

Abstract
Osteoclasts are highly specialized cells that resorb bone, and their abnormal activity is implicated in a variety of human bone diseases. In neoplastic bone metastasis, the bone destruction caused by osteoclasts is not only associated with the formation and progression of metastatic lesions, but also could contribute to frequent complications such as severe pain and pathological fractures, which greatly diminish the quality of life of patients. Bisphosphonates, potent antiresorptive drugs, have been shown to have efficacy for treating bone metastases in many types of cancer, and the development of various molecularly targeted agents is currently proceeding. Thus, inhibition of osteoclast function is now established as an important treatment strategy for bony metastases. This review focuses on promising small molecules that disrupt osteoclast function and introduces our chemical/biological approach for identifying osteoclast‐targeting small molecular inhibitors. (Cancer Sci 2009)
Bone is a dynamic organ that constantly repeats the cycle of formation by osteoblasts and destruction (resorption) by osteoclasts throughout life.( 1 ) Bone remodeling is necessary for skeletal growth and to maintain normal bone tissue. Imbalances of remodeling can result in gross perturbations in skeletal structure and function, potentially causing morbidity. Most adult skeletal diseases are due to excess osteoclastic bone resorption, including osteoporosis, rheumatoid arthritis, and neoplastic bone metastasis.( 2 )
Bone, as well as lung and liver, is one of the most preferential metastatic sites for common human malignancies, notably breast, prostate, and lung cancers.( 3 ) Bone metastases are frequently associated with severe bone pain, pathological fractures, hypercalcemia, and spinal cord compression syndromes that greatly diminish the quality of life for patients. Although bone metastases are histopathologically classified as osteolytic, osteoblastic, or mixed lesions, accumulating evidence suggests that bone resorption by osteoclasts plays a pivotal role in the development and progression of all types of metastasis.( 4 ) In fact, bone‐residing metastatic cells are not directly able to destroy the hard bone tissue to enable them to survive and grow within bone. Instead, they secrete paracrine factors, such as parathyroid hormone‐related peptide (PTHrP) and interleukin‐6 (IL‐6), which directly or indirectly stimulate osteoclast differentiation and activation. In turn, bone resorption by osteoclasts releases growth factors such as transforming growth factor‐β (TGF‐β) and insulin‐like growth factor‐1 (IGF‐1) from the bone matrix that promote tumor growth. This interaction between tumor cells and the bony microenvironment results in a vicious cycle of bone destruction and tumor growth (Fig. 1).( 4 ) Furthermore, excessive osteoclastic bone destruction could be a factor that causes the complications of metastases.
Figure 1.
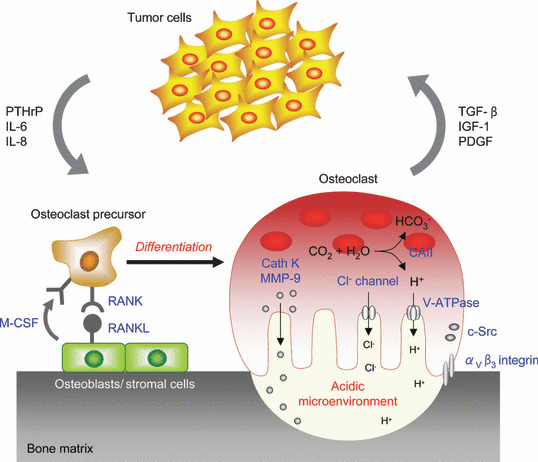
he vicious cycle of bone metastasis. Tumor cells intruding into the bone marrow produce factors such as parathyroid hormone‐related peptide (PTHrP) and interleukin‐6 (IL‐6), which stimulate osteoclastogenesis either directly or by stimulating receptor activator of nuclear factor‐κB ligand (RANKL) expression in osteoblasts. These lead to an increase of bone resorption, which in turn allows the release from the bone matrix of growth factors such as transforming growth factor‐β (TGF‐β) and insulin‐like growth factor‐1 (IGF‐1) that enhance tumor cell growth and metabolism. CAII, carbonic anhydrase II; Cath K, cathepsin K; M‐CSF, macrophage‐colony stimulating factor; MMP‐9, matrix metalloproteinase‐9; PDGF, platelet‐derived growth factor.
The concept of the vicious cycle alters the approach to the treatment of bone metastases, because it means that antiresorptive agents might also decrease the tumor burden in bone. This concept has recently been supported by the clinical performance of bisphosphonates (BPPs), which are potent antiresorptive drugs. Many clinical trials using zoledronic acid, a potent BPP, showed remarkably reduced rates of skeletal‐related events (SREs), defined as pathological fractures, spinal cord compression, radiation therapy, or surgery to bone in various tumor types. Additionally, zoledronic acid significantly increased the time to the first SRE and significantly reduced the risk of developing an SRE.( 5 ) In response, The United States Food and Drug Administration has approved zoledronic acid for use in any solid tumor with bony metastases. Currently, as the molecular mechanisms of osteoclast function and regulation become clearer, various molecularly targeted agents based on the specific inhibition of osteoclastic activity are being developed, including denosumab (AMG 162) and cathepsin K inhibitors. Denosumab is a fully human monoclonal antibody that binds and neutralizes human receptor activator of nuclear factor‐κB ligand (RANKL), which is the critical biological factor of osteoclastogenesis. A phase 1 clinical trial in patients with breast carcinoma with bone metastases or multiple myeloma showed that a single subcutaneous injection of denosumab caused rapid and sustained suppression of bone turnover markers and was well tolerated. Denosumab is currently in phase 3 trials in patients with metastatic disease in the skeleton and postmenopausal osteoporosis.( 6 )
Thus, suppressing osteoclastic bone resorption is now being established as an important treatment strategy in cancer therapy. In this review, we summarize the molecular mechanism and activity of promising antiresorptive agents with a focus on small molecule compounds. Furthermore, we describe our chemical/biological approach for identifying osteoclast‐targeting small molecule inhibitors.
