

Abstract
Cyclooxygenase (COX)‐2 is overexpressed in many human tumors including neuroblastoma (NB) and promotes tumor progression. We evaluated the antitumor effect of irinotecan (CPT‐11) treatment combined with prolonged very low‐dose administration of celecoxib, a selective COX‐2 inhibitor, against three human NB xenografts, TNB9, TS‐N‐2nu, and TS‐N‐5nu. In addition, the effects of the celecoxib‐combined treatment were examined on tumor cell proliferation, apoptosis, angiogenesis, and expression of vascular endothelial growth factor and apoptosis‐related proteins in xenografts. Celecoxib administered daily at 5 mg/kg body weight/day could not prevent the growth of any of the NB xenografts. However, the combination of daily low‐dose CPT‐11 (5.9 mg/kg body weight/day) and simultaneous very low‐dose celecoxib resulted in highly significant suppression of tumor growth in all three xenografts (P < 0.001) compared not only with low‐dose CPT‐11 therapy alone but also with the combination therapy of intermittent conventional‐dose CPT‐11 (59 mg/kg body weight) and celecoxib accompanied by decreased proliferation and increased induction of apoptosis in tumor cells. Induction of apoptosis by CPT‐11 with and without celecoxib was associated with the up‐regulation of Bax expression and the down‐regulation of Bcl‐2 expression. The enhanced antitumor effect of the combination of the two drugs against the NB xenografts might be partially COX‐2‐independent and was probably mediated through multiple factors including diminished expression of VEGF and activation of the caspase‐dependent mitochondrial apoptosis pathway. These findings demonstrate that prolonged low‐dose CPT‐11 treatment combined with very low‐dose celecoxib shows promising antitumor activity through the blockage of multiple critical targets related to NB tumor cell survival and proliferation. (Cancer Sci 2009; 00: 000–000)
Neuroblastoma (NB) is one of the most common malignant childhood solid tumors. There have been significant advances in the treatment of NB with intensive induction and consolidation chemotherapy regimens. However, the treatment results for patients aged 1 year or older with disseminated disease and for those with MYCN‐amplified NB are still unsatisfactory. Among the antitumor drugs with therapeutic activity, irinotecan (CPT‐11), a semisynthetic derivative of camptothecin, is promising for NB treatment.( 1 , 2 , 3 , 4 ) Recently, phase II studies of CPT‐11 in children with refractory solid tumors including NB and with relapsed or refractory NB concluded that CPT‐11 treatment was well tolerated, but not effective as a single agent, with the possible exception of its use against medulloblastoma.( 5 , 6 ) We have previously shown that CPT‐11 is highly effective against human NB xenografts when administered at a conventional dose (LD50 × 1/3 per dose) at 4‐day intervals according to the protocol of Battelle Columbus laboratories, and that a daily low‐dose (LD50 × 1/30 per dose) administration is equally or more effective than conventional‐dose intermittent CPT‐11 treatment.( 7 ) However, treatment with CPT‐11 alone could not eradicate the tumor in mice.
Cyclooxygenases (COXs) catalyze the conversion of arachidonic acid to prostaglandins and consist of two isoenzymes, the constitutive COX‐1 and the inducible COX‐2. COX‐1 is constitutively and ubiquitously expressed in most tissues, whereas COX‐2 is induced by various inflammatory and mitogenic stimuli such as cytokines and growth factors.( 8 ) Recent studies suggest that COX‐2 promotes tumorigenesis by stimulating cancer cell proliferation, increasing tumor angiogenesis, preventing cancer cell apoptosis, and enhancing tumor metastasis.( 9 , 10 , 11 , 12 , 13 ) COX‐2 is overexpressed in various human tumors including NB.( 14 , 15 ) Besides its anti‐inflammatory and analgesic effects, selective COX‐2 inhibitors have been shown to exert antitumor activity through many mechanisms, especially via anti‐angiogenic and pro‐apoptotic effects.( 15 , 16 , 17 , 18 ) We evaluated the inhibitory effect of very low‐dose celecoxib, a selective COX‐2 inhibitor, on tumor growth combined with prolonged low‐dose or intermittent conventional‐dose CPT‐11 treatment against three neuroblastoma xenografts in mice. We further investigated the in vivo effects of combined treatment involving low‐dose CPT‐11 and celecoxib on tumor cell proliferation, apoptosis, angiogenesis, and the expression of vascular endothelial growth factor (VEGF) and apoptosis‐related proteins in xenografts, as well as the in vitro effects of low concentrations of celecoxib on cytotoxic activity of SN‐38, an active metabolite of CPT‐11, and expression of caspases in NB cell lines.
