

Abstract
Although discovery of the tyrosine kinase inhibitor (TKI) imatinib mesylate has significantly improved the prognosis of chronic myeloid leukemia (CML) patients, a rare population of CML stem cells is known to be resistant to TKI therapy, causing recurrence of CML. However, recent progress in CML stem cell biology may present a novel therapeutic avenue for CML patients. In this review, we focus on mechanisms used by CML stem cells to maintain TKI‐resistance. Comprehensive approaches including mouse genetics, prospective identification of CML stem cells, and syngenic transplantation techniques have identified several key molecules or signaling pathways, including hedgehog (Hh)/Smo, promyelocytic leukemia (PML), 5‐lipoxygenase (5‐LO), and forkhead box class O (FOXO), that function in CML stem cell maintenance. Inhibiting some of these factors in combination with TKI administration successfully antagonized resistance of CML stem cells to TKI therapy, resulting in efficient eradication of leukemia cells in vivo. Thus, development of methods that sensitize CML stem cells to TKI therapy may lead to novel therapies to treat CML patients. (Cancer Sci 2010)
Chronic Myeloid Leukemia (CML) as a “Stem Cell Disease”
Hematopoietic stem cells (HSCs), which top the hierarchy of the normal hematopoietic system, are defined by both their ability to reproduce themselves, a property known as self‐renewal, and their capacity to give rise to all mature hematopoietic cell lineages throughout an individual’s lifetime. Recently, it was revealed that CML contains leukemia stem cells which form a hierarchy similar to that seen in normal hematopoiesis. Human CML is a biphasic myeloproliferative disease (MPD), which initially assumes a chronic phase before progressing to an accelerated phase and then to blast crisis. In CML patients, the initial chronic phase is characterized by massive expansion of myeloid cells. Fialkow et al. ( 1 ) demonstrated that chronic phase CML cells can produce functionally normal mature blood cells, indicating that CML cells in the chronic phase can differentiate into mature blood cells, like normal HSCs. Acquisition of additional genetic mutations and/or epigenetic alterations results in progression from the chronic phase to an accelerated phase and finally to blast crisis. Indeed, Jamieson et al. ( 2 ) proposed a model in which abnormal activation of β‐catenin in a granulocyte/macrophage progenitor (GMP)‐like population in CML patients promotes disease progression from chronic phase to blast crisis. Blast crisis is characterized by accumulation of myeloid or lymphoid blast cells in peripheral blood or bone marrow, indicating that CML cells in blast crisis can proliferate but that their differentiation capacity is blocked. Since CML patients in blast crisis show poor prognosis with short survival, it is critical to treat CML patients during the chronic phase.
Most CML is caused by a translocation between human chromosomes 9 and 22 that generates what is known as the Philadelphia chromosome and resultant breakpoint cluster region‐abelson (BCR–ABL) fusion gene, which encodes a constitutively active tyrosine kinase.( 3 ) Several lines of evidence indicate that CML cells emerge due to expression of BCR–ABL in normal HSCs (Fig. 1). Transplantation of multipotent murine HSCs expressing BCR–ABL into recipient mice induces CML‐like MPD.( 4 , 5 , 6 , 7 , 8 ) By contrast, CML is not induced in committed murine hematopoietic progenitor cells expressing BCR–ABL.( 9 ) These results demonstrate that chronic phase CML cells originate from multipotent HSCs. In a mouse CML‐like MPD model, CML stem cells, which are defined by the ability to induce CML in transplanted mice, can be purified in a rare c‐Kit+Lineage−Sca‐1+ (KLS+) population of mouse CML cells (i.e. in cells that exhibit the marker profile of normal HSCs).( 10 , 11 , 12 , 13 , 14 ) Interestingly, in human CML patients, CML stem cells are apparently found in a cell fraction expressing cell surface markers characteristic of primitive hematopoietic cells.( 15 , 16 ) All of these findings support the notion that CML is a “stem cell disease” (Fig. 1). In this review, we present evidence from our investigations and those of others supporting a link between molecular mechanisms regulating self‐renewal in normal HSCs and CML stem cells, and discuss current efforts aimed at developing novel therapeutics to specifically target tyrosine kinase inhibitor (TKI)‐resistant CML stem cells.
Figure 1.
