that 8OHdG and 8OHG are stable in the body and in the circulation, and so may be excreted into urine unchanged, whereas most of dG undergoes breakdown and may not be detectable in urine as intact dG. This hypothesis is supported by the finding that the quantities of 8OHdG and 8OHG in urine are greatly disproportionate to the quantity of dG in urine. 18 , 22 , 52 It is also interesting that 8OHdG and 8OHG seem not to be metabolized or reused, suggesting the presence of mechanisms that do not allow the naturally occurring damaged base to be incorporated into nucleic acids. Our discovery of the stability of 8OHdG in the presence of rat liver homogenates suggests a useful substrate that could be used to study nucleases. Because 8OHdG is a stable product of nuclease reaction and can be determined with high sensitivity, DNA with 8OHdG seems to be a better substrate than DNA without 8OHdG.
In conclusion, 8OHdG is released from DNA by rat liver homogenates in quantities that correspond with the levels of oxidative damage in the DNA. Because 8OHdG is stable in the presence of rat liver homogenates, it is likely that 8OHdG is stable enough in circulation to be excreted into urine. Thus, urinary 8OHdG, if determined at appropriate times or with 24‐h urine testing, is a useful marker of oxidative DNA damage that is induced by oxidative stress, particularly oxidative stress that leads to the organ or cell destruction, or apoptosis. Although the present results do not show the in vivo generation of 8OHdG from DNA directly, they show that 8OHdG is generated from DNA by a biological material, rat liver homogenate.
Acknowledgments
This work was supported by Grand‐in‐Aid for Scientific Research awarded by the Ministry of Education, Science, Sports and Culture of Japan (#14370140).
The authors express thanks to Professor Kanehisa Morimoto (Department of Social and Environmental Medicine, Course of Social Medicine, Osaka University Graduate School of Medicine) for his encouragement and support of this work.
References
- 1. Lindahl T. Instability and decay of the primary structure of DNA. Nature 1993; 362: 709–15. [DOI] [PubMed] [Google Scholar]
- 2. Beckman KB, Ames BN. Oxidative decay of DNA. J Biol Chem 1997; 272: 19633–6. [DOI] [PubMed] [Google Scholar]
- 3. Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. Oxford: Oxford University Press, 1999. [Google Scholar]
- 4. Halliwell B, Aruoma OI. DNA damage by oxygen‐derived species. Its mechanism and measurement in mammalian systems. FEBS Lett 1991; 281: 9–19. [DOI] [PubMed] [Google Scholar]
- 5. Ames BN. Dietary carcinogens and anticarcinogens. Oxygen radicals and degenerative diseases. Science 1983; 221: 1256–64. [DOI] [PubMed] [Google Scholar]
- 6. Zhang J, Perry G, Smith MA, Robertson D, Olson SJ, Graham DG, Montine TJ. Parkinson's disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. Am J Pathol 1999; 154: 1423–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shibutani S, Takeshita M, Grollman AP. Insertion of specific bases during DNA synthesis past the oxidation‐damaged base 8‐oxodG. Nature 1991; 349: 431–4. [DOI] [PubMed] [Google Scholar]
- 8. Cheng KC, Cahill DS, Kasai H, Nishimura S, Loeb LA. 8‐Hydroxyguanine, an abundant form of oxidative DNA damage, causes G—T and A—C substitutions. J Biol Chem 1992; 267: 166–72. [PubMed] [Google Scholar]
