

Abstract
Drug resistance is a major concern in the successful treatment of ovarian cancer. In the present study we report a combinational drug regime using arsenic trioxide (ATO) and cisplatin (CDDP) to increase therapeutic potentiality in ovarian cancer cells. ATO‐mediated growth inhibition and apoptosis in human suspension ovarian cancer COC1 cells were evaluated by MTT assay and annexin V assay using flow cytometry, respectively. cDNA arrays were performed to screen ATO‐mediated gene expression. Treatment of COC1 cells with ATO alone resulted in growth inhibition and apoptosis with a dose‐and time‐dependent fashion; further cDNA arrays showed that 34 genes (23 up‐regulated genes and 11 down‐regulated genes) may strongly associate with the antiproliferative and pro‐apoptotic effects induced by ATO. Furthermore, Chou–Talalay analysis was used to evaluate the combinational effect of ATO and CDDP as well as dose‐reduction index (DRI) in a panel of ovarian cancer cells including CDDP‐sensitive and ‐resistant cell lines. The combination index (CI) analysis indicated that the interaction effect of ATO/CDDP exhibited a wide range of synergism in all the adherent ovarian cancer cells (A2780, IGROV‐1, SKOV‐3, and R182) as well as 0.93 to 0.69 for IC50 to IC90 in suspension COC1 cells where CI < 1, =1, and >1, define synergism, additive effect, and antagonism, respectively. More intriguingly, the combination of ATO and CDDP yielded favorable DRIs ranging from 1.23‐fold to 13.51‐fold dose reduction. These results suggest that ATO and its combination with CDDP present therapeutic potential for ovarian cancer, and deserve further preclinical and clinical studies. (Cancer Sci 2009; 100: 2459–2464)
Ovarian carcinoma still ranks first as the leading cause of death among gynecological malignancies.( 1 ) Platinum compounds are the key components of chemotherapy for the treatment of ovarian cancer patients. However, drug resistance is a significant obstacle hindering the successful treatment of ovarian cancer patients. As documented, patients with ovarian cancer had a 5‐year survival rate of less than 30%.( 2 ) Therefore, new therapeutic agents and new regimens consisting of combinations of chemotherapeutic agents are needed.
Arsenic trioxide (ATO) is a natural substance that has been used medically as a traditional Chinese medicine for over a thousand years, following the ancient philosophy of “treating an evil with a toxic”.( 3 ) Approval by the Food and Drug Administration for the use of ATO in the treatment of relapsed/refractory acute promyelocytic leukemia (APL) in the 1990s was supported by the impressive clinical response rates in Chinese patients with APL( 4 ) and confirmative clinical studies conducted in the USA.( 3 ) Remarkable efficacy of ATO in the treatment of APL led to exploration of its anticancer activity and underlying mechanism in other malignancies. There has been promising laboratory and pre‐ or clinical effects observed in various hematologic malignancies( 5 , 6 ) and in a variety of solid tumors.( 7 , 8 , 9 , 10 )
We, as well as others, have demonstrated anticancer activity of ATO against human ovarian cancer both in vitro ( 11 ) and in vivo ( 12 ) indicating its potential clinical value for the treatment of ovarian cancer. These findings provide an impetus for uncovering the specific mechanism of action underlying these dramatic responses. More recently, investigators have begun to use high‐throughput screening technologies such as microarray technology to examine the global effects of ATO on gene expression against different cancer types.( 13 , 14 , 15 , 16 ) It would be important to further elucidate the molecular mechanism induced by ATO on ovarian cancer cells. In our present study, a wide range of human ovarian cancer cell lines including COC1 cells with suspension characteristics similar to hematologic malignancies, and cisplatin‐sensitive and ‐resistant adherent ovarian cancer cell lines, was used to investigate the effect of ATO on proliferation and apoptosis, in particular the combinational effect of ATO and cisplatin (CDDP) on ovarian cancer cells was also demonstrated. The resulting wide range of synergism yielded by the combination of ATO and CDDP as well as the favorable dose‐reduction index (DRI) indicated the promising potentiality of ATO as a therapeutic adjunct with CDDP in the treatment of ovarian cancer. In addition, the gene profiles screening using cDNA microarrays support further exploration on the molecule mechanism underlying ATO‐mediated cytotoxicity in ovarian cancer cells.
