

Abstract
Epidermal growth factor receptor (EGFR) is one of the most promising targets for cancer therapy. Here, we show the in vitro and in vivo anticancer effects and associated mechanisms of KO‐202125, one of the synthesized aristolactam analogs, as a novel EGFR inhibitor, in EGFR‐overexpressing cancer cell lines. KO‐202125 showed more effective growth inhibition and apoptosis induction than gefitinib, a representative EGFR inhibitor, in various EGFR‐overexpressing human cancers including estrogen receptor (ER)‐negative MDA‐MB‐231 human breast cancer cells. Epidermal growth factor receptor phosphorylation at Tyr1068 was reduced and, consequently, the association of EGFR with p85 was decreased by KO‐202125 treatment in MDA‐MB‐231 cell lines. This led to inactivation of the PI3K/Akt pathway, and consequently suppression of activation of the Wnt pathway and enhancement of the nuclear import of p27Kip1. KO‐202125 treatment in nude mice injected with MDA‐MB‐231 cells showed inhibition of tumor growth without toxicity. Collectively, our results showed the possibility of KO‐202125 as an effective therapy agent of EGFR‐overexpressing cancer cells through reduced EGFR activity and downregulation of the Akt pathway. (Cancer Sci 2011; 102: 597–604)
Epidermal growth factor receptor (EGFR) is a member of the ERBB family, which is one of the receptor tyrosine kinases (RTK).( 1 ) When the ligands such as epidermal growth factor (EGF) and transforming growth factor (TGF)‐α bind the EGFR, the receptor forms homodimer or heterodimers with other ERBB receptors and is phosphorylated at tyrosine residue in its intracellular domain. This activates their many downstream signals and regulates various biological processes including cell proliferation, apoptosis, angiogenesis and differentiation. Dysregulation of the EGFR signaling is also implicated in cancer development, and gene amplification or overexpression of EGFR is a marker of poor prognosis in many types of cancer including breast cancer.( 2 , 3 , 4 , 5 , 6 ) Therefore, many drugs that target EGFR have been developed for cancer therapy. There are two types of drugs that target EGFR: one is the humanized monoclonal antibody, such as cetuximab, matuzumab and panitumumab, which blocks the binding of the ligand with the extracellular domain of the receptor and the other is the tyrosine kinase inhibitor (TKI), such as gefitinib and erlotinib, which competes with ATP in the tyrosine‐kinase domain of the receptor.( 1 ) These drugs have an anti‐tumor effect in various EGFR‐overexpressing cancer cells and have been used in clinical trials. Gefitinib is a representative TKI that is widely used in therapy of non‐small‐cell lung cancers (NSCLC).( 7 , 8 )
Breast cancer can be divided into two types in accordance with estrogen‐receptor (ER) status. One is ER‐positive cancer and the other is ER‐negative cancer that shows ER‐dependent and ER‐independent cell growth, respectively. Anti‐estrogens such as tamoxifen have been used in the chemotherapy of ER‐positive breast cancer.( 9 ) Tamoxifen is variously used to inhibit proliferation and stimulate apoptosis of breast cancer cells by regulation of estrogen‐dependent gene expression. However, ER‐negative breast cancers show no response to estrogen‐dependent cell growth and are resistant to anti‐estrogen therapy.( 10 ) Alternatively, ER‐negative breast cancers frequently overexpress other growth factor receptors such as EGFR. It has been reported that EGFR overexpression is correlated with ER negativity.( 11 , 12 ) Moreover, EGFR is highly overexpressed in metastatic breast cancers.( 13 , 14 ) Epidermal growth factor receptor overexpression in ER‐negative breast cancer leads to acceleration of cell growth, cell survival, invasion and metastasis.( 15 ) Although some receptor tyrosine kinase inhibitors including gefitinib and lapatinib have been used in ER‐negative breast cancer therapy, their effect in breast cancer was not as powerful as it was in lung cancers.( 16 ) Therefore, discovery of a more effective EGFR inhibitor of ER‐negative human breast cancer is still required.
Several EGFR downstream signaling pathways play an important role in many cancers. In particular, the phosphatidylinositol 3‐kinase (PI3K)‐Akt pathway is the main signaling pathway of EGFR downstream and a number of EGFR‐mediated biological processes are mediated by this pathway.( 1 ) This pathway regulates cell proliferation through regulation of its many downstream effectors. Activation of the PI3K/Akt pathway inhibits GSK‐3β function and consequently leads to nuclear translocation of β‐catenin and enhancement of transcription of a TCF/LEF‐responsive gene, such as cyclin D1 and c‐Myc.( 17 , 18 ) Moreover, the reduced Akt activity in breast cancer cells results in increased nuclear p27Kip1, which may ultimately contribute to attenuated cell cycle progression.( 19 ) The PI3K/Akt pathway is also involved in cell survival through inhibition of Bad and caspase 9, several apoptotic factors, and activation of the NF‐κB pathway.( 20 , 21 ) Treatment of EGFR inhibitors such as gefitinib has been found to significantly reduce Akt activation and dysregulation of this pathway can lead to resistance to EGFR‐target therapy.( 16 )
Saururus chinensis (Lour) Baill has many potent effects on diet, atopy, antioxidation and anti‐inflammation, apoptosis and anticancer activity.( 22 , 23 , 24 , 25 , 26 ) However, Aristolactam, a component of Saururus chinensis (Lour) Baill, has cytotoxic and highly nephrotoxic activities and these restrict its effectiveness as a drug.( 27 , 28 ) We synthesized KO‐202125, a derivative of sauristolactam with its R1 side chain changed to another structure, because sauristolactam was one of the aristolactam analogues that showed the lowest toxicity, in vivo and in vitro. Recently, we have shown that KO‐202125 treatment induces apoptosis in KB human oral squamous carcinoma cells.( 29 ) In this study, we show that KO‐202125 is effective in EGFR‐overexpressing human breast cancer both in vitro and in vivo, and it opens the possibility of KO‐202125 as a novel EGFR inhibitor by inhibiting EGFR activity and the Akt pathway downstream.