- 9. Floyd RA, Watson JJ, Wong PK, Altmiller DH, Rickard RC. Hydroxyl free radical adduct of deoxyguanosine: sensitive detection and mechanisms of formation. Free Radic Res Commun 1986; 1: 163–72. [DOI] [PubMed] [Google Scholar]
- 10. Kasai H. Analysis of a form of oxidative DNA damage, 8‐hydroxy‐2′‐deoxyguanosine, as a marker of cellular oxidative stress during carcinogenesis. Mutat Res 1997; 387: 147–63. [DOI] [PubMed] [Google Scholar]
- 11. Foksinski M, Kotzbach R, Szymanski W, Olinski R. The level of typical biomarker of oxidative stress 8‐hydroxy‐2′‐deoxyguanosine is higher in uterine myomas than in control tissues and correlates with the size of the tumor. Free Radic Biol Med 2000; 29: 597–601. [DOI] [PubMed] [Google Scholar]
- 12. Floyd RA. The role of 8‐hydroxyguanine in carcinogenesis. Carcinogenesis 1990; 11: 1447–50. [DOI] [PubMed] [Google Scholar]
- 13. Kasai H, Crain PF, Kuchino Y, Nishimura S, Ootsuyama A, Tanooka H. Formation of 8‐hydroxyguanine moiety in cellular DNA by agents producing oxygen radicals and evidence for its repair. Carcinogenesis 1986; 7: 1849–51. [DOI] [PubMed] [Google Scholar]
- 14. Collins AR, Gedik CM, Olmedilla B, Southon S, Bellizzi M. Oxidative DNA damage measured in human lymphocytes: large differences between sexes and between countries, and correlations with heart disease mortality rates. FASEB J 1998; 12: 1397–400. [PubMed] [Google Scholar]
- 15. Yoshida R, Ogawa Y, Kasai H. Urinary 8‐oxo‐7,8‐dihydro‐2′‐deoxyguanosine values measured by an ELISA correlated well with measurements by high‐performance liquid chromatography with electrochemical detection. Cancer Epidemiol Biomarkers Prev 2002; 11: 1076–81. [PubMed] [Google Scholar]
- 16. Tagesson C, Kallberg M, Klintenberg C, Starkhammar H. Determination of urinary 8‐hydroxydeoxyguanosine by automated coupled‐column high performance liquid chromatography: a powerful technique for assaying in vivo oxidative DNA damage in cancer patients. Eur J Cancer 1995; 31: 934–40. [DOI] [PubMed] [Google Scholar]
- 17. Lin HS, Jenner AM, Ong CN, Huang SH, Whiteman M, Halliwell B. A high‐throughput and sensitive methodology for the quantification of urinary 8‐hydroxy‐2′‐deoxyguanosine: measurement with gas chromatography‐mass spectrometry after single solid‐phase extraction. Biochem J 2004; 380: 541–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Weimann A, Belling D, Poulsen HE. Quantification of 8‐oxo‐guanine and guanine as the nucleobase, nucleoside and deoxynucleoside forms in human urine by high‐performance liquid chromatography‐electrospray tandem mass spectrometry. Nucl Acids Res 2002; 30: E7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wu LL, Chiou CC, Chang PY, Wu JT. Urinary 8‐OHdG: a marker of oxidative stress to DNA and a risk factor for cancer, atherosclerosis and diabetics. Clin Chim Acta 2004; 339: 1–9. [DOI] [PubMed] [Google Scholar]
- 20. Loft S, Vistisen K, Ewertz M, Tjonneland A, Overvad K, Poulsen HE. Oxidative DNA damage estimated by 8‐hydroxydeoxyguanosine excretion in humans: influence of smoking, gender and body mass index. Carcinogenesis 1992; 13: 2241–7. [DOI] [PubMed] [Google Scholar]
- 21. Gackowski D, Speina E, Zielinska M et al. Products of oxidative DNA damage and repair as possible biomarkers of susceptibility to lung cancer. Cancer Res 2003; 63: 4899–902. [PubMed] [Google Scholar]