Materials and Methods
Cell treatment. The human ovarian cancer COC1 cell line, derived from ascites from a patient with poorly differentiated ovarian adenocarcinoma and made by the Chinese Academy of Medical Sciences Cancer Institute, was purchased from China Typical Culture Collection Center (CTCCC; Wuhan, China). The adherent human ovarian cancer cell lines A2780, IGROV‐1, SKOV‐3, and R182 were kindly provided by John Allen (Cancer Drug Resistance, Centenary Institute, Sydney, Australia). COC1 cells were cultured in suspension with RPMI‐1640 media (Sigma, St. Louis, MO, USA) supplemented with 10% heat‐inactivated fetal calf serum (FCS; Hyclone, Logan, UT, USA) and penicillin (100 U/mL)/streptomycin (100 μg/mL). A2780, IGROV‐1, and SKOV‐3 cells were incubated in DMEM media supplemented with 10% FCS and penicillin/streptomycin. R182 cells were maintained in RPMI‐1640 media supplemented with 20% FCS and penicillin/streptomycin. Cultures were incubated in a humidified atmosphere of 95% air and 5% CO2 at 37°C. For experiments, cells were treated with the indicated concentrations of ATO and/or CDDP.
MTT assay. After incubation for the indicated times in 96‐well tissue culture plates, 20 μL of MTT (5 mg/ml 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐dipheny tetrazolium bromide) (Sigma) solution in PBS was added to each well for an additional 4 h. The unreactive supernatants in the well were carefully aspirated after centrifugation and replaced with 150 μL DMSO, followed by the measurement of OD492 values on a scanning multi‐well spectrophotometer (Multiskan MK3; Thermo Labsystems, Shanghai, China).
Annexin V assay. Annexin V assay was carried out with an Annexin V‐FITC Apoptosis Detection Kit according to the manufacturer’s instructions (BD, San Diego, CA, USA). Briefly, 2 × 105 cells were collected, washed twice with cold PBS, resuspended in 100 μL binding buffer, and incubated with Annexin V‐FITC at room temperature for 10 min, followed by addition of 6 μL propidium iodide (PI; 20 μg/mL) for an additional 5 min. Fluorescent intensities were determined by flow cytometry (Beckman Coulter, Miami, FL, USA).
cDNA microarray. The Superarray (cat no. HS‐019) contained 96 spotted probes related to drug resistance and metabolism which were selected, and the signals were detected. All technical services were provided by Shanghai Biochip Corporation (Shanghai, China). For each set of quadruplicates, the average intensity of each set of probes was used for normalization based on the GAPDH gene. The differential gene expression was analyzed using the same cut‐off criteria (≥2.0‐ or ≤0.5‐fold changes) as described for the Superarray analysis.
Median‐effect principle for dose–effect analysis and combination index (CI) for determining synergism additivity or antagonism. The multiple drug effect analysis of Chou and Talalay based on the median‐effect principle was used to calculate combined drug effects. The CI and DRI for determining synergism and antagonism between ATO and CDDP were calculated as described by Chou et al. ( 17 , 18 ). CI < 1, CI = 1, and CI > 1, respectively, indicate synergism, additivity, and antagonism. CompuSyn software for the above drug combination analysis was applied.( 19 )
Statistical analysis. The SPSS11.0 software package was used for statistical analysis (SPSS, Chicago, IL, USA). P‐values of less than 0.05 were considered to be statistically significant.
Results
ATO produces dose‐ and time‐dependent growth inhibition in COC1 human ovarian cancer suspension cells. To explore the effect of ATO on human ovarian cancer cells, proliferation of COC1 cells was assessed by MTT assays using various concentration of ATO (from 0.5 to 8 μM) applied over a time period of 24 to 96 hours. As shown in Figure 1, time‐ and concentration‐dependent inhibition of cell growth was observed upon incubation with ATO. The estimated IC50 values of the COC1 cells treated with ATO were 1.89, 1.48, and 0.92 μM for 48 h, 72 h, and 96 h, respectively.
Figure 1.
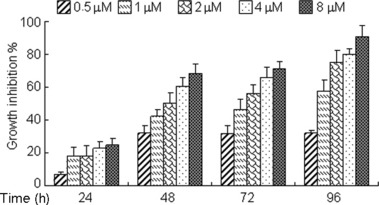
Arsenic trioxide (ATO) induces growth inhibition in ovarian cancer COC1 cells. COC1 cells were exposed to increasing concentrations of ATO for indicated hours (24–96 h). The percentages of growth inhibition were measured by MTT assay. Experiments were repeated three times with similar results; the error bar of the column indicates the SD derived from results of three experiments.