Materials and Methods
Animals and human NB xenografts. Human NB xenografts designated TNB9, TS‐N‐2nu, and TS‐N‐5nu were derived from stage 4 NBs with 80, 13, and more than 100 copies of the MYCN amplification developed in the adrenal gland of a 15‐month‐old boy, the adrenal gland of a 4‐year‐old girl, and the retroperitoneal region of a 13‐month‐old boy, respectively. Among the three xenografts, TNB9 and TS‐N‐2nu are chemosensitive and multidrug‐resistant, respectively.( 7 ) Small pieces of minced tumor were implanted subcutaneously with trochars into the unilateral or bilateral flanks of 5‐week‐old male Balb/cAJcl‐nu mice (Clea Japan, Tokyo, Japan). After tumor implantation, the mice were randomized into groups of three to five and monitored for tumor growth. Tumor weight was calculated according to the formula: tumor weight (mg) = the length of the tumor (mm) × the width of the tumor (mm)2 × 1/2. Drug treatment was initiated when the tumors weighed approximately 150 mg. Tumor size and mouse body weight were measured every 4 days. The mean relative tumor weight was calculated as follows: the mean tumor weight on day x/the mean tumor weight on day 0 (the initiation of treatment). Animal experiments were carried out in a humane manner after receiving approval from the Institutional Animal Experiment Committee of the University of Tsukuba and in accordance with the University’s Regulations for Animal Experiments as well as the Fundamental Guidelines for the Proper Conduct of Animal Experiments and Related Activities in Academic Research Institutions set forth by the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Treatment with celecoxib and CPT‐11. Celecoxib, which was supplied by Pfizer (Groton, CT, USA), was dissolved in dimethyl sulfoxide at 5 mg/mL, further diluted 10 times in 0.5% methylcellulose with 0.025% Tween 20, and administered intraperitoneally (i.p) at a dose of 5 mg/kg body weight. CPT‐11 (Tokyo Chemical Industry, Tokyo, Japan) was injected i.p. at a dose of 5.9 mg/kg body weight (low dose) or 59 mg/kg body weight (conventional dose) in 5% glucose. Celecoxib alone, low‐dose CPT‐11 alone, or celecoxib in combination with low‐dose CPT‐11 was each given once daily for 20 consecutive days. Conventional‐dose CPT‐11 was administered three times at 4‐day intervals with or without celecoxib, which was given daily for 12 consecutive days from day 0. Six to eight xenografted tumors for each treatment were used.
Neuroblastoma (NB) cell lines and cell viability assay. MYCN‐unamplified SK‐N‐AS and SK‐N‐SH, and MYCN‐amplified SK‐N‐DZ, TGW, and CHP134 cells, were maintained in RPMI‐1640 medium supplemented with 10% fetal bovine serum (BioWest, Nuaille, France) and antibiotics at 37°C in a humidified 5% CO2 atmosphere. Among the five cell lines, SK‐N‐AS showed chromosome 1p loss, 11q loss, and 17q gain; SK‐N‐SH demonstrated 17q gain; SK‐N‐DZ had 11q loss; TGW showed 1p gain, probable 11q loss, and 17q gain; and CHP134 demonstrated 1p loss and 17q gain. Cell viability was determined by a colorimetric 2‐(4‐indophenyl)‐3‐(4‐nitrophenyl)‐5‐(2,4‐disulfophenyl)‐2H‐tetrazolium (WST‐1) (Dojindo, Kumamoto, Japan) assay. The cells were plated at a density of 5000 cells/well in 96‐well tissue culture plates (Becton Dickinson, Franklin Lakes, NJ, USA). After attachment overnight, the cells were incubated with six‐step concentrations of celecoxib, SN‐38 (an active metabolite of CPT‐11) (Yakult Honsha, Tokyo, Japan) or SN‐38 combined with 10 μM or 20 μM of celecoxib for 72 hours. The absorbance was measured at 450 nm. All experiments were performed in quadruplicate and repeated three times.
Evaluation of the adverse effects of treatment using the incidence of diarrhea and white blood cell count. Daily diarrheal stool passages were counted for each animal administered with celecoxib and low‐dose CPT‐11 from days 0 to 19, and for those administered with conventional‐dose CPT‐11 with or without celecoxib from days 0 to 8. The white blood cell count in the tail vein was performed at 1, 7, and 14 days after the cessation of each therapy.
Immunohistochemistry and detection of apoptotic cell. To detect the Ki‐67 nuclear antigen, which is present throughout the cell cycle but absent in the dormant G0 phase,( 19 ) tumors were fixed in 10% neutral buffered formalin for 24–48 h prior before being embedded in paraffin. After deparaffinization, the tissue sections were heated at 121°C for 15 min in 10 mM Tris‐HCl with 1 mM EDTA (pH 9.0). Endogenous peroxidase was blocked with 3% hydrogen peroxide in methanol for 5 min at room temperature. The samples were incubated with anti‐Ki‐67 antigen, clone MIB‐1 (DakoCytomation, Carpinteria, CA, USA), for 60 min at room temperature. To detect CD31, an endothelial cell marker, the tumors were fixed in 0.1 M Tris‐HCl (pH 7.4) with 0.05% calcium acetate, 0.5% zinc acetate, and 0.5% zinc chloride for 8 h. After deparaffinization, the tissue sections were covered with 20 μg proteinase K (Wako Pure Chemical Industries, Osaka, Japan)/mL PBS(–) for 30 min at 37°C, followed by incubation with anti‐CD31, clone MEC 13.3 (BD Biosciences Pharmingen, Franklin Lakes, NJ, USA), overnight at 4°C. The bound antibodies were amplified using the standard streptavidin–biotin complex immunoperoxidase (ABC) technique (Vector Laboratories, Burlingame, CA, USA). For the coloring reaction, 3,3′‐diaminobenzidine (DAB) (Sigma, St Louis, MO, USA) was used as the chromogen, and nuclear counterstaining was carried out with hematoxylin. To assess proliferation, the number of pixels with Ki‐67 positively stained nuclei per total number of nuclear pixels was calculated using image analysis software (ImageJ) (NIH, Bethesda, MD, USA) in 15 fields per treatment group at ×150 magnification. The results are expressed as a percentage of the untreated control tumor. The number of CD31 positively stained blood vessels was counted in 20 fields per treatment group at ×300 magnification.