Materials and Methods
Chemicals and antibodies. KO‐202125 was synthesized from the Korea Research Institute of Chemical Technology (Daejeon, Korea). The antibodies against Bcl‐2, Bcl‐xL, EGFR, erbB2, erbB3, c‐Myc, p27Kip1 and sp‐1 were from Santa Cruz Biotechnologies (Santa Cruz, CA, USA); Bad, Bax, phospho‐EGFR (at Tyr1068), phospho‐erbB3 (Tyr1289), Akt, phospho‐Akt (at Ser473), GSK‐3β and phospho‐GSK‐3β (at Ser9) were from Cell Signaling Technology (Beverly, CA, USA); phospho‐erbB2 (Tyr1248) was from Upstate Biotechnology (Lake Placid, NY, USA); poly ADP ribose polymerase (PARP) and phospho‐p27Kip1 (at Thr157) were from R&D Systems (Minneapolis, MN, USA); and β‐actin was from Chemicon International (Temecula, CA, USA).
Cell culture and drug treatment. MDA‐MB‐231 and SK‐BR‐3 human breast cancer cell lines and other EGFR‐overexpressing cancer cell lines were purchased from ATCC (Manassas, VA, USA). The cell lines were maintained in phenol red‐free Dulbecco’s modified Eagle’s medium (DMEM; Welgene, Daegu, Korea) supplemented with 10% fetal bovine serum (FBS; Welgene) in an atmosphere of 5% CO2 at 37°C. For drug treatment, KO‐202125 was prepared in dimethyl sulfoxide (DMSO; Sigma‐Aldrich, St Louis, MO, USA) and cells were plated on a 100 mm dish (1 × 106 cells) in phenol red‐free DMEM with 5% FBS. The next day the cells were treated with KO‐202125 at the indicated concentration and times.
Cell proliferation assay. Cells were seeded on a 96‐well plate at 3 × 103 cells. The next day the cells were treated with KO‐202125 or gefitinib for 5 days. Cell viability was determined using the CellTiter 96 non‐radioactive cell proliferation assay kit (Promega, Madison, WI, USA) as indicated by the manufacturer. The inhibitory concentration 50% (IC50) dose of each drug was determined from the survival curve.
Analysis of DNA contents by flow cytometry. Cells were trypsinized, pelleted by centrifugation, washed with PBS and fixed in ice‐cold ethanol (75%). The relative DNA content per cell was obtained by measuring the fluorescence of DNA‐bound propidium iodide (PI) and analyzed by flow cytometry (FACSCalibur and CellQuest software; BD Biosciences, San Jose, CA, USA).
Measurement of apoptosis. DAPI staining was carried out as previously described.( 30 ) Annexin V staining was performed using an ApoScreen Annexin V Apoptosis kit (Southern Biotech, Birmingham, AL, USA) as described by the manufacturer and the annexin V‐positive cells were analyzed by flow cytometry.
Western blot analysis and co‐immunoprecipitation. Cells were lysed in RIPA buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP‐40, 0.25% sodium deoxycholate) containing phosphatase inhibitors (Phosphatase Inhibitor Cocktail I and II; Sigma‐Aldrich) and protease inhibitors (Complete, Mini, EDTA‐free; Roche Applied Science, Indianapolis, IN, USA). Each cell lysate (25–100 μg) was separated by SDS‐PAGE, transferred to nitrocellulose membranes (Whatman, Dassel, Germany) and incubated with appropriate antibodies. Protein bands were detected using an enhanced chemiluminescence Blotting Analysis System from GE Healthcare (ECL, Chalfont St Giles, UK). For co‐immunoprecipitation, an EGFR antibody was cross‐linked with protein A agarose (bead) as previously described,( 31 ) and the cross‐linked antibody was incubated with each cell lysate overnight at 4°C. The next day the immunoprecipitant was washed with RIPA buffer and subjected to immunoblotting as described above.
RNA isolation and RT‐PCR. Total RNA was isolated using Trizol reagent (Invitrogen, Carlsbad, CA, USA) as described by the manufacturer. The following primers were used for the PCR reaction: c‐myc, forward: 5′‐CAGCTGCTTAGACGCTGGATTT‐3′ and reverse: 5′‐ACCGAGTCGTAGTCGAGGTCAT‐3′, as previously described;( 32 ) GAPDH, forward: 5′‐CGGAGTCAACGGATTTGGTCGTAT‐3′ and reverse: 5′‐AGCCTTCTCCAT GGTTGGTGA AGAC‐3′.
Separation of particulate, cytosolic and nuclear fraction. Cytosolic and nuclear extracts were prepared using NE‐PER Nuclear and Cytoplasmic Extraction Reagents (Pierce, Rockford, IL, USA) as described by the manufacturer. A total of 30–100 μg cytoplasmic and nuclear proteins were subjected to western blot analysis as indicated above.
Immunocytochemistry. Cells were seeded on a four‐chamber slide glass and treated with 4 μM KO‐202125 or DMSO for 72 h. The cells were fixed with 4% paraformaldehyde, washed with 0.2% Triton X‐100 in phosphate‐buffered saline (PBS) and stained subsequently with appropriate primary‐ and phytoerythrin (PE)‐conjugated secondary antibodies. Then, 4,6′‐diamidino‐2‐phenylindole (DAPI) staining was added to visualize the nucleus. Immunofluorescence was detected by fluorescence microscopy (Nikon, Tokyo, Japan).