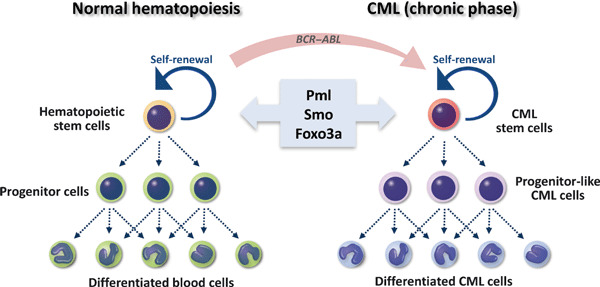
A link between molecular mechanisms regulating self‐renewal activity in normal hematopoietic stem cells (HSCs) and in chronic myeloid leukemia (CML) stem cells. HSCs, which top the hierarchy of the normal hematopoietic system, are defined by both their ability to self‐renew and to give rise to all lineages of mature hematopoietic cells throughout an individual’s lifetime. A rare population of CML stem cells also can self‐renew and generate progenitor‐like and differentiated CML cells, indicating that CML cells in the chronic phase form a hierarchy similar to that of normal HSCs. Several lines of evidence demonstrate that the emergence of CML cells requires BCR–ABL expression in normal HSCs. Recently molecules regulating normal HSC fate, including promyelocytic leukemia (Pml), Smo, and forkhead box class O (Foxo)‐3a, have also been shown to govern CML stem cell function. These observations support the idea that CML is “stem cell disease”.
Common Signaling Pathways Underlie Stem Cell Fate in Both Normal Hematopoiesis and CML
Observation that HSCs and CML stem cells share common properties suggests that signaling pathways determining normal HSC fate also govern maintenance of CML stem cell function. Several factors regulating stem cell fate in both normal hematopoiesis and CML leukemogenesis have been identified (Fig. 1). Promyelocytic leukemia (PML) protein functions in diverse cellular activities, including the DNA damage response, apoptosis, cellular senescence, and neoangiogenesis.( 17 ) Ito et al. ( 12 ) found that PML is highly expressed in normal HSCs and that Pml deficiency impairs HSC self‐renewal capacity. Importantly, whereas Pml +/+ CML stem cells maintain the ability to induce CML at least at the 4th‐serial transplantation, Pml −/− CML stem cells fail to generate disease at the 3rd‐transplantation, demonstrating that PML is essential for CML stem cell maintenance (Table 1). Since in vivo treatment with arsenic trioxide (As2O3), which can reduce PML expression, eradicates CML stem cells in combination with the anti‐leukemia drug Ara‐C, PML down‐regulation is an important candidate for CML stem cell‐targeting therapy.
Table 1.
The biology and TKI‐resistance in CML stem cells based on mouse genetic studies
| Disrupted molecule | CML stem cells | Inhibitor | Possible combined therapy | Reference |
|---|---|---|---|---|
| Pml | Defective ability of CML stem cells to develop CML at third transplantation | Arsenic trioxide | Arsenic trioxide and Ara‐C improve survival of CML‐affected mice | 12 |
| β‐Catenin | Defective ability of CML stem cells to develop CML at second transplantation | – | – | 11 |
| Alox5 | Defective ability of CML stem cells to develop CML at second transplantation | Zileuton | Zileuton and imatinib improve survival of CML‐affected mice | 45 |
| Smo | Impairment of development of CML and depletion of CML stem cells | Cyclopamin | Cyclopamin and nilotinib prolong survival of CML‐affected mice | 13, 21 |
| Foxo3a | Defective ability of CML stem cells to develop CML at third transplantation | TGF‐β inhibitor | TGF‐β inhibitor and imatinib improve survival of CML‐affected mice, and suppress infiltration of CML cells in lung | 14 |
Arsenic trioxide can reduce PML expression. Zileuton is an inhibitor of 5‐lipoxygenase. Cyclopamin stabilizes Smo in an inactive form. TGF‐β inhibitor suppresses TGF‐β–Foxo signaling. Alox5, arachidonate 5‐lipoxygenase; CML, chronic myeloid leukemia; Foxo3a, forkhead box class O 3a; PML, promyelocytic leukemia; TGF‐β, transforming growth factor‐β.