ATO induces cell apoptosis in COC1 cells. To determine whether the growth inhibitory effect of ATO is associated with cell death, annexin V/PI double staining–based flow cytometry analysis was performed on COC1 cells. Treatment of COC1 cells with ATO at the lower concentration of 1 μM or 2 μM for 24–72 h produced dose‐ and time‐dependent apoptosis, as evidenced by increased annexin V+ cells (Fig. 2a,b). With higher concentration (2 μM or 4 μM) of ATO treatment for extended exposure time, apoptosis reached the peak at 48 h or 72 h, and subsequently almost all apoptotic cells were non‐viable (Fig. 2b). The results obtained demonstrate that inhibition of cell growth by ATO is partly due to the induction of apoptotic cell death.
Figure 2.
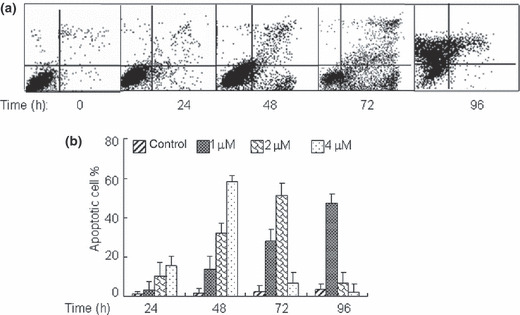
Arsenic trioxide (ATO) promotes cell apoptosis in ovarian cancer COC1 cells. COC1 cells were exposed to increasing concentrations of ATO for indicated hours. The percentages of annexin V(V)+ cells were measured respectively by flow cytometry. (a) COC1 cells were exposed to 2 μM ATO for 24–96 h. (b) The concentration‐ (1, 2, 4 μM) and time‐ (24–96 h) dependent apoptosis induced by ATO. All values represent the means with bars as SD of three samples in an independent experiment, which was repeated three times with similar results.
Effects of ATO on gene expression in COC1 cells (Superarray analysis). In order to identify early specific mechanism underlying these dramatic responses, COC1 cells were treated with or without 2 μM of ATO for 6 h following cDNA microarray, which showed that 24 genes were up‐regulated and 11 genes were down‐regulated. Of these genes (Table 1), IκBβ and c‐Myc (transcription factors and DNA‐binding proteins); Cdk4, p16INK4, and P53 (cell cycle regulators); as well as ERBB4, AR, and SRDSA2 (signal receptors), which are associated with cell proliferation and apoptosis, exhibited strong (≥10.0‐ or ≤0.1‐fold) differential expression in response to ATO treatment. On the other hand, transcription factors hypoxia inducible factor (HIF)‐1α and Rank (nuclear factor kappa B, NF‐κB activator), apoptosis‐associated gene Bax, cell cycle regulators p19INK4D, and signal receptors IGF‐1R and Met, as well as RXRA, showed moderate (2.0–10.0‐ or 0.1–0.5‐fold) difference. In addition, ATO‐responsive stress‐related genes mostly include glutathione‐S‐transferase (GST) and cytochrome P450 (CYP) families (see Supplementary Table S1). cDNA expression profiles indicated that ATO‐induced cytotoxicity in COC1 cells involves combined action of a proportionally large number of both positive and negative regulators of proliferation, which ultimately causes apoptosis and growth arrest.
Table 1.
Gene expression analysis using the cDNA Microarray System in COC1 cells
| Gene name | Gene ID | Gene description |
|---|---|---|
| Oncogene | ||
| Bax | NM_004324 | BCL2‐associated X protein |
| P53 | NM_000546 | Tumor protein p53 |
| Cell cycle | ||
| Cdk4 | NM_000075 | Cyclin‐dependent kinase 4 |
| p16INK4 | NM_000077 | Cylin‐dependent kinase inhibitoe 2A |
| p19‐INK4D | NM_001800 | Cyclin‐dependent kinase inhibitor 2D |
| p27Kip1∇ | NM_004064 | Cyclin‐dependent kinase inhibitor 1B |
| Receptors | ||
| ERBB4 | NM_005235 | v‐erb‐a erythroblastic leukemia viral oncogene homolog 4 |
| RXRA | NM_002957 | Retinoid X receptor, alpha |
| IGF‐1R∇ | NM_000875 | Insulin‐like growth factor 1 receptor |
| Met∇ | NM_000245 | Met proto‐oncogene (hepatocyte growth factor receptor) |
| AR∇ | NM_000044 | Androgen receptor |
| SRD5A∇ | NM_000348 | Steroid‐5‐alpha‐reductase, alpha polypeptide 2 |
| Transcription factors | ||
| IKBβ | NM_002503 | Nuclear factor of kappa light polypeptide gene enhancer in B‐cells inhibitor, beta |
| c‐Myc | NM_002467 | v‐myc myelocytomatosis viral oncogene homolog |
| HIF1A∇ | NM_001530 | Hypoxia‐inducible factor 1, alpha subunit |
| Rank∇ | NM_003839 | Tumor necrosis factor receptor superfamily, member 11a, NFκB activator |
The table shows data for selected 16 genes with fold ≥2.0 or ≤0.5. ∇indicates mRNA levels of genes down‐regulated by arsenic trioxide (ATO).