Apoptotic cells in tumor tissues were detected using the TUNEL assay. After deparaffinization, the tissue sections were covered with 20 μg proteinase K/mL PBS(–) for 15 min at room temperature, followed by blocking of endogenous peroxidase activity. The samples were then incubated with TdT enzyme (Takara Bio, Shiga, Japan) and biotin‐16‐dUTP (Roche Applied Science, Mannheim, Germany) in TdT buffer (Takara Bio) containing 0.01% bovine serum albumin for 1.5 h at 37°C in a humidity chamber. Biotin‐16‐dUTP nucleotides that had been incorporated into DNA fragments were detected using the ABC method with DAB as the chromogen. The number of TUNEL‐positive cells was counted in 20 fields per treatment group at ×150 magnification.
Immunoblotting. Frozen tumor samples were crushed in liquid nitrogen and homogenized in a sample buffer consisting of 125 mM Tris‐HCl (pH 6.8), 20 mM dithiothreitol, 4% SDS, 10% glycerol, and 0.5% protease inhibitor cocktail (Complete Mini; Roche Applied Science). The extracts were sonicated for 10 s and centrifuged at 17 000 g for 10 min at 4°C to remove debris. NB cells were lysed in the sample buffer. The protein concentrations were determined using the Bio‐Rad DC protein assay (Bio‐Rad Laboratories, Hercules, CA, USA). Samples containing the same amounts of protein were separated by 12.5% or 15% SDS‐PAGE, electroblotted on polyvinylidene fluoride membranes (Millipore, Bedford, MA, USA), and probed with antibodies. The primary antibodies used were against COX‐2 (Rockland, Gilbertsville, PA, USA), VEGF clone A‐20 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), Bcl‐2 clone 8C8, Bax clone 2D2, p53 clone DO‐1 (Calbiochem, Darmstadt, Germany), caspase‐8 clone E7 (Epitomics, Burlingame, CA, USA), caspase‐9 clone 5B4 (Medical & Biological Laboratories, Nagoya, Japan), cleaved caspase‐3 (Asp175) clone 5A1, phospho‐p53 (Ser15) clone 16G8, caspase‐7 clone C7 (Cell Signaling Technology, Danvers, MA, USA), and β‐actin clone AC‐74 (Sigma). Horseradish peroxidase–conjugated antimouse (Amersham Biosciences, Buckinghamshire, UK) or antirabbit antibody (Stressgen, Victoria, BC, Canada) served as secondary antibodies. An enhanced chemiluminescence system (PerkinElmer Life Sciences, Boston, MA, USA) was used for detection.
Statistical analysis. The responses of NB xenografts to celecoxib alone, CPT‐11 alone, and CPT‐11 in combination with celecoxib were evaluated with respect to tumor growth delay by measuring the mean interval in days required to reach twice the original tumor weight for individual tumors (TD2). The incidence of diarrhea, the white blood cell count in mice, the proportion of Ki‐67 positively stained cells, the number of TUNEL‐positive apoptotic cells, and the number of CD31 positively stained blood vessels in tumor sections were expressed as the mean ± SD. The Student’s t‐test or Welch’s t‐test was used to assess the significance of differences between the treatment groups.
Results
Antitumor effects of celecoxib and CPT‐11. Celecoxib administered i.p. once daily for 20 consecutive days at 5 mg/kg body weight did not prevent the growth of any of the NB xenografts tested (Fig. 1a, Table 1). Treatment with low‐dose or conventional‐dose CPT‐11 alone demonstrated significant tumor growth inhibition against the three NB xenografts (n = 6 in each) (P < 0.01 compared with the untreated control, Student’s t‐test). Furthermore, administration of low‐dose daily CPT‐11 combined simultaneously with very low‐dose celecoxib resulted in highly significant suppression of tumor growth in all three xenografts compared not only with low‐dose CPT‐11 therapy alone but also with the combination therapy of conventional‐dose CPT‐11 and celecoxib (P < 0.001) (Fig. 1, Table 1). In combination therapy involving low‐dose CPT‐11 and celecoxib, one of six TNB9 tumors, one of six TS‐N‐5nu tumors, and two of eight TS‐N‐2nu tumors weighed less than their original tumor weight at the end of the experiment, at day 100 in TNB9 and TS‐N‐2nu and at day 70 in TS‐N‐5nu. These results demonstrate that prolonged very low‐dose administration of celecoxib enhances the antitumor activity of low‐dose CPT‐11 against NB xenografts, despite the fact that very low‐dose celecoxib administration alone has no significant antitumor effect.
Figure 1.

Responses of TNB9, TS‐N‐5nu, and TS‐N‐2nu xenografts to celecoxib and (a) low‐dose irinotecan (CPT‐11) and (b) conventional‐dose CPT‐11. Treatment was initiated on day 0, when the tumors weighed approximately 150 mg. Tumors were measured every 4 days. The results are presented as mean relative tumor weight ± SD to that on day 0. Celecoxib administered i.p. once daily for 20 consecutive days at 5 mg/kg body weight did not significantly prevent the growth of any neuroblastoma (NB) xenograft. Each treatment schedule for CPT‐11 alone demonstrated significant tumor growth inhibition against the three NB xenografts. Administration of low‐dose daily CPT‐11 combined simultaneously with celecoxib resulted in highly significant suppression of tumor growth in all three xenografts compared not only with low‐dose CPT‐11 therapy alone but also with the combination therapy of conventional‐dose CPT‐11 and celecoxib. The arrowheads and crosses indicate drug administration.
Table 1.