Osteoclast Differentiation and Bone Resorption
Osteoclasts, multinucleated giant cells, are derived from hematopoietic stem cells of the monocyte/macrophage lineage. It is well known that two critical factors supplied by osteoblasts and stromal cells, RANKL, a member of the tumor necrosis factor (TNF) superfamily, and macrophage‐colony stimulating factor (M‐CSF), are essential for the differentiation and maturation of osteoclast precursors in bone.( 7 ) The binding of RANKL to the RANK receptor on the precursor cells, mainly through recruiting TNF receptor‐associated factor 6 (TRAF6), results in the activation of the mitogen‐activated protein kinase (MAPK) signaling pathway, several transcriptional factors such as NF‐κB and activator protein‐1 (AP‐1), and the calcium signaling cascade. These activating processes are mediated by immunoglobulin (Ig)‐like receptors associated with the immunoreceptor tyrosine‐based activation motif (ITAM)‐harboring adaptor molecules. Then, these signals concertedly induce the activation and expression of the nuclear factor of activated T cells c1 (NFATc1), which is the master transcription factor of osteoclastogenesis.( 8 ) Osteoclasts that develop into multinucleated giant cells by the fusion of osteoclast precursor cells are highly polarized; they form tightly sealed compartments on the surface of bone, and secrete protons and proteases such as cathepsin K from the ruffled border, resulting in the degradation of bone matrix that is composed of mineral and proteins, consisting of type I collagen (Fig. 1).
Accordingly, possible targets for small molecule compounds to selectively suppress osteoclast function would include the following: (i) the interaction of RANKL with RANK, and transduction molecules for the RANK signaling pathway; (ii) proteases responsible for bone resorption and molecules involved in the production of protons; (iii) molecules involved in cellular attachment to the bone matrix; and (iv) specific functions and environments of osteoclasts. Promising small molecules that disrupt osteoclast function are described in Table 1 and Figure 2.
Table 1.
Promising small molecules that disrupt osteoclast function
| Principal small molecules | Target molecule | Stage of development | Reference |
|---|---|---|---|
| Etidronate, Clodronate | ATP‐requiring molecules | Marketed | ( 9 ) |
| Alendronate, Zoledronate | FPP synthase | Marketed | ( 9 ) |
| MK‐0822, AAE581 | Cathepsin K | Clinical | ( 12 ) |
| AZD0530, CGP76030 | c‐Src | Clinical, Preclinical | ( 19 ) |
| L‐000845704, SB265123 | αVβ3 integrin | Clinical, Preclinical | ( 23, 57 ) |
| FR202126, Iejimalide A, B | V‐ATPase | Preclinical | ( 26, 30 ) |
| NS3736 | ClC‐7 Cl− channel | Preclinical | ( 32 ) |
| KN‐93 | CaMKIV | Preclinical | ( 33 ) |
| LFM‐A13 | Btk/Tec | Preclinical | ( 34 ) |
| (‐)‐DHMEQ, PDTC | NF‐κB | Preclinical | ( 35, 36 ) |
| Reveromycin A | IleRS | Preclinical | ( 43, 44 ) |
CaMKIV, Ca2+/calmodulin‐dependent kinase IV; (‐)‐DHMEQ, (‐)‐dehydroxymethylepoxyquinomicin; FPP, farnesyl pyrophosphate; IleRS, isoleucyl‐tRNA synthetase; PDTC, pyrrolidine dithiocarbamate.
Figure 2.
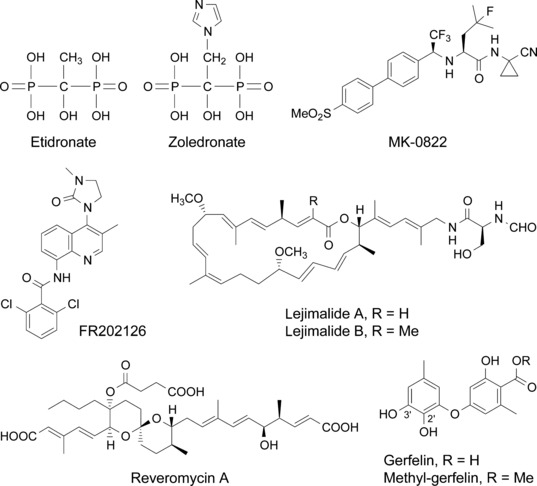
Chemical structure of the principal osteoclast‐targeting small molecule inhibitors discussed in the text.
Small Molecules that Selectively Inhibit Osteoclast Function
Bisphosphonates. BPPs are currently the major drugs used for the treatment of bone diseases that are characterized by enhanced osteoclastic bone resorption. They are structural analogs of pyrophosphate but contain a carbon instead of an oxygen atom. The phosphorus‐carbon‐phosphorus (P‐C‐P) structure allows a great number of chemical designs, which are achieved by changing the two side chains of carbon atoms.( 9 ) The effect of BBPs on osteoclasts is exerted through their accumulation and deposition in the bone: BBPs have a high affinity for bone mineral (hydroxyapatite), where they appear to be internalized selectively by bone‐resorbing osteoclasts and inhibit osteoclast function. BPPs can be grouped into two pharmacological classes with distinct molecular mechanisms of action.