Wnt/β‐catenin signaling is implicated in both normal HSC homeostasis and CML leukemogenesis. β‐Catenin reportedly functions to regulate normal mouse HSC self‐renewal.( 11 , 18 , 19 ) Interestingly, Zhao et al.,( 11 ) reported that conditional deletion of β‐catenin impairs long‐term maintenance of CML stem cells in the chronic phase, whereas loss of β‐catenin allows acute lymphoblastic leukemia (ALL) to proceed unimpaired (Table 1). Loss of β‐catenin can suppress infiltration of CML cells into the lung and liver in mice injected with CML stem cells. Thus, Wnt/β‐catenin signaling likely plays a role in maintaining CML stem cells.
Hedgehog (Hh) signaling reportedly regulates self‐renewal of normal HSCs and CML stem cells. In the absence of Hh ligands, the 12‐pass transmembrane domain receptor Patched (Ptch) inhibits Smo, a seven‐pass transmembrane domain receptor.( 20 ) Binding of Hh ligands, including Sonic hedgehog (Shh), Indian hedgehog (Ihh), and Desert hedgehog (Dhh), to Ptch activates Smo, which in turn activates signaling via Gli transcriptional effectors. To understand the role of Hh signaling in normal HSCs and CML leukemogenesis, Dierks et al. and Zhao et al. ( 13 , 21 ) in parallel studies analyzed the effects of conditional Smo deletion in the hematopoietic system. Although the frequency of normal HSCs was unchanged in Smo‐deficient mice,( 13 , 21 ) Smo was required for long‐term maintenance of HSCs in vivo.( 13 ) Interestingly, Smo loss causes CML stem cell depletion and impairs CML development,( 13 , 21 ) whereas expression of constitutively active Smo increases the frequency of CML stem cells and accelerates CML development.( 13 ) These findings demonstrate that Hh signaling plays an essential role to maintain CML stem cell function.
Forkhead box class O (FOXO) transcription factors also play an important role in the maintenance of normal HSCs and CML stem cells. The FOXO factors include FOXO1, FOXO3a, FOXO4, and FOXO6, all of which are downstream targets of Phosphatidylinositol 3 kinase (PI3K)–AKT signaling.( 22 ) In the absence of stimulation by growth factors or insulin, FOXOs are present in the nucleus and activate their transcriptional targets. When a growth factor or insulin binds to the appropriate cell surface receptor, AKT is activated and directly phosphorylates FOXOs, resulting in their nuclear exclusion and degradation in the cytoplasm. Tothova et al. ( 23 ) reported that in mice with triple conditional deletion of Foxo1, Foxo3a, and Foxo4, the HSC population markedly increases. We and others have reported that Foxo3a alone is essential to maintain the HSC pool,( 24 , 25 ) indicating that FOXOs are essential to maintain normal HSC self‐renewal. Recently, we demonstrated an essential role for Foxo3a in the maintenance of CML stem cells.( 14 ) Previous studies using CML cell lines indicate that BCR–ABL likely activates PI3K–AKT signaling, leading to FOXO nuclear export and suppression of FOXO transcriptional activity.( 26 , 27 , 28 ) However, using a mouse CML‐like MPD model we found that whereas non‐CML stem cells showed high levels of Akt phosphorylation and cytoplasmic localization of Foxo3a, cells with decreased Akt phosphorylation and nuclear localization of Foxo3a were enriched in the CML stem cell population, despite expression of BCR–ABL. We also showed that the ability of CML stem cells to promote disease at the 3rd‐transplantation is significantly decreased by Foxo3a deficiency in vivo. In particular, cells deficient in Foxo3a lose the potential to generate malignancies in multiple lineages.( 14 ) Thus, Foxo3a is essential for long‐term maintenance of leukemia‐initiating potential in CML stem cells.
Resistance of Quiescent CML Stem Cells to TKI Therapy
The development of imatinib mesylate, an inhibitor of ABL kinase, has represented a breakthrough in CML treatment.( 29 ) However, most CML patients require additional therapy due to resistance or intolerance.( 30 , 31 , 32 , 33 ) Recent studies report that TKIs including imatinib, nilotinib, and dasatinib are potent TKI inhibitors in differentiated CML cells but are not as effective in quiescent primitive CML stem cells (Fig. 2).( 34 , 35 , 36 , 37 , 38 , 39 ) In addition, mathematical modeling has shown that successful imatinib therapy is characterized by a bi‐phasic, exponential decline in the number of CML cells, but that imatinib likely does not deplete CML stem cells in patients in the chronic phase.( 40 , 41 ) Since residual CML stem cells are the source of disease recurrence, drugs capable of eradicating CML stem cells would provide markedly improved therapeutic benefits to CML patients.( 42 )
Figure 2.