Tumor doubling times of TNB9, TS‐N‐5nu, and TS‐N‐2nu xenografts treated with irinotecan (CPT‐11) and celecoxib

Effects of celecoxib and SN‐38 on the survival of SK‐N‐AS, SK‐N‐DZ, and TGW cells in vitro. The IC50s of celecoxib were 50 μM for SK‐N‐AS, 52 μM for SK‐N‐DZ, and 60 μM for TGW (Fig. 2, left panels). Combined treatment with 10 μM or 20 μM celecoxib did not enhance cell killing by SN‐38 in any cell line irrespective of MYCN amplification status and pattern of chromosome gains and losses (Fig. 2 right panels).
Figure 2.

Effects of celecoxib and SN‐38 on the survival of SK‐N‐AS, SK‐N‐DZ, and TGW cells in vitro. The IC50s of celecoxib were 50 μM for SK‐N‐AS, 52 μM for SK‐N‐DZ, and 60 μM for TGW (left panels). Combined treatment with 10 μM or 20 μM celecoxib did not enhance cell killing by SN‐38 in any cell line (right panels). The cells were incubated with six‐step concentrations of celecoxib, SN‐38, or SN‐38 combined with 10 μM or 20 μM of celecoxib for 72 hours. Cell viability was determined by the 2‐(4‐indophenyl)‐3‐(4‐nitrophenyl)‐5‐(2,4‐disulfophenyl)‐2H‐tetrazolium (WST‐1) assay. The results are presented as the mean ± SD.
Effects of celecoxib and CPT‐11 on body weight gain, incidence of diarrhea, and white blood cell count. The body weight of the nude mice did not fall below that at the initiation of treatment for any treatment schedule (Fig. 3). Celecoxib alone caused no diarrhea in the mice (Table 2). CPT‐11 induced slight diarrhea with wet and soft stools within 2 h of the injection irrespective of the administered dose. The diarrhea stopped within 6 h. No CPT‐11‐induced late diarrhea was observed. The percentage of incidence of acute diarrheal episode after the administration of CPT‐11 with or without celecoxib was calculated for each animal. Interestingly, daily co‐treatment with celecoxib significantly suppressed the incidence of diarrhea induced by daily low‐dose CPT‐11 (P = 0.033 compared with low‐dose CPT‐11 alone, Student’s t‐test) (Table 2). The white blood cell count was significantly reduced 24 h after the cessation of intermittent conventional‐dose therapy with CPT‐11 with or without celecoxib (P < 0.01 compared with the untreated control, Student’s t‐test). No mice died during the experiments.
Figure 3.

Body weight changes of mice during the treatments. The body weight of the mice did not fall below that at day 0 for any treatment schedule. The body weights of five untreated mice without tumors were measured every 7 days. Data are presented as the mean body weight relative to that at 6 weeks of age. The mean relative body weight (RBW) for tumor‐bearing mice was calculated as follows: RBW = Bx/Bo, where Bx and Bo are the mean values of observed body weight minus the calculated tumor weight of animals that received the same treatment of celecoxib and CPT‐11 on days x and day 0, respectively. The results are presented as the mean body weight relative to that on day 0. For simplicity, SD bars are not shown.
Table 2.
Effects of celecoxib and irinotecan (CPT‐11) on the incidence of diarrhea and white blood cell count

Tumor cell proliferation in NB xenografts. Tumors from untreated mice and from animals treated with celecoxib alone showed no significant difference in tumor cell proliferation in each NB xenograft (4, 5, panels a and b). Daily administration of low‐dose CPT‐11 alone for 10 consecutive days significantly inhibited cell proliferation in both TNB9 and TS‐N‐5nu compared with the untreated control (P < 0.0001, Student’s t‐test). TNB9 is sensitive to many antitumor drugs including CPT‐11. At 24 h after the cessation of the therapy, there was no significant difference in proliferation inhibition between TNB9 tumors treated with CPT‐11 alone and those treated with CPT‐11 and celecoxib (data not shown). At 96 h after the cessation of the therapy, TNB9 from mice administered with CPT‐11 alone was in its re‐growth phase and showed increased proliferation of tumor cells. In contrast, in TNB9 treated with CPT‐11 and celecoxib, tumor cell proliferation was significantly inhibited compared with CPT‐11 treatment alone, even at 96 h after the removal of the drugs (P < 0.0001, Welch’s t‐test) (4, 5, panels c and d). Tumor cell proliferation in TS‐N‐5nu tumor was significantly decreased by the combination therapy of CPT‐11 and celecoxib compared with CPT‐11 alone (P < 0.0001 Student’s t‐test) (4, 5, panels e and f). In TS‐N‐2nu, a multidrug resistant xenograft, no significant decrease in tumor cell proliferation was observed in spite of the longer administration of low‐dose CPT‐11 (once daily for 15 consecutive days) (P = 0.780, Welch’s t‐test). On the other hand, the combination therapy of CPT‐11 and celecoxib resulted in a significant inhibition of TS‐N‐2nu tumor cell proliferation when compared not only to the untreated control (P < 0.0001, Welch’s t‐test), but also to CPT‐11 therapy alone (P = 0.00036, Student’s t‐test) (4, 5, panels g and h). These findings show that prolonged very low‐dose administration of celecoxib enhances the inhibitory effect of low‐dose CPT‐11 on tumor cell proliferation.
Figure 4.