ATO acts synergistically with CDDP to inhibit growth of COC1 ovarian cancer cells. Platinum compounds are the key components of chemotherapy for the treatment of ovarian cancer patients; however, tumors that are resistant to CDDP‐induced cytotoxicity represent a significant obstacle to successful treatment outcome. Hence, we evaluated the combinational effect of ATO with CDDP on ovarian cancer cells. For this purpose, increasing doses (0.375 to 3 μM for ATO and 0.53 to 4.24 μM for CDDP) of the compounds were applied to COC1 cells. As shown in Figure 3a, combination treatment yielded greater growth inhibition than either agent alone in COC1 cells. The mass‐action law parameters (Dm, m, and r values), representing the potency, shape of dose–effect curve, and the conformity to the mass‐action law, respectively for each drug, and their combinations are given in Table 2. These Dm and m values were then used to calculate the CI values based on the Chou–Talalay combination index equation,( 17 , 18 ) where CI < 1, =1, and >1, indicate synergism, additive effect, and antagonism, respectively. The CI–fa curve given in Figure 3b indicated that ATO and CDDP combinations yielded a synergistic effect with CI values ranging from 0.93 to 0.69 at different effect levels from IC50 to IC90. Both ATO and CDDP also had a favorable DRI ranging from 1.99‐fold to 5.13‐fold dose reduction (Table 3). These projected dose reduction at each given effect levels have clinical implications since reduced dose for a given effect leads to reduced toxicity in chemotherapy.
Figure 3.
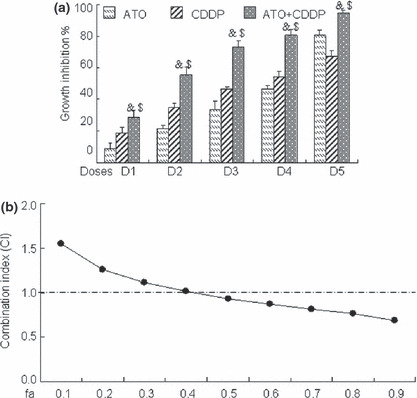
Combinational effect of arsenic trioxide (ATO) and cisplatin (CDDP) on human ovarian cancer COC1 cells. (a) COC1 cells were exposed to graded concentrations of ATO (0.375, 0.75, 1.125, 1.5, 3 μM, respectively, for D1–D5) and/or CDDP (0.53, 1.07, 1.59, 2.12, 4.24 μM, respectively, for D1‐D5) for 72 h. The percentages of growth inhibition were measured by MTT assay. Symbols & and $ represent P < 0.05 respectively compared with ATO and CDDP treatment alone using the paired t‐test. All values represent the means with bars as SD of three samples in an independent experiment, which was repeated three times with similar results. (b) Combination index (CI) of ATO with CDDP in COC1 cells was calculated as described.( 17 ) Here fa on the x‐axis denotes fraction affected (e.g. fa of 0.5 is equivalent to a 50% reduction of cell growth.
Table 2.
Dose–effect relationship parameters of ATO or/and CDDP against COC1 cells in vitro
| Compound | m | Dm (μM) | r | n |
|---|---|---|---|---|
| ATO | 1.055 | 1.38 | 0.994 | 5 |
| CDDP | 1.945 | 1.96 | 0.980 | 5 |
| ATO + CDDP | 1.818 | 1.56 | 0.998 | 5 |
The parameters m, Dm, and r signify the shape of the dose–effect curve, and the potency (IC50) and conformity of the data to the mass‐action law. n is the number of sets of dose–effect relationship experiments that were carried out. m and Dm values were calculated as described. ATO, arsenic trioxide; CDDP, cisplatin.( 17 )
Table 3.
DRI analysis of CDDP and ATO combination in COC1 cells
| fa | 0.2 | 0.3 | 0.4 | 0.5 | 0.6 | 0.7 | 0.8 | 0.9 |
|---|---|---|---|---|---|---|---|---|
| CDDP | 2.26 | 2.22 | 2.18 | 2.15 | 2.12 | 2.09 | 2.05 | 1.99 |
| ATO | 1.23 | 1.53 | 1.82 | 2.14 | 2.51 | 3.00 | 3.71 | 5.13 |
DRI represents the order of magnitude (fold) of dose reduction that is allowed in combination for a given degree of effects as compared with the dose of each drug alone.( 18 )“fa” denotes fraction affected (e.g. fa of 0.5 is equivalent to a 50% reduction of cell growth. ATO, arsenic trioxide; CDDP, cisplatin.