Promoter assay. MDA‐MB‐231 cells were seeded onto 12‐well plates at 1 × 105 cells/well, and then transfected with pTOP‐FLASH, a TCF4 promoter, or pFOP‐FLASH, a TOP mutant form, and β‐gal cDNA using FuGENE6 reagent (Roche Diagnostics Corp., Indianapolis, IN, USA). After incubation for 24 h, the cells were treated with the indicated dose of KO‐202125 for 48 h. Luciferase activity was measured as previously described.( 31 )
Xenograft in murine model. Six‐ to 7‐week‐old female CAnN.Cg‐Foxn1 nu/CrlCrlj nu mice (Charles River, Yokohama, Japan) were cared for in accordance with the Hanyang University Committee Guidelines for Experimental Animal Welfare (Seoul, Korea). MDA‐MB‐231 cells (1 × 106 cells/150 μL) were injected into the flank of the mice. When the tumor width reached 2.5–5 mm, 20 animals were randomly selected and placed in groups. After regrouping, the mice were treated with the indicated doses of KO‐202125 daily p.o. for 28 days, and the tumor size and bodyweight were monitored three times per week. Tumors were measured along two diameters with calipers to permit calculation of the tumor volume, V = π/6([[D + d]/2]3), where D and d were the largest and smallest diameters, respectively.( 30 , 33 )
Results
Attenuation of cell growth and induction of apoptosis by KO‐202125 in EGFR‐overexpressing cancer cell lines. To determine the anti‐proliferative effect of KO‐202125, it was chemically synthesized (Fig. 1a), and the IC50 value of KO‐202125 was determined from the growth curve in various EGFR‐overexpressing cancer cell lines, which have no EGFR mutation, including lung, ovarian, skin, colorectal and breast cancers. As shown in Table 1, KO‐202125 showed a growth inhibition effect in all tested cell lines. Moreover, when compared with gefitinib, a representative EGFR inhibitor, KO‐202125 was shown to be more effective at a lower dose. In particular, in MDA‐MB‐231, the IC50 of KO‐202125 was 11‐fold lower than gefitinib (1.17 μM vs 13.36 μM). Next, we determined the apoptosis effect of KO‐202125 in MDA‐MB‐231 and SK‐BR‐3 human breast cancer cell lines. As shown in Figure 1b, the subG1 population was increased by KO‐202125 treatment as measured by DNA contents analysis. Moreover, apoptosis was induced in KO‐202125‐treated cells as determined by DAPI staining and annexin V analysis (Fig. 1c,d). To examine how KO‐202125 induced apoptosis, changes in the apoptosis‐related proteins by KO‐202125 was investigated. In MDA‐MB‐231 cells, KO‐202125 treatment decreased the expression of anti‐apoptotic Bcl‐2 and Bcl‐xL proteins and increased the expression of pro‐apoptotic protein Bax, and consequently increased PARP cleavage (Fig. 1e). In SK‐BR‐3 cells, PARP cleavage and Bax expression were enhanced by KO‐202125 treatment. Together, these data suggest that KO‐202125 effectively inhibits cell growth and induces apoptosis in EGFR‐overexpressing cancer cell lines.
Figure 1.
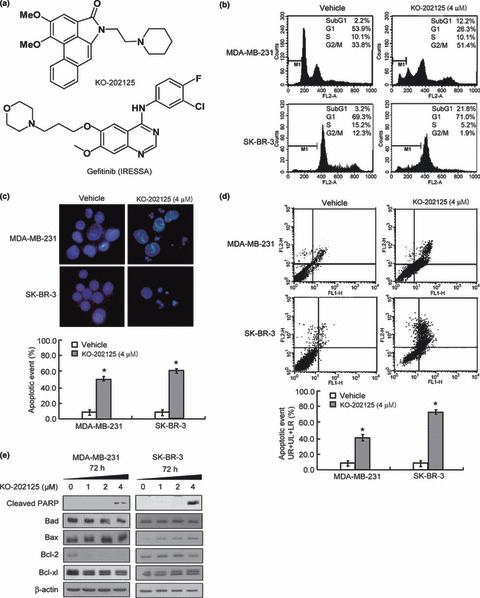
The effect of KO‐202125 on cell growth and apoposis in MDA‐MB‐231 and SK‐BR‐3 human breast cancer cells. (a) Chemical structures of KO‐202125 (top) and gefitinib (bottom). (b) Cells were treated with vehicle or the indicated dose of KO‐202125 for 72 h and the sub‐G1 population cells were measured by FACS analysis. (c,d) Cells were treated with vehicle or KO‐202125 as above and the apoptotic body was measured by DAPI staining and annexin V staining, respectively. LR, lower right quadrant; UL, upper left quadrant; UR, upper right quadrant. (e) Cells prepared under the same conditions as above were subjected to western blot analysis with the indicated antibodies. All of the procedures were performed in triplicate and statistically analyzed with the Student’s t‐test. Data are presented as mean ± SD. *Statistically significant difference of P < 0.05.
Table 1.
IC50 value in various EGFR‐overexpressing cancer cell lines
| Compound | IC50 (μM) | |||||
|---|---|---|---|---|---|---|
| A549 | SK‐OV‐3 | HCT15 | A431 | MDA‐MB‐231 | SK‐BR‐3 | |
| KO‐202125 | 1.0 | 1.3 | 1.4 | 1.15 | 1.17 | 1.79 |
| Gefitinib | 7.5 | 1.8 | 5.9 | 1.46 | 13.36 | 0.93 |
A431, skin cancer; A549, lung cancer; EGFR, epidermal growth factor receptor; HCT15, colorectal cancer; MDA‐MB‐231 and SK‐BR‐3, breast cancer; SK‐OV‐3, ovarian cancer.
Downregulation of RTK signals by KO‐202125. Because the KO‐202125 treatment was effective in the EGFR family overexpressing cancer cells as shown in Table 1, we investigated the effect of KO‐202125 on the expression and activity of EGFR in MDA‐MB‐231 cell lines. After KO‐202125 treatment for 72 h, EGFR phosphorylation at Tyr1068 expression was significantly decreased, but total EGFR expression was unchanged in a dose‐dependent manner (Fig. 2a). Moreover, short‐term treatment of KO‐202125 could block EGF‐mediated EGFR phosphorylaion (data not shown). We also checked the expression and activity of other EGFR family kinases, ErbB2 and ErbB3, but their expressions were not detected in these cells (data not shown). In ErbB2 overexpressing SK‐BR‐3 cells, KO‐202125 did not affect ErbB2 expression, but decreased EGFR and HER3 expression (data not shown). Because the Tyr1068 site of EGFR can associate with p85, a regulatory subunit of PI3K, and this association led to activation of the PI3K/Akt pathway, 34 we determined the association of p85 with EGFR in KO‐202125‐treated MDA‐MB‐231 cells. As a result of the KO‐202125 treatment, the binding of p85 with EGFR was decreased as measured by co‐immunoprecipitation. Consequently, Akt activity was reduced as evidenced by reduced Akt phosphorylation at Ser473, but ERK activity was unchanged by KO‐202125 (Fig. 2b). However, treatment of more than 8 μM KO‐202125 also decreased the ERK phosphorylation in these cells (data not shown). Taken together, these results proposed that KO‐202125 could inhibit the phosphorylation of EGFR and the activation of the EGFR‐mediated PI3K/Akt pathway through inhibition of the binding of EGFR with p85.