Prolonged very low‐dose administration of celecoxib enhanced the inhibitory effect of low‐dose irinotecan (CPT‐11) on (a) tumor cell proliferation and (b) CPT‐11‐induced apoptosis, but might not be associated with (c) its anti‐angiogenic effects. Celecoxib alone, low‐dose CPT‐11 alone, or low‐dose CPT‐11 in combination with celecoxib were given once daily for 10 consecutive days to mice with the TNB9 or TS‐N‐5nu xenografts and for 15 consecutive days to mice with the TS‐N‐2nu xenograft. To assess tumor cell proliferation, the number of pixels with Ki‐67 positively stained nuclei per total number of nuclear pixels was calculated for 15 fields per treatment group at ×150 magnification. The results are expressed as the mean percentage ± SD of the untreated control tumor. The numbers of TUNEL‐positive apoptotic cells and CD31‐positive blood vessels in the tumor sections were counted in 20 fields per treatment group at ×150 magnification and ×300 magnification, respectively. The results are presented as the mean ± SD.
Figure 5.
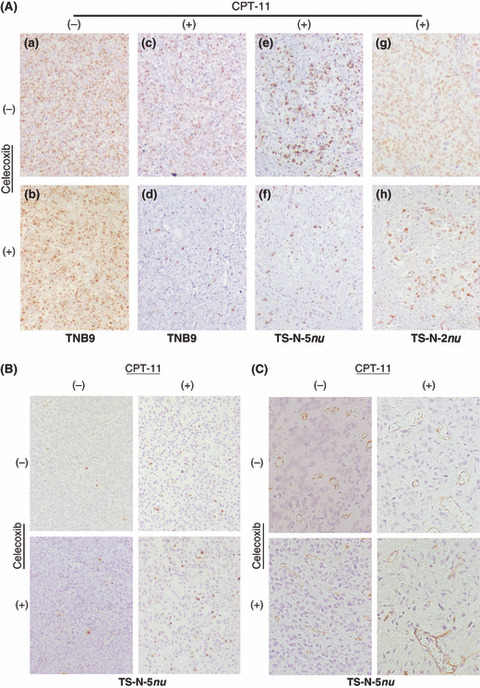
Immunohistochemistry for Ki‐67 antigen, TUNEL, and CD31. (Original magnification, ×150 for A and B; ×300 for C.) (A) (a,b) TNB9 tumors from untreated mice and from animals administered with celecoxib alone showed no difference in tumor cell proliferation. (c,d) At 96 h after the cessation of the therapy, TNB9 tumors from mice administered with low‐dose irinotecan (CPT‐11) alone were in their regrowth phase and showed increased proliferation of tumor cells. In contrast, in TNB9 treated with low‐dose CPT‐11 combined with celecoxib, tumor cell proliferation remained inhibited at 96 h after the removal of the drugs. (e,f and g,h) The Ki‐67‐positive cells in TS‐N‐5nu and TS‐N‐2nu were fewer in number after the combination therapy of low‐dose CPT‐11 and celecoxib compared with after CPT‐11 alone. (B) There was no difference in the number of TUNEL‐positive apoptotic cells between the untreated control TS‐N‐5nu tumors and those administered with celecoxib alone. The number of apoptotic cells induced by combined treatment involving low‐dose CPT‐11 and celecoxib was increased compared with that for CPT‐11 alone. (C) Blood vessels in TS‐N‐5nu tumors treated with low‐dose CPT‐11 were fewer in number than those in untreated and celecoxib‐treated tumors. There was no difference in the number of blood vessels between the tumors treated with CPT‐11 alone and those treated with CPT‐11 and celecoxib.
Apoptotic cells and blood vessels in NB xenografts. There was no significant difference between the untreated control tumors and those administered with celecoxib alone in both the number of TUNEL‐positive apoptotic cells (Fig. 4b) and the number of CD31‐positive blood vessels (Fig. 4c) for any of the NB xenografts. The combination of low‐dose CPT‐11 and celecoxib significantly induced apoptosis in TS‐N‐5nu compared with CPT‐11 therapy alone (P < 0.0001, Student’s t‐test) (4, 5). Apoptotic cell number was significantly different between TNB9 tumors treated with CPT‐11 alone and those treated with CPT‐11 and celecoxib 96 h after the cessation of the therapy (P < 0.0001, Student’s t‐test). Tumor vascularity was significantly decreased in all xenografts treated with CPT‐11 alone compared to the untreated control (P = 0.0046 for TNB9, P = 0.0092 for TS‐N‐5nu, and P = 0.012 for TS‐N‐2nu). There was no significant difference in the number of blood vessels between the tumors treated with CPT‐11 alone and those treated with CPT‐11 and celecoxib in any of three xenografts (4, 5). These results demonstrate that prolonged very low‐dose celecoxib augments CPT‐11‐induced apoptosis, but may not be associated with the inhibitory effects of CPT‐11 on angiogenesis in NB xenografts.