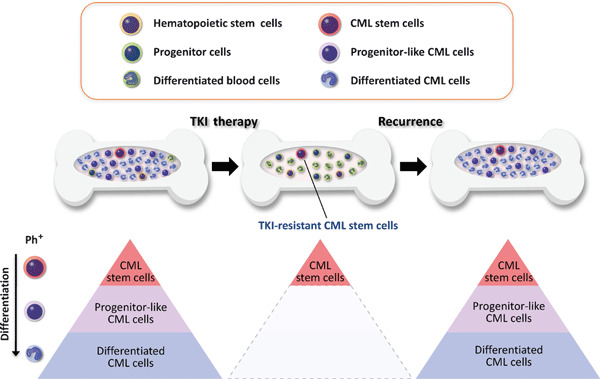
Residual CML stem cells are responsible for TKI resistance in CML patients. CML arises in HSCs due to activity of the oncogenic BCR–ABL fusion protein. The TKI imatinib, which inhibits ABL tyrosine kinase activity, is now standard therapy for CML. Imatinib can eradicate numerous CML cells, such as progenitor‐like CML cells and differentiated CML cells, and significantly improves the prognosis of CML patients in the chronic phase. However, it does not deplete CML stem cells, which top the CML hierarchy and are responsible for disease recurrence. Thus, effective human CML therapy requires measures to eradicate TKI‐resistant CML stem cells. Ph+, Philadelphia chromosome‐positive CML cells.
Molecular Mechanisms of TKI‐Resistance of CML Stem Cells
Several potential mechanisms underlying TKI‐resistance of CML stem cells have been proposed. It has been shown that CML stem cells are quiescent and undifferentiated, resulting in resistance to chemotherapy. CML stem cell properties are possibly regulated by association with a niche. Several lines of evidence indicate that inactivation of key molecules for CML stem cell maintenance possibly contributes to development of CML therapy. For example, pharmacological blockade of Hh signaling by cyclopamine, which stabilizes Smo in an inactive form, impairs CML development by CML stem cells (Table 1).( 13 , 21 ) Furthermore, combining a TKI (nilotinib) with cyclopamine increased the amount of time to disease recurrence more effectively than did treatment with a TKI alone.( 21 )
BCR–ABL drives robust AKT activation, which represses FOXO function. TKIs block this activity and promote FOXO nuclear localization and activation, which induces apoptosis or cell cycle arrest. However, as noted above we observed unexpected nuclear localization of FOXO in CML stem cells in mouse models.( 14 ) Since CML stem cells resist TKI therapy, it is suggested that this “stem cell paradox” regarding FOXO activation may reflect stem cell status. Komatsu et al. previously reported that introduction of an activated form of FOXO into the CML line KCL22 led to cell cycle arrest, concluding that FOXO3a is a downstream effector of TKI‐induced cell cycle arrest in CML cells. This study also reported the interesting finding that FOXO inactivation sensitizes cells to TKI treatment in vitro, suggesting that FOXO contributes to resistance to TKI therapy. To address whether FOXO functions as a TKI effector or an inhibitor of TKI in CML stem cells in vivo, we investigated roles of Foxo3a in response to TKI therapy using a CML mouse model.( 14 ) We found that Foxo3a deficiency enhanced sensitivity of CML stem cells to TKI therapy and proposed that Foxo3a plays distinct roles in CML stem cells versus non‐CML stem cells.( 14 ) In our model, FOXO activation protects CML stem cells against various stresses, including TKI therapy, whereas it induces apoptosis or cell cycle arrest in non‐CML stem cells in response to TKI therapy.