Non‐nitrogen‐containing BPPs such as etidronate (Fig. 2) and clodronate can be metabolically incorporated into non‐hydrolysable analogs of adenosine 5′‐triphosphate (ATP) that may inhibit ATP‐dependent intracellular enzymes. On the other hand, the more potent, nitrogen‐containing BPPs such as alendronate and zoledronate (Fig. 2) inhibit a key enzyme, farnesyl pyrophosphate (FPP) synthase, in the mevalonate pathway, thereby preventing the biosynthesis of isoprenoid compounds that are essential for the post‐translational modification of small GTP‐binding proteins such as Rab, Rho, and Rac. The inhibition of protein prenylation, especially protein geranylgeranylation, leads to dysfunction of the cytoskeletal rearrangement and membrane ruffling, and ultimately causes apoptosis.( 9 ) The recently elucidated X‐ray crystal structure of the human FPP synthase in complexes with risedronate and zoledronate, and kinetic analyses, reveal the detailed molecular mechanisms through which these agents bind to the dimethylallyl/geranyl pyrophosphate ligand pocket and exhibit a competitive‐type inhibition pattern against geranyl pyrophosphate.( 10 )
Thus, the selectivity of BPPs to osteoclasts is attributed not to the specificity of their intracellular target molecules, but to their physicochemical properties and their high affinity to bone.
Cathepsin K inhibitors. Cathepsin K is a lysosomal cysteine proteinase that is highly expressed in osteoclasts and has been identified as the crucial enzyme in collagen breakdown during bone resorption. The essential role that cathepsin K plays in bone resorption is shown by data obtained from cathepsin K null mice, which display an osteopetrotic phenotype associated with a severe impairment of the resorptive activity of osteoclasts in the absence of any other overt pathological signs.( 11 ) Many highly selective and potent cathepsin K inhibitors have been developed recently, and preclinical studies have been shown that cathepsin K inhibitors reduce tumor‐induced osteolysis and the skeletal tumor burden.( 12 , 13 ) Several cathepsin K inhibitors, including MK‐0822 (odanacatib, Fig. 2), AAE581 (balicatib), ONO‐5334, and SB462795 (relacatib), are currently in clinical trials for osteoporosis, osteoarthritis, and neoplastic bone metastasis.
c‐Src inhibitors. c‐Src is a non‐receptor tyrosine kinase that is ubiquitously expressed in all cells. When this gene was deleted in mice through homologous recombination, osteoclast inactivation was the only detectable phenotypic change.( 14 ) c‐Src is activated in osteoclasts after integrin binding when the cells attach to the bone matrix to initiate bone resorption( 15 ); it mediates the complex intracellular cytoskeletal reorganization. c‐Src is also activated in response to the RANKL/RANK interaction after the recruitment of TRAF6 to the intracellular domain of RANK.( 16 ) It binds to TRAF6 and recruits several signaling proteins, including Cbl, Pyk‐2, and cortactin, which mediate polarization of the cell and the formation of the actin ring and ruffled border, in a process that is, as yet, incompletely understood.
In addition to its role in osteoclasts, c‐Src also has been involved in various elements of human cancer development and progression.( 17 ) Accordingly, c‐Src inhibitors are expected to target both tumor cells and osteoclasts, thus disrupting their mutual interactions in bone metastases. CGP76030, a c‐Src inhibitor, decreased the morbidity and lethality and also suppressed the metastasis‐induced osteolysis of the mice inoculated with MDA‐MB‐231 breast cancer cells.( 18 ) By now, numerous c‐Src inhibitors have been developed,( 19 ) and clinical trials are in progress for some of them. Such c‐Src inhibitors include pyrimidinylaminothiazole‐based BMS‐354825 (dasatinib), quinazoline‐based AZD0530, quinoline‐based SKI‐606 (bosutinib), pyridopyrimidinone‐based PD180970, pyrazolopyrimidine‐based PP1, and pyrrolopyrimidine‐based CGP76030.