Protein expression analysis in NB xenografts and NB cells. Protein expression was analyzed in xenografts that had been untreated or treated with CPT‐11 and celecoxib once daily for 10 consecutive days for TS‐N‐2nu and for 7 consecutive days for TNB9 and TS‐N‐5nu. The proteins were also analyzed in NB cells untreated or treated in vitro with 2 nM SN‐38 (for CHP134), 5 nM SN‐38 (for SK‐N‐SH), and 20 μM celecoxib for 72 h. COX‐2 protein expression was down‐regulated in TS‐N‐2nu administered with CPT‐11 and celecoxib, a selective COX‐2 inhibitor (Fig. 6a). In the SK‐N‐AS cells and the TNB9 and TS‐N‐5nu tumors, the expression level of COX‐2 protein was low. Caspase‐8, which represents the apical caspase in the death receptor pathway, was inactivated during apoptosis in all three xenografts (Fig. 6b). We then investigated the expression of VEGF and apoptosis‐related proteins in the mitochondrial pathway in the chemosensitive TNB9 and the multidrug resistant TS‐N‐2nu. VEGF protein expression, which may contribute to tumor angiogenesis, was inhibited by the combination of CPT‐11 and celecoxib (Fig. 6c). Administration of CPT‐11 with and without celecoxib caused up‐regulation of Bax, one of the pro‐apoptotic members of Bcl‐2 family, and marked down‐regulation of Bcl‐2, an inhibitor of apoptosis. Combination therapy involving low‐dose CPT‐11 and celecoxib induced caspase‐9 and ‐7 activation in TNB9 and caspase‐9, ‐7, and ‐3 activation in TS‐N‐2nu. p53 seemed to be independent of the observed apoptosis. Combined treatment with low concentrations of SN‐38 and celecoxib activated caspase 9/7‐ and 9/3‐dependent pathways in the SK‐N‐SH and CHP134 cells, respectively (Fig. 6D). These results demonstrate that the enhanced antitumor effects of combination therapy involving low‐dose CPT‐11 and very low‐dose celecoxib against NB xenografts may be partially COX‐2‐independent and are probably mediated through multiple mechanisms including diminished VEGF expression, and activation of the caspase‐dependent mitochondrial apoptosis pathway.
Figure 6.
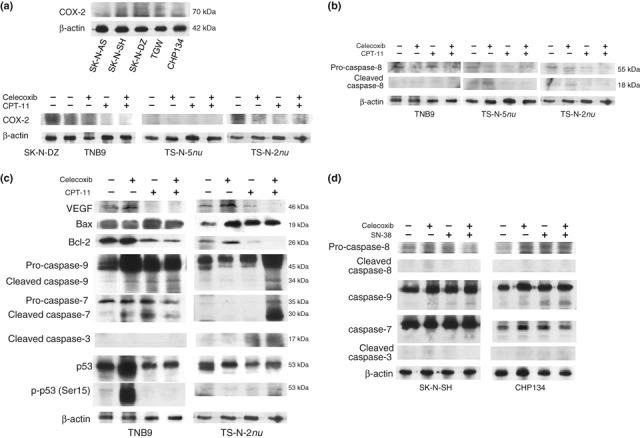
Immunoblot analysis in neuroblastoma (NB) xenografts and NB cell lines. Tumors that had been untreated or treated with drugs once daily for 10 consecutive days for TS‐N‐2nu and for 7 consecutive days for TNB9 and TS‐N‐5nu were crushed in liquid nitrogen and lysed in a sample buffer. NB cells were lysed in the sample buffer. Western blot analysis was performed as described in “Materials and Methods”. Immunoblotting for β‐actin demonstrated equivalent protein loading. (a) Cyclooxygenase (COX)‐2 protein expression in NB cells and NB xenografts. The expression of COX‐2 in SK‐N‐DZ cells was used as a positive control. (b) Caspase‐8 protein expression in TNB9, TS‐N‐5nu, and TS‐N‐2nu xenografts. (c) Expression of VEGF and apoptosis‐related proteins in the mitochondrial pathway in the chemosensitive TNB9 and the multidrug resistant TS‐N‐2nu. VEGF, vascular endothelial growth factor; Bax, Bcl‐2‐associated X protein; Bcl‐2, B cell lymphoma/leukemia‐2; p‐p53 (Ser15), phospho‐p53 at Ser15. (d) Protein expression of caspase‐8, ‐9, ‐7, and ‐3 in SK‐N‐SH and CHP134 cells. The cells were incubated with 5 nM SN‐38 (for SK‐N‐SH), 2 nM SN‐38 (for CHP134), and 20 μM celecoxib for 72 h.
Discussion
This is the first in vivo report that demonstrates that the antitumor effects of combination therapy involving prolonged low‐dose CPT‐11 and very low‐dose celecoxib against NB xenografts are apparently synergistic without substantial side effects in mice, i.e. much greater than the sum of the effects of each drug administered alone. Daily celecoxib alone administered at a very low dose could not prevent the growth of any of the NB xenografts tested. However, the combination of prolonged low‐dose CPT‐11 and simultaneous very low‐dose celecoxib resulted in highly significant suppression of tumor growth in all three xenografts compared not only with low‐dose CPT‐11 therapy alone, but also with the combination therapy of intermittent conventional‐dose CPT‐11 and celecoxib accompanied by decreased proliferation and increased induction of apoptosis in tumor cells. A number of studies have shown additive antitumor efficacy when celecoxib was used in combination with CPT‐11 in mouse and rat tumor models.( 20 , 21 ) In each of these studies, celecoxib alone administered daily at doses five to 10 times as high as that we used caused tumor growth inhibition. Our results indicate that the administration of prolonged low‐dose CPT‐11 is essential for achieving enhanced antitumor effects against NB xenografts, and that it is sufficient for celecoxib to be administered daily at a very low dose when combined with prolonged low‐dose CPT‐11. The in vitro WST‐1 cell viability assay revealed that low concentrations of celecoxib did not enhance the cytotoxic activity of SN‐38 in any of the three cell lines irrespective of their MYCN amplification status, patterns of chromosome gains and losses, and COX‐2 expression levels. The findings obtained from human NB xenografts in nude mice indicate realistic preclinical data and may be useful in developing treatment for NB. The in vivo relationship between genetic background of the NB xenografts and sensitivity to the combined therapy with CPT‐11 and celecoxib should further be investigated.