Figure 2.
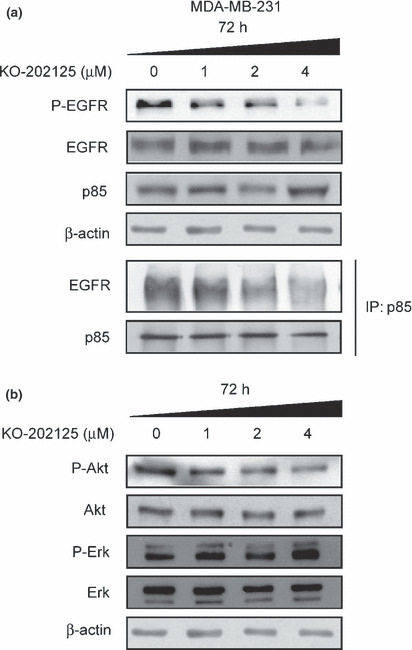
Suppression of the epidermal growth factor receptor (EGFR)/Akt signaling pathway by KO‐202125 in MDA‐MB‐231 human breast cancer cells. Cells were treated with vehicle or indicated doses of KO‐202125 for 72 h. (a) The effect of KO‐202125 on the expression of each protein was determined by western blot analysis. Cells were immunoprecipitated with p85 antibody and the association of EGFR with p85 was determined by western blot analysis. (b) The effect of KO‐202125 on the activity and expression of Akt was confirmed by western blot analysis. Representatives of three independent experiments with similar results are shown.
Downregulation of Akt‐mediated canonical Wnt signaling pathway by KO‐202125. Next, we explored the changes in Akt downstream signaling by KO‐202125. As the Akt‐mediated canonical Wnt pathway has an important role in regulation of cancer cell growth and survival, we checked the effect of KO‐202125 on the Akt‐mediated Wnt pathway. Reduced phosphorylation of GSK‐3β at Ser9 was identified by immunoblotting in KO‐202125‐treated MDA‐MB‐231 cells (Fig. 3a). Consequently, β‐catenin expression was downregulated and both mRNA and protein levels of c‐Myc were decreased by KO‐202125. To confirm the effect of KO‐202125 on this signaling, whether the accumulation of nuclear β‐catenin was inhibited by KO‐202125 was tested. As shown in Figure 3b, nuclear β‐catenin expression was downregulated by KO‐202125 treatment in MDA‐MB‐231 cell lines. Consistently, KO‐202125 decreased the nuclear translocation of β‐catenin in these cell lines as assessed by immunocytochemistry (Fig. 3c). Moreover, the TCF4 promoter activity was decreased as measured by promoter assay (Fig. 3d). Taken together, these suggest that KO‐202125 reduces c‐Myc expression through inhibition of the Akt‐mediated Wnt pathway and this may lead to KO‐202125‐mediated inhibition of cell growth.
Figure 3.
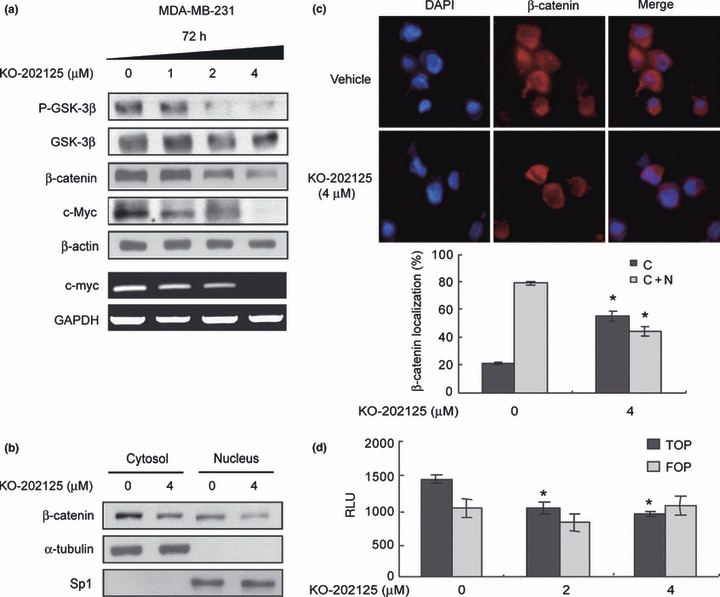
Downregulation of the Akt‐mediated Wnt/β‐catenin pathway by KO‐202125 in MDA‐MB‐231 human breast cancer cells. Cells were treated with vehicle or 4 μM KO‐202125 for 72 h. (a) The effect of KO‐202125 on the Akt‐mediated canonical Wnt signaling pathway was determined by immunoblotting. (b) The effect of KO‐202125 on cytoplasmic/nuclear localization of β‐catenin was examined by biochemical fractionation. (c) The effect of KO‐202125 on subcellular localization of β‐catenin was assessed by immunofluorescence microscopy and the result was quantified by counting 1000 total cells each in three independent experiments (d) The effect of KO‐202125 on TCF4 promoter activity was determined by a promoter assay as described in the Materials and Methods. FOP, reporter assay with FOP‐FLASH construct; TOP, reporter assay with TOP‐FLASH construct; RLU, relative light unit. Each data was statistically analyzed with the Student’s t‐test. Data are presented as mean ± SD. *Statistically significant difference of P < 0.05.