- 22. Shigenaga MK, Gimeno CJ, Ames BN. Urinary 8‐hydroxy‐2′‐deoxyguanosine as a biological marker of in vivo oxidative DNA damage. Proc Natl Acad Sci USA 1989; 86: 9697–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gackowski D, Rozalski R, Roszkowski K, Jawien A, Foksinski M, Olinski R. 8‐Oxo‐7,8‐dihydroguanine and 8‐oxo‐7,8‐dihydro‐2′‐deoxyguanosine levels in human urine do not depend on diet. Free Radic Res 2001; 35: 825–32. [DOI] [PubMed] [Google Scholar]
- 24. Lunec J, Holloway KA, Cooke MS, Faux S, Griffiths HR, Evans MD. Urinary 8‐oxo‐2′‐deoxyguanosine. redox regulation of DNA repair in vivo? Free Radic Biol Med 2002; 33: 875–85. [DOI] [PubMed] [Google Scholar]
- 25. Demple B, Harrison L. Repair of oxidative damage to DNA, enzymology and biology. Annu Rev Biochem 1994; 63: 915–48. [DOI] [PubMed] [Google Scholar]
- 26. Hazra TK, Izumi T, Maidt L, Floyd RA, Mitra S. The presence of two distinct 8‐oxoguanine repair enzymes in human cells: their potential complementary roles in preventing mutation. Nucl Acids Res 1998; 26: 5116–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bessho T, Tano K, Kasai H, Ohtsuka E, Nishimura S. Evidence for two DNA repair enzymes for 8‐hydroxyguanine (7,8‐dihydro‐8‐oxoguanine) in human cells. J Biol Chem 1993; 268: 19416–21. [PubMed] [Google Scholar]
- 28. Dianov G, Bischoff C, Piotrowski J, Bohr VA. Repair pathways for processing of 8‐oxoguanine in DNA by mammalian cell extracts. J Biol Chem 1998; 273: 33811–6. [DOI] [PubMed] [Google Scholar]
- 29. Hayakawa H, Taketomi A, Sakumi K, Kuwano M, Sekiguchi M. Generation and elimination of 8‐oxo‐7,8‐dihydro‐2′‐deoxyguanosine 5′‐triphosphate, a mutagenic substrate for DNA synthesis, in human cells. Biochemistry 1995; 34: 89–95. [DOI] [PubMed] [Google Scholar]
- 30. Maki H, Sekiguchi M. MutT protein specifically hydrolyses a potent mutagenic substrate for DNA synthesis. Nature 1992; 355: 273–5. [DOI] [PubMed] [Google Scholar]
- 31. Cooke MS, Evans MD, Herbert KE, Lunec J. Urinary 8‐oxo‐2′‐deoxyguanosine – source, significance and supplements. Free Radic Res 2000; 32: 381–97. [DOI] [PubMed] [Google Scholar]
- 32. Takeuchi T, Nakajima M, Morimoto K. Establishment of a human system that generates O2‐ and induces 8‐hydroxydeoxyguanosine, typical of oxidative DNA damage, by a tumor promoter. Cancer Res 1994; 54: 5837–40. [PubMed] [Google Scholar]
- 33. Takeuchi T, Nakajima M, Ohta Y, Mure K, Takeshita T, Morimoto K. Evaluation of 8‐hydroxydeoxyguanosine, a typical oxidative DNA damage, in human leukocytes. Carcinogenesis 1994; 15: 1519–23. [DOI] [PubMed] [Google Scholar]
- 34. Loft S, Poulsen HE. Estimation of oxidative DNA damage in man from urinary excretion of repair products. Acta Biochim Pol 1998; 45: 133–44. [PubMed] [Google Scholar]
- 35. Bergtold DS, Simic MG, Alessio H, Cutler RG. Urine biomarkers for oxidative DNA damage. In: Simic MG, Taylor KA, Ward JF, Von Sontag C, eds. Oxygen Radicals in Biology and Medicine. New York: Plenum Press, 1988; 483–90. [DOI] [PubMed] [Google Scholar]
- 36. Kasai H, Nishimura S. Hydroxylation of deoxyguanosine at the C‐8 position by ascorbic acid and other reducing agents. Nucl Acids Res 1984; 12: 2137–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Greenberg MM. In vitro and in vivo effects of oxidative damage to deoxyguanosine. Biochem Soc Trans 2004; 32: 46–50. [DOI] [PubMed] [Google Scholar]