αVβ3 integrin inhibitors. Another relatively selective osteoclast‐specific structure that seems to play a rate‐limiting role in osteoclast activity is the αVβ3 integrin. Integrins, a family of cell‐surface adhesion receptors that mediate cell–cell and cell–matrix interactions, are transmembrane glycoproteins composed of non‐covalently linked α and β subunits. The adhesion of the osteoclasts to bony surfaces is an important initial step in bone resorption. Various studies have indicated that the most abundant integrin in osteoclasts, αVβ3, interacts in bone with the extracellular matrix proteins that contain the arginine‐glycine‐aspartic acid (RGD) sequence, mediating bone resorption.( 20 ) In addition, integrins, like c‐Src, are known to contribute to tumor growth and metastasis in a variety of ways.( 21 ) Harms et al. demonstrated that an antagonist of the αVβ3 integrin suppressed the development of osteolytic breast cancer metastases and could inhibit the vicious cycle established between tumor cells and osteoclasts during the development of bone metastases.( 22 ) Moreover, clinical studies in women with postmenopausal osteoporosis have confirmed the usefulness of L‐000845704, an αVβ3 integrin antagonist, as an antiresorptive drug.( 23 )
Vacuolar H+‐ATPase inhibitors. An essential component of osteoclastic bone resorption is the acidic extracellular microenvironment formed on the surface of bone that dissolves the mineral content of bone. A vacuolar H+‐ATPase (V‐ATPase) in the osteoclast membrane plays a key role in this process. Bafilomycin, an inhibitor of this enzyme, has been shown to inhibit osteoclastic bone resorption in vitro and in vivo.( 24 ) We also showed that iejimalide (Fig. 2),( 25 ) a 24‐membered macrolide obtained from a marine tunicate, targeted V‐ATPase and induced osteoclast cell death.( 26 ) It has been reported that the V‐ATPase, an enzyme present in virtually every cell, may have a unique 116‐kD subunit in the osteoclast that is necessary for osteoclast‐mediated extracellular acidification.( 27 ) Recently, osteoclast‐selective V‐ATPase inhibitors such as SB242784, FR167356, and FR202126 (Fig. 2) were shown to inhibit osteoclastic bone resorption in experimental animal models of osteoporosis and metastatic bone disease.( 28 , 29 , 30 )
Others. Chloride channels as well as V‐ATPase play an essential role in the acidification of the resorption lacuna. Mice with a deficiency of the chloride channel, ClC7, show severe osteopetrosis resulting from impairment of chloride ion extrusion during osteoclastic bone resorption.( 31 ) Schaller et al. demonstrated that NS3736, a chloride channel inhibitor, suppressed bone loss in ovariectomized rats.( 32 )
Other possible compounds that specifically inhibit osteoclast function include inhibitors of transduction molecules within the RANK signaling pathway, such as Ca2+/calmodulin‐dependent kinase IV (CaMKIV), Btk/Tec tyrosine kinases, and NF‐κB.( 33 , 34 , 35 , 36 )
Forward Chemical Genetics of Natural Products
Reveromycin A. Reveromycin A (RM‐A; Fig. 2) is a polyketide natural compound produced by Streptomyces sp. that we previously found to be an inhibitor of the mitogenic activity of epidermal growth factor (EGF) in mouse keratinocyte lines.( 37 , 38 , 39 , 40 , 41 ) Yeast genetic approaches revealed that the intracellular molecular target of RM‐A is isoleucyl‐tRNA synthetase (IleRS),( 42 ) which is one of the aminoacyl‐tRNA synthetases that catalyze the attachment of specific amino acids to their cognate tRNAs, thereby ensuring the faithful translation of genetic code. We recently found a unique biological activity: RM‐A‐induced cell death occurred specifically in osteoclasts with a several hundred‐fold lesser concentration of RM‐A than that which inhibited the cell growth of various other cells, including osteoblastic cells and osteoclast precursor cells.( 43 ) It is unlikely that such selectivity is due to the target molecule of RM‐A. Chemical analysis of RM‐A showed that it is an acidic compound with three carboxylic groups in its structure (Fig. 2). This compound is normally in a highly polar form, which has poor cell permeability, with dissociation of protons from carboxylic acid moieties in blood or cell culture medium. On the other hand, osteoclasts, unlike other cells, form an acidic extracellular microenvironment to aid the resorption of bone matrix (Fig. 1). Therefore, it is likely that RM‐A would be converted into a non‐polar form, which is more permeable into cells, by suppressing the dissociation of protons under the acidic microenvironment of osteoclasts, thereby being selectively incorporated into osteoclasts (Fig. 3). This hypothesis was supported by the following experimental results: (i) tritium‐labeled RM‐A ([3H]RM‐A) was markedly and selectively incorporated into osteoclasts among various cell types in culture; (ii) the incorporation of [3H]RM‐A into osteoclasts was suppressed by alkalinizing their acidic microenvironment formation; and (iii) acidic culture medium (pH 5.5) induced incorporation of [3H]RM‐A into osteoclast precursor cells and enhanced sensitivity of osteoclast precursor cells (inhibition of protein synthesis and cell growth) to RM‐A, which do not have an acidic extracellular microenvironment. In osteoclasts, RM‐A inhibited IleRS activity and protein synthesis, resulting in the induction of apoptosis (Fig. 3). In vivo experiments also showed a marked decrease in osteoclast number in the bone of RM‐A‐treated animals with increased bone resorption. RM‐A significantly suppressed bone loss in ovariectomized mice without a loss in weight. In addition, RM‐A significantly inhibited bone metastasis formation through inhibition of osteoclastic bone destruction in various animal models of tumor bone metastasis including lung and prostate cancer metastatic cells.( 44 , 45 , 46 )
Figure 3.
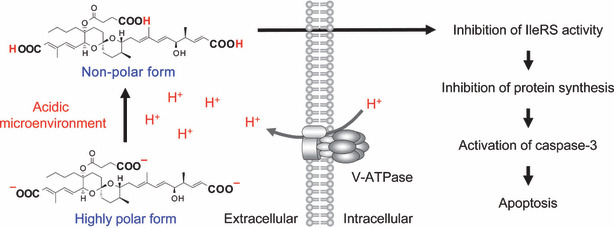
Possible mechanism of action of reveromycin A (RM‐A) in osteoclasts. The specific sensitivity of osteoclasts to RM‐A is due to their acidic microenvironment, which increases cell permeability of RM‐A by suppressing the dissociation of protons from their carboxylic acid moieties. In osteoclasts, RM‐A induces apoptosis through inhibition of isoleucyl‐tRNA synthetase (IleRS) activity.
As described above, the selectivity of RM‐A to osteoclasts, like that of BPPs, is not attributed to the specificity of its target molecule, but to its physicochemical properties.