Enhanced nuclear localization of p27Kip1 by KO‐202125. Nuclear p27Kip1 leads to cell cycle arrest through inhibition of cyclin‐dependent kinase (CDK) and Akt blocks its nuclear localization through its phosphorylation.( 19 ) Therefore, we determined the expression and phosphorylation levels of p27Kip1 in KO‐202125‐treated MDA‐MB‐231 cells. KO‐202125 treatment reduced p27Kip1 phosphorylation at Thr157 in a dose‐dependent manner (Fig. 4a). We then tested whether this change affected p27Kip1 subcellular localization. As shown in Figure 4b, nuclear p27Kip1 expression was upregulated by KO‐202125 treatment in MDA‐MB‐231 cells. Increased nuclear localization of p27Kip1 was confirmed by immunocytochemistry (Fig. 4c). These data suggest that p27Kip1 protein is translocated to the nucleus as a result of reduced Akt activity and p27Kip1 phosphorylation by KO‐202125 and this result may lead to attenuated cell growth.
Figure 4.
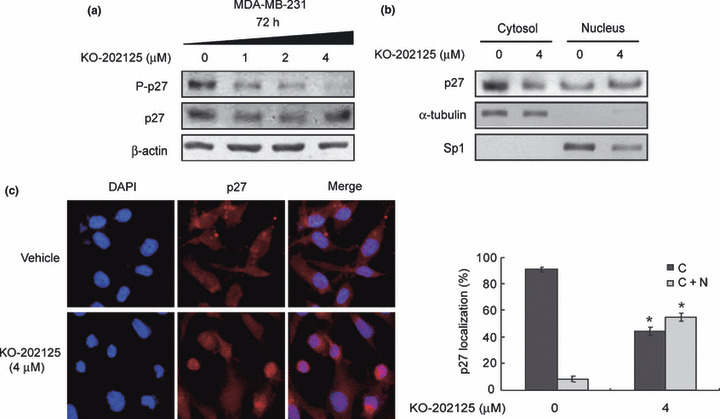
Enhanced nuclear localization of p27Kip1 by KO‐202125 in MDA‐MB‐231 human breast cancer cells. Cells were treated with vehicle or 4 μM KO‐202125 for 72 h. (a) The effect of KO‐202125 on p27Kip1 phosphorylation and total protein expression was determined by western blot analysis. (b) The effect of KO‐202125 on cytoplasmic/nuclear localization of p27Kip1 was examined by biochemical fractionation. (c) The effect of KO‐202125 on subcellular localization of p27Kip1 was assessed by immunofluorescence microscopy (left). The result was quantified by counting 1000 total cells each in three independent experiments and statistically analyzed with the Student’s t‐test (right). Data are presented as mean ± SD. *Statistically significant difference of P < 0.05.
Inhibition of breast cancer growth by KO‐202125 in mouse xenograft models. To explore whether the diminished cell count observed with KO‐202125 treatment in vitro could be found in vivo, we transplanted MDA‐MB‐231 cells into nude mice and treated them with KO‐202125. Upon oral administration of KO‐202125 for 28 days, tumor growth was inhibited in a dose‐ and time‐dependent manner (Fig. 5a,b). There was no evidence of subacute toxicity such as loss of bodyweight in the mouse xenograft model (Fig. 5c). Therefore, these results suggest an anti‐tumor effect of KO‐202125 both in vitro and in vivo.
Figure 5.
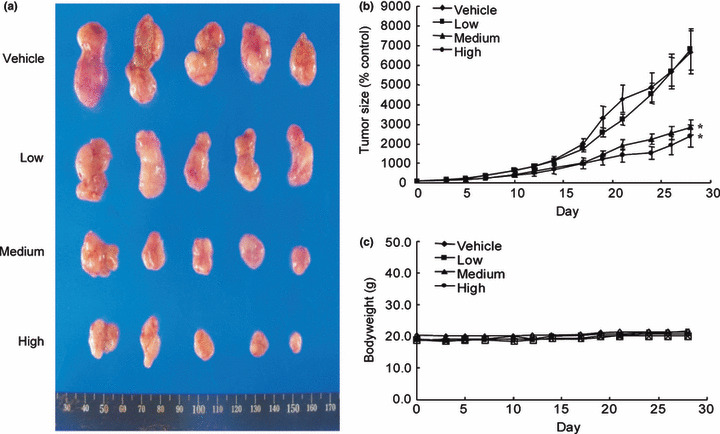
Inhibition of breast cancer growth by KO‐202125 in mouse xenograft models. MDA‐MB‐231 cells were engrafted s.c. into nude mice. When the tumor reached a width of 2.5–5 mm, the mice were treated daily p.o. with corn oil or KO‐202125 for 28 days (day 0 set as the time of starting drug treatment). The dose of KO‐202125 is as follows: low, 0.078 mg/kg; medium, 0.39 mg/kg; high, 1.95 mg/kg. The tumor volume was measured as described in the Materials and Methods. Results are from at least four animals and presented as mean ± SD. P values indicate statistical significance of inhibition of tumor growth by KO‐202125 (post‐hoc test using Scheffe). *P < 0.005.
Discussion
In the present study we showed that KO‐202125 repressed the growth of EGFR‐overexpressing cancer cells both in vitro and in vivo. Moreover, upon KO‐202125 treatment, EGFR phosphorylation and association of EGFR with p85 were reduced in MDA‐MB‐231 cells. Akt, its downstream effector, was inactivated. The Akt‐mediated canonical Wnt signaling pathway was downregulated by KO‐202125, as evidenced by reduced GSK‐3β phosphorylation, nuclear β‐catenin and c‐Myc expressions. Moreover, nuclear localization of p27Kip1 was enhanced by KO‐202125.