In order to determine whether combined or sequential administration of the compounds affected their efficacy, several different drug regimes were examined. Administration of ATO and CDDP were added simultaneously for 72 h. In addition, 24‐h treatment with CDDP was followed by 48 h treatment with ATO and CDDP; alternatively, 24‐h treatment with ATO was followed by 48‐h treatment with ATO and CDDP. We observed no statistical difference in drug response between different treatment regimens (Fig. 4), indicating that order of drug administration had no effect on the efficacy of combination treatment by ATO and CDDP.
Figure 4.
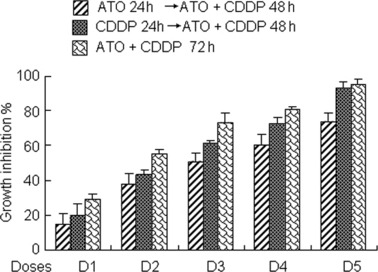
Effects on cell inhibition induced by the sequential or combined treatment of arsenic trioxide (ATO) and cisplatin (CDDP). Cells were exposed to three different drug regimes (the concentrations used for ATO and CDDP treatment were the same as in Fig. 3), respectively, and MTT assays were performed. Experiments were repeated three times with similar results; the error bar of the column indicates the SD derived from results of three experiments. Statistical difference among the three groups was determined by one‐way anova.
ATO produces growth inhibition alone as well as synergistic effect combined with CDDP in a panel of adherent ovarian cancer cell lines. The promising results that ATO mediated growth inhibition and apoptosis stimulation – especially in synergistic combination with CDDP in suspension COC1 cells – led us to confirm whether a similar effect could be observed in other adherent ovarian cancer cells. We then investigated CDDP‐sensitive cells A2780 and IGROV‐1 and CDDP‐resistant cells SKOV‐3 and R182 (the differential susceptibility of these adherent ovarian cancer cell lines to CDDP has been confirmed by MTT assay; data not shown). The broad range of ATO‐induced cytotoxicity in the panel of ovarian cancer cells was demonstrated in a time‐ and concentration‐dependent manner (Fig. 5a–d); and subsequently, the combinational effect of ATO with CDDP in those cell lines was investigated by Chou–Talalay analysis. The broad range of synergism exerted by the combination of ATO and CDDP was observed in both sensitive and resistant ovarian cancer cells with CI ranging from 0.44 to 0.97 for IC20 to IC60 (Fig. 6a–d) as well as DRI ranging from 1.51‐fold to 13.51‐fold dose reduction for IC10 to IC80 (Table 4).
Figure 5.
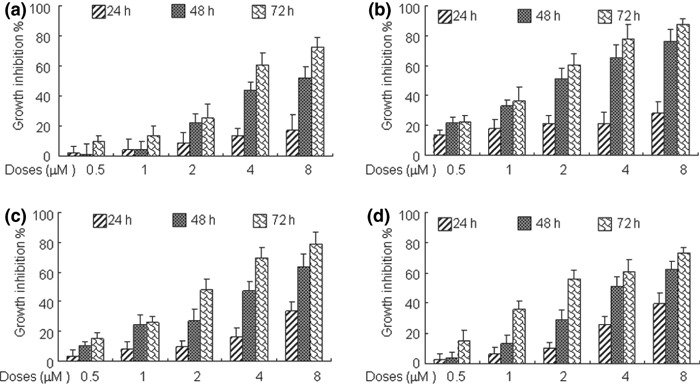
Arsenic trioxide (ATO) induces growth inhibition in a panel of adherent ovarian cancer cells. The various adherent ovarian cancer cells A2780 (a), IGROV‐1 (b), SKOV‐3 (c), and R182 (d) with appropriate seeding density were exposed to increasing concentrations of ATO for indicated hours (24–72 h). The percentages of growth inhibition were measured by MTT assay. Experiments were repeated three times with similar results; the error bar of the column indicates the SD derived from results of three experiments.
Figure 6.