Methyl‐gerfelin. In order to obtain new small molecules that inhibit osteoclast function, we performed cellular phenotype‐based screening from the chemical library of RIKEN Natural Products Depository (NPDepo).( 47 ) As a result, we found fungus‐derived gerfelin (GFN)( 48 , 49 ) and its methyl ester derivative, methyl‐gerfelin (M‐GFN), as hit compounds (Fig. 2).( 50 ) M‐GFN, which was stronger than GFN, suppressed RANKL‐induced osteoclastogenesis with no influence on the survival and phagocytic ability of mouse bone marrow–derived macrophages (BMMs).
We next planned to identify the molecular target of M‐GFN in inhibiting osteoclastogenesis by using M‐GFN‐immobilized beads, which were made by using our recently developed photocross‐linking method (Fig. 4a).( 51 , 52 , 53 ) This photocross‐linking method is a coupling technique that enables the introduction of a variety of small molecules onto solid supports through a photoaffinity reaction. In this method, aryldiazirine groups covalently attach to solid supports and are transformed by ultraviolet (UV) irradiation into highly reactive carbenes, which bind to or irreversibly insert into proximal small molecules in a functional group‐independent manner. This would allow the small molecules immobilized in a functional group‐independent manner to bind to all potential intracellular binding proteins because all surfaces of the small molecules are virtually presented on beads. Affinity purification experiments using M‐GFN‐immobilized beads showed that M‐GFN bound directly to glyoxalase I (GLO1) and sterol carrier protein 2 (SCP2). Knockdowns of GLO1, but not SCP2, inhibited RANKL‐induced osteoclast formation, which suggests that GLO1 is involved in osteoclastogenesis. GLO1 (EC 4.4.1.5) is an essential component in pathways leading to the detoxification of α‐oxoaldehydes, especially methylglyoxal (MG), which is continuously generated as a side product of glycolysis.( 54 ) The substrate for GLO1 is hemithioacetal, which is formed through the nonenzymatic conjugation of MG with glutathione (GSH). The product of the GLO1‐catalyzed reaction is S‐d‐lactoylglutathione, which is then hydrolyzed by GLO2 (EC 3.1.2.6) to d‐lactate. An enzyme kinetics analysis showed that M‐GFN inhibited mouse GLO1 activity in a competitive manner (K i = 0.23 μm). Moreover, increased intracellular MG levels were observed in M‐GFN‐treated BMMs, and the addition of MG to BMMs suppressed osteoclastogenesis, which strongly suggests that M‐GFN inhibits GLO1 activity, thereby allowing the accumulation of MG, resulting in the inhibition of osteoclastogenesis (Fig. 4b).
Figure 4.
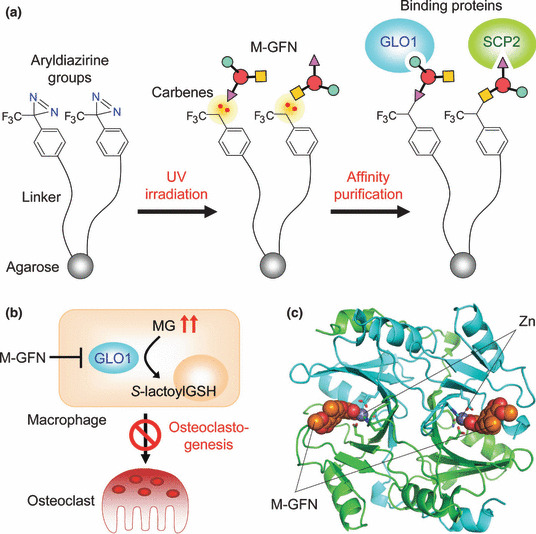
Target identification of methyl‐gerfelin (M‐GFN) that inhibits osteoclastogenesis. (a) Identification of M‐GFN‐binding proteins by using a photocross‐linked M‐GFN affinity matrix. (b) Possible mechanism of action of M‐GFN in osteoclast precursor cells. M‐GFN targets glyoxalase I (GLO1), resulting in the inhibition of osteoclastogenesis. (c) Cocrystal structure of mouse GLO1 complexed with M‐GFN. M‐GFN binds to GLO1 in the active site by coordination bonding via the zinc ion. Carbon atoms in one molecule of the GLO1 dimer and the other molecule (stick and ribbon model) are drawn in green and cyan, respectively. Orange and gray spheres represent M‐GFN and the zinc ion, respectively. MG, methylglyoxal; SCP2, sterol carrier protein 2.
To clarify the binding mode of M‐GFN, we determined the crystal structure of mouse GLO1 complexed with M‐GFN at 1.7‐Å resolution (Fig. 4c). This structural model contains one protein homodimer, two zinc ions, and two M‐GFN molecules in an asymmetrical unit. In the active site, which is located at the dimer interface, the zinc ion is in an octahedral coordination and is bound by four amino acid residues and two hydroxyl groups (2´OH and 3´OH) of M‐GFN. This binding mode of M‐GFN seems to mimic that of the intermediate reaction. Thus, it was revealed that M‐GFN binds to GLO1 at the active site, mainly by coordination bonding via the zinc ion and a hydrophobic interaction with the hydrophobic region.
Although the validation of GLO1 as a target of antiresorptive agents remains to be elucidated, these findings can facilitate the study of osteoclast biology and the development of therapeutic agents.