- 38. Higuchi Y. Chromosomal DNA fragmentation in apoptosis and necrosis induced by oxidative stress. Biochem Pharmacol 2003; 66: 1527–35. [DOI] [PubMed] [Google Scholar]
- 39. Simon HU, Haj‐Yehia A, Levi‐Schaffer F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000; 5: 415–18. [DOI] [PubMed] [Google Scholar]
- 40. Hengartner MO, Apoptosis. DNA, destroyers. Nature 2001; 412: 27–9. [DOI] [PubMed] [Google Scholar]
- 41. Davies AM, Hershman S, Stabley GJ, Hoek JB, Peterson J, Cahill A. A Ca2+‐induced mitochondrial permeability transition causes complete release of rat liver endonuclease G activity from its exclusive location within the mitochondrial intermembrane space. Identification of a novel endo‐exonuclease activity residing within the mitochondrial matrix. Nucl Acids Res 2003; 31: 1364–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Barry ME, Pinto‐Gonzalez D, Orson FM, McKenzie GJ, Petry GR, Barry MA. Role of endogenous endonucleases and tissue site in transfection and CpG‐mediated immune activation after naked DNA injection. Hum Gene Ther 1999; 10: 2461–80. [DOI] [PubMed] [Google Scholar]
- 43. Hibino Y, Iwakami N, Sugano N. A nuclease from rat‐liver nuclei with endo‐ and exonucleolytic activity. Biochim Biophys Acta 1991; 1088: 305–7. [DOI] [PubMed] [Google Scholar]
- 44. Cordis GA, Goldblatt PJ, Deutscher MP. Purification and characterization of a major endonuclease from rat liver nuclei. Biochemistry 1975; 14: 2596–603. [DOI] [PubMed] [Google Scholar]
- 45. Toh Y, Yamamoto M, Endo H, Misumi Y, Ikehara Y. Isolation and characterization of a rat liver alkaline phosphatase gene. A single gene with two promoters. Eur J Biochem 1989; 182: 231–7. [DOI] [PubMed] [Google Scholar]
- 46. Alpers DH, Eliakim R, DeSchryver‐Kecskemeti K. Secretion of hepatic and intestinal alkaline phosphatases: similarities and differences. Clin Chim Acta 1990; 186: 211–23. [DOI] [PubMed] [Google Scholar]
- 47. Yusifov TN, Abduragimov AR, Gasymov OK, Glasgow BJ. Endonuclease activity in lipocalins. Biochem J 2000; 347: 815–19. [PMC free article] [PubMed] [Google Scholar]
- 48. Pan CQ, Lazarus. RA. Hyperactivity of human DNase I variants. Dependence on the number of positively charged residues and concentration, length, and environment of DNA. J Biol Chem 1998; 273: 11701–8. [DOI] [PubMed] [Google Scholar]
- 49. Fraga CG, Shigenaga MK, Park JW, Degan P, Ames BN. Oxidative damage to DNA during aging. 8‐hydroxy‐2′‐deoxyguanosine in rat organ DNA and urine. Proc Natl Acad Sci USA 1990; 87: 4533–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tchou J, Bodepudi V, Shibutani S, Antoshechkin I, Miller J, Grollman AP, Johnson F. Substrate specificity of Fpg protein. Recognition and cleavage of oxidatively damaged DNA. J Biol Chem 1994; 269: 15318–24. [PubMed] [Google Scholar]
- 51. David SS, Williams SD. Chemistry of glycosylases and endonucleases involved in base‐excision repair. Chem Rev 1998; 98: 1221–62. [DOI] [PubMed] [Google Scholar]
- 52. Weimann A, Riis B, Poulsen HE. Oligonucleotides in human urine do not contain 8‐oxo‐7,8‐dihydrodeoxyguanosine. Free Radic Biol Med 2004; 36: 1378–82. [DOI] [PubMed] [Google Scholar]