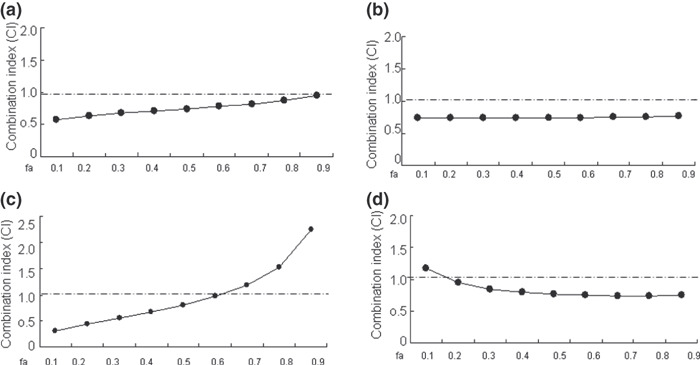
Combinational effect of arsenic trioxide (ATO) and cisplatin (CDDP) on adherent ovarian cancer cells. The ovarian cancer cells A2780 (a) and IGROV‐1 (b) were exposed to graded concentrations of ATO (0.25, 0.5, 1.0, 2, 4 μM) and/or CDDP (0.5, 1.0, 2.0, 4.0, 8 μM) for 72 h; SKOV‐3 (c) and R182 (d) were exposed to graded concentrations of ATO (0.5, 1.0, 2, 4, 8 μM) and/or CDDP (0.75, 1.5, 3.0, 6.0, 12 μM) for 72 h, followed by MTT assay and Chou–Talalay analysis as described previously.
Table 4.
DRI analysis of CDDP and ATO combination in adherent ovarian cancer cells
| fa | 0.2 | 0.3 | 0.4 | 0.5 | 0.6 | 0.7 | 0.8 |
|---|---|---|---|---|---|---|---|
| CDDP/A2780 | 2.29 | 2.15 | 2.03 | 1.93 | 1.84 | 1.74 | 1.63 |
| ATO/A2780 | 5.11 | 4.86 | 4.66 | 4.49 | 4.33 | 4.15 | 3.95 |
| CDDP/IGROV1 | 3.84 | 4.02 | 4.18 | 4.35 | 4.55 | 4.8 | 5.21 |
| ATO/IGROV1 | 2.1 | 2.05 | 2 | 1.95 | 1.9 | 1.84 | 1.75 |
| CDDP/SKOV3 | 3.44 | 2.85 | 2.43 | 2.11 | 1.83 | 1.56 | 1.29 |
| ATO/SKOV3 | 6.7 | 4.88 | 3.77 | 2.97 | 2.34 | 1.81 | 1.32 |
| CDDP/R182 | 2.45 | 3.42 | 4.48 | 5.75 | 7.38 | 9.69 | 13.51 |
| ATO/R182 | 1.88 | 1.8 | 1.74 | 1.68 | 1.63 | 1.57 | 1.51 |
ATO, arsenic trioxide; CDDP, cisplatin; DRI, dose‐reduction index.
Discussion
The high success rate of ATO in inducing complete remission in both newly diagnosed and relapsed patients with APL( 3 , 4 ) provides an impetus for uncovering its broad antitumor activity in other hematologic malignancies and solid tumors. In this report we compared the efficacy of a traditional Chinese compound, ATO, alone and in combination with CDDP, a compound traditionally used for the treatment of ovarian cancer. We established that ATO induced antiproliferative and pro‐apoptotic effects in human suspension ovarian cancer COC1 cells as well as obvious cytotoxicity in a panel of adherent ovarian cancer cells including CDDP‐sensitive and ‐resistant cells, indicating its broad anticancer activity. Further, this work proposed that ATO exerted antagonistic effects on an ovarian cancer cell line on multiple levels at least with inhibition of growth, induction of apoptosis, and regulation of transcription factors NF‐κB and HIF‐1α; apoptosis‐associated targets c‐Myc, P53, and Bax; and cell cycle regulators cdk4, p16INK4, and P27Kip1.