Concluding Remarks
According to the clinical performances of BPPs, suppression of the functions of osteoclasts could be established as an important treatment strategy for bone metastases. c‐Src and αVβ3 integrin inhibitors are expected to have a beneficial effect on bone metastasis treatment because they target both osteoclasts and tumor cells. In addition, a combined approach of chemical biology/chemical genetics, which is aimed at understanding and regulating vital functions using small molecules, could lead to further elucidation of osteoclast function and development of new antiresorptive agents. For example, our photocross‐linking method is useful in the preparation of chemical probes for fishing binding proteins (Fig. 4a) and for chemical arrays for high‐throughput ligand screening for proteins of interest.( 55 , 56 ) Since this method is able to immobilize uniformly any compounds that have a diversity of structure on solid supports, it is possible to prepare a chemical array that has several thousand compounds on a single glass slide. Indeed, by high‐throughput ligand screening of carbonic anhydrase II (CAII), which is involved in proton generation in osteoclasts (Fig. 1) using the chemical array, we have obtained a small molecule compound that inhibits CAII activity more strongly than the known inhibitor, acetazolamide.( 55 )
Since the discovery of the RANKL, our understanding of the osteoclast in bone biology has advanced considerably, but there are still questions to solve. Especially, the molecular mechanisms involving in the balanced coupling between bone resorption and bone formation remain poorly understood. Furthermore, little is known of the mechanisms that are responsible for the distortion of this process that occurs in metastatic cancer growing within bone. Further understanding of osteoclast functions and of the pathophysiology of bone metastases will lead to the availability of new therapeutic agents to treat bone metastases.
Acknowledgments
We thank Drs. J.‐T. Woo (Chubu University), T. Shinki (Nihon Pharmaceutical University), J. Kobayashi (Hokkaido University), M. Imoto (Keio University), and H. Okumura (RIKEN) for their collaborative studies. Work in our laboratory was supported in part by a Grant‐in‐Aid from the Ministry of Education, Culture, Sports, Science, and Technology of Japan and by the Bioarchitect and Chemical Biology projects (RIKEN).
References
- 1. Karsenty G, Wagner EF. Reaching a genetic and molecular understanding of skeletal development. Dev Cell 2002; 2: 389–406. [DOI] [PubMed] [Google Scholar]
- 2. Rodan GA, Martin TJ. Therapeutic approaches to bone diseases. Science 2000; 289: 1508–14. [DOI] [PubMed] [Google Scholar]
- 3. Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer 2002; 2: 584–93. [DOI] [PubMed] [Google Scholar]
- 4. Roodman GD. Mechanisms of bone metastasis. N Engl J Med 2004; 350: 1655–64. [DOI] [PubMed] [Google Scholar]
- 5. Body J‐J. Bisphosphonates for malignancy‐related bone disease: current status, future developments. Support Care Cancer 2006; 14: 408–18. [DOI] [PubMed] [Google Scholar]
- 6. Geusens P. Emerging treatments for postmenopausal osteoporosis – focus on denosumab. Clin Interv Aging 2009; 4: 241–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature 2003; 423: 337–42. [DOI] [PubMed] [Google Scholar]
- 8. Takayanagi H. Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat Rev Immunol 2007; 7: 292–304. [DOI] [PubMed] [Google Scholar]
- 9. Roelofs AJ, Thompson K, Gordon S, Rogers MJ. Molecular mechanisms of action of bisphosphonates: current status. Clin Cancer Res 2006; 12: 6222s–30s. [DOI] [PubMed] [Google Scholar]
- 10. Kavanagh KL, Guo K, Dunford JE et al. The molecular mechanism of nitrogen‐containing bisphosphonates as antiosteoporosis drugs. Proc Natl Acad Sci USA 2006; 103: 7829–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Saftig P, Hunziker E, Wehmeyer O et al. Impaired osteoclastic bone resorption leads to osteopetrosis in cathepsin‐K‐deficient mice. Proc Natl Acad Sci USA 1998; 95: 13453–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Le Gall C, Bonnelye E, Clézardin P. Cathepsin K inhibitors as treatment of bone metastasis. Curr Opin Support Palliat Care 2008; 2: 218–22. [DOI] [PubMed] [Google Scholar]
- 13. Le Gall C, Bellahcène A, Bonnelye E et al. A cathepsin K inhibitor reduces breast cancer‐induced osteolysis and skeletal tumor burden. Cancer Res 2007; 67: 9894–902. [DOI] [PubMed] [Google Scholar]
- 14. Soriano P, Montgomery C, Geske R, Bradley A. Targeted disruption of the c‐src proto‐oncogene leads to osteopetrosis in mice. Cell 1991; 64: 693–702. [DOI] [PubMed] [Google Scholar]