EGFR‐mediated signaling mainly functions as an accelerator of cell proliferation and survival in ER‐negative breast cancer that shows ER‐independent cell growth.( 15 , 35 ) Because traditional hormonal therapy cannot be used for ER‐negative breast cancer, the need for alternative therapies such as treatment of EGFR family target drugs is well recognized. It has been reported that some EGFR target drugs act as receptor‐binding blockers and other drugs function as kinase inhibitors. Lapatinib, which targets both EGFR and c‐ErbB2 and inhibits their kinase activity, is more effectively used for breast cancer patients who have EGFR family overexpression.( 36 ) Gefitinib, which targets EGFR as a tyrosine kinase inhibitor and is widely used for lung cancer and patients, is also used for breast cancer patients.( 16 ) However, the discovery of a more efficient and preventive therapeutic agent is still required. In this view, KO‐202125 may become an attractive candidate as it suppressed EGFR and was more effective as an anti‐tumor drug than gefitinib (Table 1). Our results show that a 13‐fold lower concentration of KO‐202125 than gefitinib can effectively block breast cancer cell growth and induce apoptosis. According to our data, KO‐202125‐mediated cell growth inhibition was more effective in EGFR‐overexpressing breast cancers than EGFR‐negative breast cancers (data not shown). Therefore, the major mechanism of the anti‐tumor effect of KO‐202125 may be the EGFR/Akt signaling downregulation in human breast cancer cells. Moreover, KO‐202125 is effective both in vitro and in vivo with no toxicity. These data suggest the possibility of the use of KO‐202125 in clinical trials. Although we found that KO‐202125 was very effective in EGFR inhibition, it is still unclear how KO‐202125 regulates EGFR activity. It is possible that KO‐202125 might directly regulate EGFR activity like other tyrosine kinase inhibitors such as gefitinib or erlotinib. On the other hand, it is assumed that KO‐202125 could indirectly inhibit EGFR activity through modulating EGFR regulators such as cyclooxygenase 2 (COX‐2), because several studies have shown the induction of EGFR activity by COX‐2 and we have previously shown that COX‐2 is a target of KO‐202125.( 29 , 37 , 38 ) To clarify the exact mechanism of EGFR regulation by KO‐202125, further studies are needed.
It is of significance that the anti‐tumor effect of KO‐202125 in EGFR‐overexpressing breast cancer is mediated by downregulation of Akt activity through EGFR inactivation. When comparing the inhibitory effect of KO‐202125 with those of gefitinib regarding the EGFR downstream signaling pathway, there are several differences between them. It has been reported that gefitinib could not effectively block the Akt and MAPK pathways despite reduction of EGFR activity in MDA‐MB‐231 cells.( 39 , 40 ) However, KO‐202125 could inhibit the EGFR/PI3K/Akt signal cascade, but not the ERK pathway. A reason for why both KO‐202125 and gefitinib could not affect the ERK pathway in MDA‐MB‐231 cells might be the mutational status of K‐Ras. It has been previously reported that MDA‐MB‐231 cells have a K‐Ras G13D mutation that leads to constitutive binding of GTP and consequent activation of downstream signaling.( 41 ) Therefore, insensitivity to the MAPK pathway by KO‐202125 or gefitinib might result in constant activation of the RAS/MAPK pathway by a K‐Ras mutation in the MDA‐MB‐231 cells. Taken together, we suggest that a more powerful inhibitory effect of KO‐202125 than gefitinib might be caused by downregulation of Akt signaling. Furthermore, KO‐202125 regulated the Akt downstream effector, Wnt‐related molecules including GSK‐3β, β‐catenin and c‐Myc. The Wnt pathway is important to many biological processes including development, proliferation and differentiation, and aberrant activation of the Wnt pathway has been found to be related to the development of cancers.( 42 ) A major molecule of the canonical Wnt pathway, β‐catenin, is upregulated during cancer development in cancer cells and tissues. β‐catenin, stabilized by inhibition of GSK‐3β, enters the nucleus and enhances expression of Wnt target genes including cyclin D1 and c‐Myc. These Wnt target genes play a very important role in cell growth.( 43 ) Therefore, our data suggest that the Wnt pathway may be part of the mechanism of the cell growth inhibition effect of KO‐202125 in ER‐negative breast cancer. Moreover, the Wnt/β‐catenin pathway is involved in the epithelial mesenchymal transition (EMT).( 44 ) The EMT has been proposed as a crucial step for progression of invasion and metastasis in human cancer. Therefore, it is possible that KO‐202125 may also be useful in cancer therapy through inhibition of the Wnt pathway‐mediated EMT. Our data also show that the reduced Akt activity by KO‐202125 in EGFR‐overexpressing breast cancer cells resulted in reduced phosphorylation of p27Kip1, a CDK inhibitor, and enhanced expression of nuclear p27Kip1, which may lead to G1 arrest during cell cycle progression. The relationship between the p27Kip1 expression level and tumor progression has been reported. The p27Kip1 protein expression level is reduced during tumor development and progression in some tissues including epithelial tissues.( 45 ) Moreover, Liang et al. ( 46 ) showed that a high level of nuclear p27Kip1 expression is involved in the high survival rate in breast cancer patients. Therefore, it could be suggested that KO‐202125 treatment may improve the survival of EGFR‐overexpressing breast cancer patients with a poor prognosis as KO‐202125 increased nuclear p27Kip1 expression through Akt inhibition. Collectively, our data strongly suggest that KO‐202125 may effectively inhibit cell proliferation through regulation of the Wnt pathway and p27Kip1 localization in Akt activity overexpressing cancer cells.
In summary, KO‐202125 was shown to have anti‐proliferative and apoptotic activity in EGFR‐overexpressing breast cancers both in vitro and in vivo. As possible molecular mechanisms responsible for the antitumor effects of KO‐202125, downregulation of EGFR activity and particularly the Akt signaling pathway was implicated. Therefore, KO‐202125 is suggested as a potential anti‐cancer drug for the more aggressive ER‐negative and EGFR‐overexpressing breast cancers.
Disclosure Statement
The authors have no conflict of interest.