Although recent disappointing studies of the first phase II trial combining ATO with conventional chemotherapeutic agents in solid tumor (advanced melanoma) have shown no expected efficacy,( 20 ) the significant effects of ATO on CDDP‐resistant ovarian cancer cells at a very close concentration and time range when compared with parental CDDP‐sensitive cells,( 21 ) and the encouraging report of ATO/ATRA combination therapy for APL,( 22 , 23 , 24 ) provide a potential rationale for combination ATO and CDDP regimens for ovarian cancer treatment. The integration of new drugs into the treatment resulted in leaps in survival when highly active drugs such as CDDP and paclitaxel were added to primary therapy. Although most cases respond to initial chemotherapy, many cases relapse within 3 years. Such relapsed and persistent cases become resistant to first‐line chemotherapy and require second‐line chemotherapy.( 25 ) Various combinational regimes have become a major strategy for overcoming drug resistance and improving response and cure rates.( 26 , 27 , 28 )
It is not possible to “determine” synergism in humans for scientific, practical, and ethical reasons. Thus preclinical drug combination studies in vitro and/or in animals are essential to obtain the basis and rationale for studies in humans prior to drug combination clinical trials.( 29 ) As such, here we investigated the possible effects of ATO in combination with CDDP on a panel of human ovarian cancer cells using the media–effect equation and the CI method.( 12 ) The CI analysis demonstrated that the interaction effect of ATO/CDDP exhibited a broad range of synergism in the suspension COC1 cells with CI ranging from 0.93 to 0.69 for IC50 to IC90 in both CDDP‐sensitive and ‐resistant adherent ovarian cancer cells, with CI ranging from 0.44 to 0.97 for IC20 to IC60 where CI < 1, =1, and >1 define synergism, additive effect, and antagonism, respectively. More intriguingly, the combination of ATO and CDDP yielded favorable DRIs ranging from 1.23‐fold to 13.51‐fold dose reduction. These projected dose reductions at each given effect levels have clinical implications since reduced dose for a given effect leads to reduced toxicity in chemotherapy.( 18 ) Together, these results suggest that combination of ATO and CDDP have significant clinical potential for treatment of ovarian cancers, and deserve further preclinical and clinical studies.
Supporting information
Table S1. Gene expression analysis using the cDNA Microarray System in COC1 cells.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Acknowledgments
The authors consulted with Dr T.C. Chou of the Memorial Sloan‐Kettering Cancer Center, New York, USA, the co‐developer of the combination index method, on the present drug combination data analysis. We are very much grateful for his critical reading and valuable comments. This work has been supported by grants from the International Collaborative Items of Ministry of Science and Technology of China (no. 2006DFA32780) and the Science and Technology Commission of Shanghai municipality (no. N0.02JC14028).
References
- 1. Salzberg M, Thurlimann B, Bonnefois H et al. Current concepts of treatment strategies in advanced or recurrent ovarian cancer. Oncology 2005; 68: 293–8. [DOI] [PubMed] [Google Scholar]
- 2. Memarzadeh S, Berek JS. Advances in the management of epithelial ovarian cancer. J Reprod Med 2001; 46: 621–9; discussion 9–30. [PubMed] [Google Scholar]
- 3. Sanz MA, Fenaux P, Lo Coco F. Arsenic trioxide in the treatment of acute promyelocytic leukemia. A review of current evidence. Haematologica 2005; 90: 1231–5. [PubMed] [Google Scholar]
- 4. Douer D, Tallman MS. Arsenic trioxide: new clinical experience with an old medication in hematologic malignancies. J Clin Oncol 2005; 23: 2396–410. [DOI] [PubMed] [Google Scholar]
- 5. Berenson JR, Yeh HS. Arsenic compounds in the treatment of multiple myeloma: a new role for a historical remedy. Clin Lymphoma Myeloma 2006; 7: 192–8. [DOI] [PubMed] [Google Scholar]
- 6. Litzow MR, Lee S, Bennett JM et al. A phase II trial of arsenic trioxide for relapsed and refractory acute lymphoblastic leukemia. Haematologica 2006; 91: 1105–8. [PubMed] [Google Scholar]
- 7. Park JH, Kim EJ, Jang HY et al. Combination treatment with arsenic trioxide and sulindac enhances apoptotic cell death in lung cancer cells via activation of oxidative stress and mitogen‐activated protein kinases. Oncol Rep 2008; 20: 379–84. [PubMed] [Google Scholar]
- 8. Wang X, Wang G, Dong D, Fu S, Yang B. Inhibition on LS‐174T cell growth and activity of telomerase in vitro and in vivo by arsenic trioxide. Exp Toxicol Pathol 2008; 60: 481–8. [DOI] [PubMed] [Google Scholar]