- 15. Insogna KL, Sahni M, Grey AB et al. Colony‐stimulating factor‐1 induces cytoskeletal reorganization and c‐src‐dependent tyrosine phosphorylation of selected cellular proteins in rodent osteoclasts. J Clin Invest 1997; 100: 2476–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wong BR, Besser D, Kim N et al. TRANCE, a TNF family member, activates Akt/PKB through a signaling complex involving TRAF6 and c‐Src. Mol Cell 1999; 4: 1041–9. [DOI] [PubMed] [Google Scholar]
- 17. Ishizawar R, Parsons SJ. c‐Src and cooperating partners in human cancer. Cancer Cell 2004; 6: 209–14. [DOI] [PubMed] [Google Scholar]
- 18. Rucci N, Recchia I, Angelucci A et al. Inhibition of protein kinase c‐Src reduces the incidence of breast cancer metastases and increases survival in mice: implications for therapy. J Pharmacol Exp Ther 2006; 318: 161–72. [DOI] [PubMed] [Google Scholar]
- 19. Rucci N, Susa M, Teti A. Inhibition of protein kinase c‐Src as a therapeutic approach for cancer and bone metastases. Anticancer Agents Med Chem 2008; 8: 342–9. [DOI] [PubMed] [Google Scholar]
- 20. Nakamura I, Duong le T, Rodan SB, Rodan GA. Involvement of αVβ3 integrins in osteoclast function. J Bone Miner Metab 2007; 25: 337–44. [DOI] [PubMed] [Google Scholar]
- 21. Felding‐Habermann B. Integrin adhesion receptors in tumor metastasis. Clin Exp Metastasis 2003; 20: 203–13. [DOI] [PubMed] [Google Scholar]
- 22. Harms JF, Welch DR, Samant RS et al. A small molecule antagonist of the αVβ3 integrin suppresses MDA‐MB‐435 skeletal metastasis. Clin Exp Metastasis 2004; 21: 119–28. [DOI] [PubMed] [Google Scholar]
- 23. Murphy MG, Cerchio K, Stoch SA et al. Effect of L‐000845704, an αVβ3 integrin antagonist, on markers of bone turnover and bone mineral density in postmenopausal osteoporotic women. J Clin Endocrinol Metab 2005; 90: 2022–8. [DOI] [PubMed] [Google Scholar]
- 24. Farina C, Gagliardi S. Selective inhibitors of the osteoclast vacuolar proton ATPase as novel bone antiresorptive agents. Drug Discov Today 1999; 4: 163–72. [DOI] [PubMed] [Google Scholar]
- 25. Kobayashi J, Cheng J, Ohta T et al. Iejimalides A and B, novel 24‐membered macrolides with potent antileukemic activity from the Okinawan marine tunicate Eudistoma cf. rigida . J Org Chem 1988; 53: 6147–50. [Google Scholar]
- 26. Kazami S, Muroi M, Kawatani M et al. Iejimalides show anti‐osteoclast activity via V‐ATPase inhibition. Biosci Biotechnol Biochem 2006; 70: 1364–70. [DOI] [PubMed] [Google Scholar]
- 27. Li Y‐P, Chen W, Liang Y, Li E, Stashenko P. Atp6i‐deficient mice exhibit severe osteopetrosis due to loss of osteoclast‐mediated extracellular acidification. Nat Genet 1999; 23: 447–51. [DOI] [PubMed] [Google Scholar]
- 28. Visentin L, Dodds RA, Valente M et al. A selective inhibitor of the osteoclastic V‐H+‐ATPase prevents bone loss in both thyroparathyroidectomized and ovariectomized rats. J Clin Invest 2000; 106: 309–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Niikura K, Takeshita N, Takano M et al. A vacuolar ATPase inhibitor, FR167356, prevents bone resorption in ovariectomized rats with high potency and specificity: potential for clinical application. J Bone Miner Res 2005; 20: 1579–88. [DOI] [PubMed] [Google Scholar]
- 30. Niikura K. Effect of a V‐ATPase inhibitor, FR202126, in syngeneic mouse model of experimental bone metastasis. Cancer Chemother Pharmacol 2007; 60: 555–62. [DOI] [PubMed] [Google Scholar]
- 31. Kornak U, Kasper D, Bösl MR et al. Loss of the ClC‐7 chloride channel leads to osteopetrosis in mice and man. Cell 2001; 104: 205–15. [DOI] [PubMed] [Google Scholar]
- 32. Schaller S, Henriksen K, Sveigaard C et al. The chloride channel inhibitor NS3736 prevents bone resorption in ovariectomized rats without changing bone formation. J Bone Miner Res 2004; 19: 1144–53. [DOI] [PubMed] [Google Scholar]
- 33. Sato K, Suematsu A, Nakashima T et al. Regulation of osteoclast differentiation and function by the CaMK‐CREB pathway. Nat Med 2006; 12: 1410–6. [DOI] [PubMed] [Google Scholar]
- 34. Shinohara M, Koga T, Okamoto K et al. Tyrosine kinases Btk and Tec regulate osteoclast differentiation by linking RANK and ITAM signals. Cell 2008; 132: 794–806. [DOI] [PubMed] [Google Scholar]
- 35. Takatsuna H, Asagiri M, Kubota T et al. Inhibition of RANKL‐induced osteoclastogenesis by (‐)‐DHMEQ, a novel NF‐κB inhibitor, through downregulation of NFATc1. J Bone Miner Res 2005; 20: 653–62. [DOI] [PubMed] [Google Scholar]
- 36. Strait K, Li Y, Dillehay DL, Weitzmann MN. Suppression of NF‐κB activation blocks osteoclastic bone resorption during estrogen deficiency. Int J Mol Med 2008; 21: 521–5. [PubMed] [Google Scholar]
- 37. Osada H, Koshino H, Isono K, Takahashi H, Kawanishi G. Reveromycin A, a new antibiotic which inhibits the mitogenic activity of epidermal growth factor. J Antibiot (Tokyo) 1991; 44: 259–61. [DOI] [PubMed] [Google Scholar]