References
- 1. Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer 2005; 5: 341–54. [DOI] [PubMed] [Google Scholar]
- 2. Sibilia M, Kroismayr R, Lichtenberger BM, Natarajan A, Hecking M, Holcmann M. The epidermal growth factor receptor: from development to tumorigenesis. Differentiation 2007; 75: 770–87. [DOI] [PubMed] [Google Scholar]
- 3. Lafky JM, Wilken JA, Baron AT, Maihle NJ. Clinical implications of the ErbB/epidermal growth factor (EGF) receptor family and its ligands in ovarian cancer. Biochim Biophys Acta 2008; 1785: 232–65. [DOI] [PubMed] [Google Scholar]
- 4. Hirsch FR, Scagliotti GV, Langer CJ, Varella‐Garcia M, Franklin WA. Epidermal growth factor family of receptors in preneoplasia and lung cancer: perspectives for targeted therapies. Lung Cancer 2003; 41 (Suppl 1): S29–42. [DOI] [PubMed] [Google Scholar]
- 5. Rajkumar T, Gullick WJ. The type I growth factor receptors in human breast cancer. Breast Cancer Res Treat 1994; 29: 3–9. [DOI] [PubMed] [Google Scholar]
- 6. Cho EY, Choi YL, Han JJ, Kim KM, Oh YL. Expression and amplification of Her2, EGFR and cyclin D1 in breast cancer: immunohistochemistry and chromogenic in situ hybridization. Pathol Int 2008; 58: 17–25. [DOI] [PubMed] [Google Scholar]
- 7. Janne PA, Gurubhagavatula S, Yeap BY et al. Outcomes of patients with advanced non‐small cell lung cancer treated with gefitinib (ZD1839, “Iressa”) on an expanded access study. Lung Cancer 2004; 44: 221–30. [DOI] [PubMed] [Google Scholar]
- 8. Cascone T, Morelli MP, Ciardiello F. Small molecule epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors in non‐small cell lung cancer. Ann Oncol 2006; 17 (Suppl 2): ii46–8. [DOI] [PubMed] [Google Scholar]
- 9. Katzenellenbogen BS, Frasor J. Therapeutic targeting in the estrogen receptor hormonal pathway. Semin Oncol 2004; 31: 28–38. [DOI] [PubMed] [Google Scholar]
- 10. Massarweh S, Schiff R. Resistance to endocrine therapy in breast cancer: exploiting estrogen receptor/growth factor signaling crosstalk. Endocr Relat Cancer 2006; 13 (Suppl 1): S15–24. [DOI] [PubMed] [Google Scholar]
- 11. Mori T, Morimoto T, Komaki K, Monden Y. Comparison of estrogen receptor and epidermal growth factor receptor content of primary and involved nodes in human breast cancer. Cancer 1991; 68: 532–7. [DOI] [PubMed] [Google Scholar]
- 12. Long B, McKibben BM, Lynch M, van den Berg HW. Changes in epidermal growth factor receptor expression and response to ligand associated with acquired tamoxifen resistance or oestrogen independence in the ZR‐75‐1 human breast cancer cell line. Br J Cancer 1992; 65: 865–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. van Agthoven T, Timmermans M, Dorssers LC, Henzen‐Logmans SC. Expression of estrogen, progesterone and epidermal growth factor receptors in primary and metastatic breast cancer. Int J Cancer 1995; 63: 790–3. [DOI] [PubMed] [Google Scholar]
- 14. Halterman PA. Lapatinib and ixabepilone for the treatment of metastatic breast cancer. Pharmacotherapy 2008; 28: 1255–66. [DOI] [PubMed] [Google Scholar]
- 15. Nicolini A, Carpi A, Tarro G. Biomolecular markers of breast cancer. Front Biosci 2006; 11: 1818–43. [DOI] [PubMed] [Google Scholar]
- 16. Ferrer‐Soler L, Vazquez‐Martin A, Brunet J, Menendez JA, De Llorens R, Colomer R. An update of the mechanisms of resistance to EGFR‐tyrosine kinase inhibitors in breast cancer: gefitinib (Iressa)‐induced changes in the expression and nucleo‐cytoplasmic trafficking of HER‐ligands (review). Int J Mol Med 2007; 20: 3–10. [PubMed] [Google Scholar]
- 17. Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase‐3 by insulin mediated by protein kinase B. Nature 1995; 378: 785–9. [DOI] [PubMed] [Google Scholar]
- 18. Sharma M, Chuang WW, Sun Z. Phosphatidylinositol 3‐kinase/Akt stimulates androgen pathway through GSK3beta inhibition and nuclear beta‐catenin accumulation. J Biol Chem 2002; 277: 30935–41. [DOI] [PubMed] [Google Scholar]
- 19. Shin I, Yakes FM, Rojo F et al. PKB/Akt mediates cell‐cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nat Med 2002; 8: 1145–52. [DOI] [PubMed] [Google Scholar]
- 20. Datta SR, Dudek H, Tao X et al. Akt phosphorylation of BAD couples survival signals to the cell‐intrinsic death machinery. Cell 1997; 91: 231–41. [DOI] [PubMed] [Google Scholar]
- 21. Cardone MH, Roy N, Stennicke HR et al. Regulation of cell death protease caspase‐9 by phosphorylation. Science 1998; 282: 1318–21. [DOI] [PubMed] [Google Scholar]
- 22. Yu MH, Im HG, Lee JW et al. Effects of ethanol extract from Saururus chinensis (Bour.) Baill on lipid and antioxidant metabolisms in rats fed a high‐fat diet. Nat Prod Res 2008; 22: 275–83. [DOI] [PubMed] [Google Scholar]
- 23. Choi MS, Kim EC, Lee HS et al. Inhibitory effects of Saururus chinensis (LOUR.) BAILL on the development of atopic dermatitis‐like skin lesions in NC/Nga mice. Biol Pharm Bull 2008; 31: 51–6. [DOI] [PubMed] [Google Scholar]
- 24. Cho HY, Cho CW, Song YS. Antioxidative and anti‐inflammatory effects of Saururus chinensis methanol extract in RAW 264.7 macrophages. J Med Food 2005; 8: 190–7. [DOI] [PubMed] [Google Scholar]