- 9. Liu B, Pan S, Dong X et al. Opposing effects of arsenic trioxide on hepatocellular carcinomas in mice. Cancer Sci 2006; 97: 675–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Woo SY, Lee MY, Jung YJ et al. Arsenic trioxide inhibits cell growth in SH‐SY5Y and SK‐N‐AS neuroblastoma cell lines by a different mechanism. Pediatr Hematol Oncol 2006; 23: 231–43. [DOI] [PubMed] [Google Scholar]
- 11. Bornstein J, Sagi S, Haj A, Harroch J, Fares F. Arsenic Trioxide inhibits the growth of human ovarian carcinoma cell line. Gynecol Oncol 2005; 99: 726–9. [DOI] [PubMed] [Google Scholar]
- 12. Zhang J, Wang B. Arsenic trioxide (As(2)O(3)) inhibits peritoneal invasion of ovarian carcinoma cells in vitro and in vivo. Gynecol Oncol 2006; 103: 199–206. [DOI] [PubMed] [Google Scholar]
- 13. Nasr R, Rosenwald A, El‐Sabban ME et al. Arsenic/interferon specifically reverses 2 distinct gene networks critical for the survival of HTLV‐1‐infected leukemic cells. Blood 2003; 101: 4576–82. [DOI] [PubMed] [Google Scholar]
- 14. Wang HY, Liu SX, Zhang M. Gene expression profile changes in NB4 cells induced by arsenic trioxide. Acta Pharmacol Sin 2003; 24: 646–50. [PubMed] [Google Scholar]
- 15. Ahn WS, Bae SM, Lee KH et al. Comparison of effects of As2O3 and As4O6 on cell growth inhibition and gene expression profiles by cDNA microarray analysis in SiHa cells. Oncol Rep 2004; 12: 573–80. [PubMed] [Google Scholar]
- 16. Zhao S, Zhang J, Zhang X, Dong X, Sun X. Arsenic trioxide induces different gene expression profiles of genes related to growth and apoptosis in glioma cells dependent on the p53 status. Mol Biol Rep 2008; 35: 421–9. [DOI] [PubMed] [Google Scholar]
- 17. Chou TC, Talalay P. Quantitative analysis of dose‐effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 1984; 22: 27–55. [DOI] [PubMed] [Google Scholar]
- 18. Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 2006; 58: 621–81. [DOI] [PubMed] [Google Scholar]
- 19. Chou T.‐C, Martin N. CompuSyn Software for Drug Combinations and for General Dose‐Effect Analysis, and User’s Guide. Paramus, NJ: ComboSyn, 2007. [Google Scholar]
- 20. Bael TE, Peterson BL, Gollob JA. Phase II trial of arsenic trioxide and ascorbic acid with temozolomide in patients with metastatic melanoma with or without central nervous system metastases. Melanoma Res 2008; 18: 147–51. [DOI] [PubMed] [Google Scholar]
- 21. Kong B, Huang S, Wang W et al. Arsenic trioxide induces apoptosis in cisplatin‐sensitive and ‐resistant ovarian cancer cell lines. Int J Gynecol Cancer 2005; 15: 872–7. [DOI] [PubMed] [Google Scholar]
- 22. Wang G, Li W, Cui J et al. An efficient therapeutic approach to patients with acute promyelocytic leukemia using a combination of arsenic trioxide with low‐dose all‐trans retinoic acid. Hematol Oncol 2004; 22: 63–71. [DOI] [PubMed] [Google Scholar]
- 23. Shen ZX, Shi ZZ, Fang J et al. All‐trans retinoic acid/As2O3 combination yields a high quality remission and survival in newly diagnosed acute promyelocytic leukemia. Proc Natl Acad Sci U S A 2004; 101: 5328–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Estey E, Garcia‐Manero G, Ferrajoli A et al. Use of all‐trans retinoic acid plus arsenic trioxide as an alternative to chemotherapy in untreated acute promyelocytic leukemia. Blood 2006; 107: 3469–73. [DOI] [PubMed] [Google Scholar]
- 25. Kikuchi Y, Kita T, Takano M, Kudoh K, Yamamoto K. Treatment options in the management of ovarian cancer. Expert Opin Pharmacother 2005; 6: 743–54. [DOI] [PubMed] [Google Scholar]
- 26. Delaloge S, Laadem A, Taamma A et al. Pilot study of the paclitaxel, oxaliplatin, and cisplatin combination in patients with advanced/recurrent ovarian cancer. Am J Clin Oncol 2000; 23: 569–74. [DOI] [PubMed] [Google Scholar]
- 27. Gordinier ME, Kudelka AP, Kavanagh JJ, Wharton JT, Freedman RS. Thiotepa in combination with cisplatin for primary epithelial ovarian cancer: a phase II study. Int J Gynecol Cancer 2002; 12: 710–4. [DOI] [PubMed] [Google Scholar]
- 28. Serova M, Calvo F, Lokiec F et al. Characterizations of irofulven cytotoxicity in combination with cisplatin and oxaliplatin in human colon, breast, and ovarian cancer cells. Cancer Chemother Pharmacol 2006; 57: 491–9. [DOI] [PubMed] [Google Scholar]
- 29. Chou TC. Preclinical versus clinical drug combination studies. Leuk Lymphoma 2008; 49: 2059–80. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Gene expression analysis using the cDNA Microarray System in COC1 cells.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item