- 38. Takahashi H, Osada H, Koshino H et al. Reveromycins, new inhibitors of eukaryotic cell growth. I. Producing organism, fermentation, isolation and physico‐chemical properties. J Antibiot (Tokyo) 1992; 45: 1409–13. [DOI] [PubMed] [Google Scholar]
- 39. Takahashi H, Osada H, Koshino H et al. Reveromycins, new inhibitors of eukaryotic cell growth. II. Biological activities. J Antibiot (Tokyo) 1992; 45: 1414–9. [DOI] [PubMed] [Google Scholar]
- 40. Koshino H, Takahashi H, Osada H, Isono K. Reveromycins, new inhibitors of eukaryotic cell growth. III. Structures of reveromycins A, B, C and D. J Antibiot (Tokyo) 1992; 45: 1420–7. [DOI] [PubMed] [Google Scholar]
- 41. Takahashi H, Yamashita Y, Takaoka H, Nakamura J, Yoshihama M, Osada H. Inhibitory action of reveromycin A on TGF‐α‐dependent growth of ovarian carcinoma BG‐1 in vitro and in vivo. Oncol Res 1997; 9: 7–11. [PubMed] [Google Scholar]
- 42. Miyamoto Y, Machida K, Mizunuma M et al. Identification of Saccharomyces cerevisiae isoleucyl‐tRNA synthetase as a target of the G1‐specific inhibitor reveromycin A. J Biol Chem 2002; 277: 28810–4. [DOI] [PubMed] [Google Scholar]
- 43. Woo J‐T, Kawatani M, Kato M et al. Reveromycin A, an agent for osteoporosis, inhibits bone resorption by inducing apoptosis specifically in osteoclasts. Proc Natl Acad Sci USA 2006; 103: 4729–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Muguruma H, Yano S, Kakiuchi S et al. Reveromycin A inhibits osteolytic bone metastasis of small‐cell lung cancer cells, SBC‐5, through an antiosteoclastic activity. Clin Cancer Res 2005; 11: 8822–8. [DOI] [PubMed] [Google Scholar]
- 45. Hiraoka K, Zenmyo M, Watari K et al. Inhibition of bone and muscle metastases of lung cancer cells by a decrease in the number of monocytes/macrophages. Cancer Sci 2008; 99: 1595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yano A, Tsutsumi S, Soga S et al. Inhibition of Hsp90 activates osteoclast c‐Src signaling and promotes growth of prostate carcinoma cells in bone. Proc Natl Acad Sci USA 2008; 105: 15541–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tomiki T, Saito T, Ueki M et al. RIKEN natural products encyclopedia (RIKEN NPEdia), a chemical database of RIKEN natural products depository (RIKEN NPDepo). J Comp Aided Chem 2006; 7: 157–62. [Google Scholar]
- 48. Zenitani S, Tashiro S, Shindo K, Nagai K, Suzuki K, Imoto M. Gerfelin, a novel inhibitor of geranylgeranyl diphosphate synthase from Beauveria felina QN22047. I. Taxonomy, fermentation, isolation, and biological activities. J Antibiot (Tokyo) 2003; 56: 617–21. [DOI] [PubMed] [Google Scholar]
- 49. Zenitani S, Shindo K, Tashiro S et al. Gerfelin, a novel inhibitor of geranylgeranyl diphosphate synthase from Beauveria felina QN22047. II. Structural elucidation. J Antibiot (Tokyo) 2003; 56: 658–60. [DOI] [PubMed] [Google Scholar]
- 50. Kawatani M, Okumura H, Honda K et al. The identification of an osteoclastogenesis inhibitor through the inhibition of glyoxalase I. Proc Natl Acad Sci USA 2008; 105: 11691–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kanoh N, Kumashiro S, Simizu S et al. Immobilization of natural products on glass slides by using a photoaffinity reaction and the detection of protein‐small‐molecule interactions. Angew Chem Int Ed Engl 2003; 42: 5584–7. [DOI] [PubMed] [Google Scholar]
- 52. Kanoh N, Honda K, Simizu S, Muroi M, Osada H. Photo‐cross‐linked small‐molecule affinity matrix for facilitating forward and reverse chemical genetics. Angew Chem Int Ed Engl 2005; 44: 3559–62. [DOI] [PubMed] [Google Scholar]
- 53. Kanoh N, Asami A, Kawatani M et al. Photo‐cross‐linked small‐molecule microarrays as chemical genomic tools for dissecting protein‐ligand interactions. Chem Asian J 2006; 1: 789–97. [DOI] [PubMed] [Google Scholar]
- 54. Thornalley PJ. Glutathione‐dependent detoxification of α‐oxoaldehydes by the glyoxalase system: involvement in disease mechanisms and antiproliferative activity of glyoxalase I inhibitors. Chem Biol Interact 1998; 111–112: 137–51. [DOI] [PubMed] [Google Scholar]
- 55. Miyazaki I, Simizu S, Ichimiya H, Kawatani M, Osada H. Robust and systematic drug screening method using chemical arrays and the protein library: identification of novel inhibitors of carbonic anhydrase II. Biosci Biotechnol Biochem 2008; 72: 2739–49. [DOI] [PubMed] [Google Scholar]
- 56. Miyazaki I, Simizu S, Ishida K, Osada H. On‐chip fragment‐based approach for discovery of high‐affinity bivalent inhibitors. Chembiochem 2009; 10: 838–43. [DOI] [PubMed] [Google Scholar]
- 57. Lark MW, Stroup GB, Hwang SM et al. Design and characterization of orally active Arg‐Gly‐Asp peptidomimetic vitronectin receptor antagonist SB 265123 for prevention of bone loss in osteoporosis. J Pharmacol Exp Ther 1999; 291: 612–7. [PubMed] [Google Scholar]