Celecoxib, the first COX‐2 specific inhibitor approved for use against osteoarthritis and rheumatoid arthritis, has similar efficacy to conventional nonsteroidal anti‐inflammatory drugs in relieving pain and improving functional status, but is associated with a lower incidence of upper gastrointestinal ulceration and complications.( 22 , 23 ) Recently, data suggesting an increased cardiovascular risk associated with the use of celecoxib have been reported.( 24 , 25 ) However, other papers have demonstrated that both COX‐specific and non‐specific inhibitors may increase the risk of serious cardiovascular events, but that celecoxib therapy is associated with increased cardiovascular risk only when used at doses substantially higher than those recommended for the treatment of arthritis.( 26 , 27 , 28 , 29 ) COX‐2 is overexpressed in many human tumors and promotes tumor progression, suggesting that COX‐2 inhibition may be beneficial for cancer treatment, while recent studies have demonstrated that dimethyl‐celecoxib, a derivative of celecoxib that lacks its COX‐2‐inhibitory function, potently mimics the antitumor effects of celecoxib on Burkitt’s lymphoma, multiple myeloma, and glioblastoma in vitro and in vivo.( 30 , 31 , 32 ) The sensitivity of NB cells to celecoxib in vitro seemed to be independent of their COX‐2 protein expression levels. The combination of low‐dose CPT‐11 and very low‐dose celecoxib highly suppressed tumor growth irrespective of the expression level of COX‐2 protein compared with CPT‐11 therapy alone. The enhanced antitumor effect of the combination of the two drugs against NB xenografts is at least partially COX‐2‐independent and is probably mediated through multiple factors including diminished expression of VEGF and activation of the caspase‐dependent mitochondrial apoptosis pathway.
Treatment with low‐dose CPT‐11 alone resulted in significantly decreased tumor vascularity, whereas the combination of low‐dose CPT‐11 and very low‐dose celecoxib led to no more inhibition of angiogenesis. Eggert et al. ( 33 ) showed that high gene expression levels of seven angiogenic factors, including VEGF, are strongly correlated with advanced‐stage NB. They suggested that the inhibition of VEGF bioactivity alone might not be sufficient for anti‐angiogenic therapy for human NB.
Acknowledgments
We thank Dr. Akira Nakagawara, Dr. Miki Ohira, and Dr. Yohko Nakamura in the Chiba Cancer Center Research Institute for providing chromosomal data on the NB cell lines. This work was supported in part by a Grant‐in‐Aid for Exploratory Research (KAKENHI no. 19659456) and a Grant‐in‐Aid for Scientific Research (A) (KAKENHI no. 19209054) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
References
- 1. Inagaki J, Yasui M, Sakata N et al. Successful treatment of chemoresistant stage 3 neuroblastoma using irinotecan as a single agent. J Pediatr Hematol Oncol 2005; 27: 604–6. [DOI] [PubMed] [Google Scholar]
- 2. Shitara T, Shimada A, Hanada R et al. Irinotecan for children with relapsed solid tumors. Pediatr Hematol Oncol 2006; 23: 103–10. [DOI] [PubMed] [Google Scholar]
- 3. Kushner BH, Kramer K, Modak S et al. Irinotecan plus temozolomide for relapsed or refractory neuroblastoma. J Clin Oncol 2006; 24: 5271–6. [DOI] [PubMed] [Google Scholar]
- 4. Kiyota N, Tahara M, Fujii S et al. Nonplatinum‐based chemotherapy with irinotecan plus docetaxel for advanced or metastatic olfactory neuroblastoma: a retrospective analysis of 12 cases. Cancer 2008; 112: 885–91. [DOI] [PubMed] [Google Scholar]
- 5. Bomgaars LR, Bernstein M, Krailo M et al. Phase II trial of irinotecan in children with refractory solid tumors: a Children’s Oncology Group Study. J Clin Oncol 2007; 25: 4622–7. [DOI] [PubMed] [Google Scholar]
- 6. Vassal G, Giammarile F, Brooks M et al. A phase II study of irinotecan in children with relapsed or refractory neuroblastoma: a European cooperation of the Société Françaīse d’Oncologie Pédiatrìque (SFOP) and the United Kingdom Children Cancer Study Group (UKCCSG). Eur J Cancer 2008; 44: 2453–60. [DOI] [PubMed] [Google Scholar]
- 7. Kaneko S, Ishibashi M, Kaneko M. Vascular endothelial growth factor expression is closely related to irinotecan‐mediated inhibition of tumor growth and angiogenesis in neuroblastoma xenografts. Cancer Sci 2008; 99: 1209–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Taketo MM. Cyclooxygenase‐2 inhibitors in tumorigenesis (Part 1). J Natl Cancer Inst 1998; 90: 1529–36. [DOI] [PubMed] [Google Scholar]
- 9. Hung WC. Anti‐metastatic action of non‐steroidal anti‐inflammatory drugs. Kaohsiung J Med Sci 2008; 24: 392–7. [DOI] [PubMed] [Google Scholar]
- 10. Pai R, Soreghan B, Szabo IL et al. Prostaglandin E2 transactivates EGF receptor: a novel mechanism for promoting colon cancer growth and gastrointestinal hypertrophy. Nat Med 2002; 8: 289–93. [DOI] [PubMed] [Google Scholar]