- 25. Choi SK, Seo BR, Lee KW, Cho W, Jeong SH, Lee KT. Saucernetin‐7 isolated from Saururus chinensis induces caspase‐dependent apoptosis in human promyelocytic leukemia HL‐60 cells. Biol Pharm Bull 2007; 30: 1516–22. [DOI] [PubMed] [Google Scholar]
- 26. Song SY, Lee I, Park C, Lee H, Hahm JC, Kang WK. Neolignans from Saururus chinensis inhibit PC‐3 prostate cancer cell growth via apoptosis and senescence‐like mechanisms. Int J Mol Med 2005; 16: 517–23. [PubMed] [Google Scholar]
- 27. Mei N, Arlt VM, Phillips DH, Heflich RH, Chen T. DNA adduct formation and mutation induction by aristolochic acid in rat kidney and liver. Mutat Res 2006; 602: 83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stiborova M, Sopko B, Hodek P, Frei E, Schmeiser HH, Hudecek J. The binding of aristolochic acid I to the active site of human cytochromes P450 1A1 and 1A2 explains their potential to reductively activate this human carcinogen. Cancer Lett 2005; 229: 193–204. [DOI] [PubMed] [Google Scholar]
- 29. Leem DH, Choi KH, Han HS et al. KO‐202125, a sauristolactam derivate, induces apoptosis to prevent KB human oral squamous carcinoma cells through inhibition of cyclooxygenase‐2 expression. Eur J Cancer Prev 2010; 19: 23–30. [DOI] [PubMed] [Google Scholar]
- 30. Chung H, Jung JY, Cho SD et al. The antitumor effect of LJ‐529, a novel agonist to A3 adenosine receptor, in both estrogen receptor‐positive and estrogen receptor‐negative human breast cancers. Mol Cancer Ther 2006; 5: 685–92. [DOI] [PubMed] [Google Scholar]
- 31. Lee JY, Jang KS, Shin DH et al. Mel‐18 negatively regulates INK4a/ARF‐independent cell cycle progression via Akt inactivation in breast cancer. Cancer Res 2008; 68: 4201–9. [DOI] [PubMed] [Google Scholar]
- 32. Mitas M, Mikhitarian K, Walters C et al. Quantitative real‐time RT‐PCR detection of breast cancer micrometastasis using a multigene marker panel. Int J Cancer 2001; 93: 162–71. [DOI] [PubMed] [Google Scholar]
- 33. Farhan H, Schuster C, Klinger M et al. Inhibition of xenograft tumor growth and down‐regulation of ErbB receptors by an antibody directed against Lewis Y antigen. J Pharmacol Exp Ther 2006; 319: 1459–66. [DOI] [PubMed] [Google Scholar]
- 34. Ono M, Kuwano M. Molecular mechanisms of epidermal growth factor receptor (EGFR) activation and response to gefitinib and other EGFR‐targeting drugs. Clin Cancer Res 2006; 12: 7242–51. [DOI] [PubMed] [Google Scholar]
- 35. Averbuch S, Kcenler M, Morris C, Wakeling A. Therapeutic potential of tyrosine kinase inhibitors in breast cancer. Cancer Invest 2003; 21: 782–91. [DOI] [PubMed] [Google Scholar]
- 36. McNeil C. Two targets, one drug for new EGFR inhibitors. J Natl Cancer Inst 2006; 98: 1102–3. [DOI] [PubMed] [Google Scholar]
- 37. Han C, Michalopoulos GK, Wu T. Prostaglandin E2 receptor EP1 transactivates EGFR/MET receptor tyrosine kinases and enhances invasiveness in human hepatocellular carcinoma cells. J Cell Physiol 2006; 207: 261–70. [DOI] [PubMed] [Google Scholar]
- 38. Han C, Wu T. Cyclooxygenase‐2‐derived prostaglandin E2 promotes human cholangiocarcinoma cell growth and invasion through EP1 receptor‐mediated activation of the epidermal growth factor receptor and Akt. J Biol Chem 2005; 280: 24053–63. [DOI] [PubMed] [Google Scholar]
- 39. Moasser MM, Basso A, Averbuch SD, Rosen N. The tyrosine kinase inhibitor ZD1839 (“Iressa”) inhibits HER2‐driven signaling and suppresses the growth of HER2‐overexpressing tumor cells. Cancer Res 2001; 61: 7184–8. [PubMed] [Google Scholar]
- 40. Corkery B, Crown J, Clynes M, O’Donovan N. Epidermal growth factor receptor as a potential therapeutic target in triple‐negative breast cancer. Ann Oncol 2009; 20: 862–7. [DOI] [PubMed] [Google Scholar]
- 41. Torbett NE, Luna‐Moran A, Knight ZA et al. A chemical screen in diverse breast cancer cell lines reveals genetic enhancers and suppressors of sensitivity to PI3K isoform‐selective inhibition. Biochem J 2008; 415: 97–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cadigan KM, Nusse R. Wnt signaling: a common theme in animal development. Genes Dev 1997; 11: 3286–305. [DOI] [PubMed] [Google Scholar]
- 43. Prasad CP, Gupta SD, Rath G, Ralhan R. Wnt signaling pathway in invasive ductal carcinoma of the breast: relationship between beta‐catenin, dishevelled and cyclin D1 expression. Oncology 2007; 73: 112–7. [DOI] [PubMed] [Google Scholar]
- 44. Neth P, Ries C, Karow M, Egea V, Ilmer M, Jochum M. The Wnt signal transduction pathway in stem cells and cancer cells: influence on cellular invasion. Stem Cell Rev 2007; 3: 18–29. [DOI] [PubMed] [Google Scholar]
- 45. Lloyd RV, Erickson LA, Jin L et al. p27kip1: a multifunctional cyclin‐dependent kinase inhibitor with prognostic significance in human cancers. Am J Pathol 1999; 154: 313–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liang J, Zubovitz J, Petrocelli T et al. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27‐mediated G1 arrest. Nat Med 2002; 8: 1153–60. [DOI] [PubMed] [Google Scholar]