- 11. Tsujii M, Kawano S, Tsuji S et al. Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell 1998; 93: 705–16. [DOI] [PubMed] [Google Scholar]
- 12. Lin MT, Lee RC, Yang PC et al. Cyclooxygenase‐2 inducing Mcl‐1‐dependent survival mechanism in human lung adenocarcinoma CL1.0 cells. Involvement of phosphatidylinositol 3‐kinase/Akt pathway. J Biol Chem 2001; 276: 48997–9002. [DOI] [PubMed] [Google Scholar]
- 13. Costa C, Soares R, Reis‐Filho JS et al. Cyclo‐oxygenase 2 expression is associated with angiogenesis and lymph node metastasis in human breast cancer. J Clin Pathol 2002; 55: 429–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Koki AT, Masferrer JL. Celecoxib: a specific COX‐2 inhibitor with anticancer properties. Cancer Control 2002; 9: 28–35. [DOI] [PubMed] [Google Scholar]
- 15. Johnsen JI, Lindskog M, Ponthan F et al. Cyclooxygenase‐2 is expressed in neuroblastoma, and nonsteroidal anti‐inflammatory drugs induce apoptosis and inhibit tumor growth in vivo . Cancer Res 2004; 64: 7210–5. [DOI] [PubMed] [Google Scholar]
- 16. Thun MJ, Henley SJ, Patrono C. Nonsteroidal anti‐inflammatory drugs as anticancer agents: mechanistic, pharmacologic and clinical issues. J Natl Cancer Inst 2002; 94: 252–66. [DOI] [PubMed] [Google Scholar]
- 17. Gasparini G, Longo R, Sarmiento R et al. Inhibitors of cyclo‐oxygenase 2: a new class of anticancer agents? Lancet Oncol 2003; 4: 605–15. [DOI] [PubMed] [Google Scholar]
- 18. Gately S, Kerbel R. Therapeutic potential of selective cyclooxygenase‐2 inhibitors in the management of tumor angiogenesis. Prog Exp Tumor Res 2003; 37: 179–92. [DOI] [PubMed] [Google Scholar]
- 19. Gerdes J, Lemke H, Baisch H et al. Cell cycle analysis of a cell proliferation‐associated human nuclear antigen defined by the monoclonal antibody Ki‐67. J Immunol 1984; 133: 1710–5. [PubMed] [Google Scholar]
- 20. Trifan OC, Durham WF, Salazar VS et al. Cyclooxygenase‐2 inhibition with celecoxib enhances antitumor efficacy and reduces diarrhea side effect of CPT‐11. Cancer Res 2002; 62: 5778–84. [PubMed] [Google Scholar]
- 21. Ponthan F, Wickström M, Gleissman H et al. Celecoxib prevents neuroblastoma tumor development and potentiates the effect of chemotherapeutic drugs in vitro and in vivo. Clin Cancer Res 2007; 13: 1036–44. [DOI] [PubMed] [Google Scholar]
- 22. Emery P, Zeidler H, Kvien TK et al. Celecoxib versus diclofenac in long‐term management of rheumatoid arthritis: randomized double‐blind comparison. Lancet 1999; 354: 2106–11. [DOI] [PubMed] [Google Scholar]
- 23. Clemett D, Goa KL. Celecoxib: a review of its use in osteoarthritis, rheumatoid arthritis and acute pain. Drugs 2000; 59: 957–80. [DOI] [PubMed] [Google Scholar]
- 24. Solomon SD, McMurray JJ, Pfeffer MA et al. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med 2005; 352: 1071–80. [DOI] [PubMed] [Google Scholar]
- 25. Solomon SD, Wittes J, Finn PV et al. Cardiovascular risk of celecoxib in 6 randomized placebo‐controlled trials: the cross trial safety analysis. Circulation 2008; 117: 2104–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. White WB, Faich G, Borer JS et al. Cardiovascular thrombotic events in arthritis trials of the cyclooxygenase‐2 inhibitor celecoxib. Am J Cardiol 2003; 92: 411–8. [DOI] [PubMed] [Google Scholar]
- 27. Caldwell B, Aldington S, Weatherall M et al. Risk of cardiovascular events and celecoxib: a systematic review and meta‐analysis. J R Soc Med 2006; 99: 132–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McGettigan P, Henry D. Cardiovascular risk and inhibition of cyclooxygenase: a systematic review of the observational studies of selective and nonselective inhibitors of cyclooxygenase 2. JAMA 2006; 296: 1633–44. [DOI] [PubMed] [Google Scholar]
- 29. Howes LG. Selective COX‐2 inhibitors, NSAIDs and cardiovascular events – is celecoxib the safest choice? Ther Clin Risk Manag 2007; 3: 831–45. [PMC free article] [PubMed] [Google Scholar]
- 30. Kardosh A, Wang W, Uddin J et al. Dimethyl‐celecoxib (DMC), a derivative of celecoxib that lacks cyclooxygenase‐2‐inhibitory function, potently mimics the anti‐tumor effects of celecoxib on Burkitt’s lymphoma in vitro and in vivo. Cancer Biol Ther 2005; 4: 571–82. [DOI] [PubMed] [Google Scholar]
- 31. Kardosh A, Soriano N, Liu YT et al. Multitarget inhibition of drug‐resistant multiple myeloma cell lines by dimethyl‐celecoxib (DMC), a non‐COX‐2 inhibitory analog of celecoxib. Blood 2005; 106: 4330–8. [DOI] [PubMed] [Google Scholar]
- 32. Pyrko P, Soriano N, Kardosh A et al. Downregulation of survivin expression and concomitant induction of apoptosis by celecoxib and its non‐cyclooxygenase‐2‐inhibitory analog, dimethyl‐celecoxib (DMC), in tumor cells in vitro and in vivo. Mol Cancer 2006; 5: 19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Eggert A, Ikegaki N, Kwiatkowski J et al. High‐level expression of angiogenic factors is associated with advanced tumor stage in human neuroblastomas. Clin Cancer Res 2000; 6: 1900–8. [PubMed] [Google Scholar]