We have also searched for chemical compounds that alter Akt–Foxo status in CML stem cells and found that treatment of CML stem cells with Ly364947, an inhibitor of transforming growth factor (TGF)‐β signaling, efficiently activates Akt and suppresses Foxo. Although administration of Ly364947 alone did not extend the survival of CML‐affected mice, Ly364947 combined with imatinib significantly reduced recipient lethality and decreased CML stem cell frequency (Table 1).( 14 ) These in vivo data demonstrate a critical role for the TGF‐β/FOXO axis in maintenance of imatinib‐resistant CML stem cells. Interestingly, Yamazaki et al. ( 43 ) reported that TGF‐β is a candidate niche factor to maintain HSC dormancy. Thus the TGF‐β pathway may be common to both normal HSCs and CML stem cells. However, we found that inhibition of TGF‐β had a more potent effect on CML stem cells than on normal stem/progenitor cells.( 14 ) Supporting this finding, it has been reported that TGF‐β signaling is enhanced by the presence of BCR–ABL.( 44 ) These data indicate that inhibition of TGF‐β signaling may lead to efficient eradication of residual CML stem cells.
Chen et al. ( 45 ) propose an alternate mechanism underlying TKI resistance of CML stem cells. The Arachidonate 5‐lipoxygenase (5‐LO) gene (Alox5) encodes a lipoxygenase, which plays a role in the synthesis of leukotrienes from arachidonic acid and functions in numerous physiological and pathological processes, including oxidative stress, inflammation, and cancer.( 46 , 47 ) Gene expression profiling revealed that Alox5 expression is specifically up‐regulated by BCR–ABL.( 45 ) Whereas recipient mice transplanted with Alox5 +/+ CML stem cells died of CML, mice transplanted with Alox5 −/− CML stem cells survived, indicating that Alox5 deficiency underlies impaired CML stem cell function. Combined administration of the 5‐LO inhibitor Zileuton with imatinib prolonged survival of CML‐affected mice (Table 1).( 45 ) Since loss of Alox5 does not affect normal HSC self‐renewal, combined therapy of TKIs and 5‐LO inhibitors may bring therapeutic benefits to CML patients in the chronic phase. Interestingly, Alox5 up‐regulation was not inhibited by TKI treatment, which may explain why imatinib does not inhibit CML stem cells. Further investigation regarding BCR–ABL functions not affected by TKI treatment are required in order to understand mechanisms of TKI‐resistance in CML stem cells.
Although interferon (IFN)‐α was a first line treatment for CML, imatinib has replaced IFN‐α therapy due to the higher response rate. However, it was reported that patients previously exposed to IFN‐α showed a high rate of long‐term complete remission.( 48 ) Interestingly, it has been recently demonstrated that type I IFNs act directly on HSCs to induce cell cycle progression,( 49 , 50 ) suggesting the possibility that IFN‐α pre‐treatment sensitized the CML stem cells to TKI therapy. Investigation of the IFN signaling on CML stem cells may influence the future treatment of CML.
Future Perspectives
In this review, we have focused on recent advances in our understanding of CML stem cell maintenance. Notably, factors regulating normal HSCs also govern TKI‐resistance in CML stem cells, indicating that identifying molecular mechanisms governing normal HSC fate could shed light on activities of CML stem cells. For instance, inhibiting the association between CML stem cells and their microenvironment, or niche, could constitute one therapeutic strategy whereby CML patients could be treated by specific suppression of TKI‐resistant CML stem cells (Fig. 3). Since normal HSCs are supported within a niche, TKI‐resistant CML stem cells may also be protected and maintained in a dormant, undifferentiated state by interaction with their niche. Thus, inhibitors of niche signals could promote TKI‐sensitive phenotypes in CML stem cells by activating cell cycling or inducing differentiation, leading to eradication of CML cells. Continued investigations of differences between normal HSCs and CML stem cells are also critical for development of CML therapy that does not promote severe side effects due to damage to normal HSCs. Further progress in stem cell biology offers promising directions for development of successful CML therapies.
Figure 3.
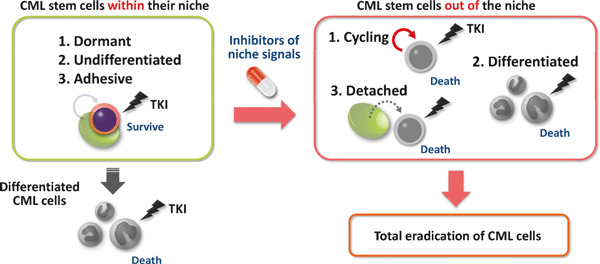
Possible strategy to eradicate chronic CML cells by inhibiting association of CML stem cells with their niche. Although imatinib can eradicate numerous TKI‐sensitive differentiated CML cells, it does not deplete CML stem cells responsible for disease recurrence. Interaction of TKI‐resistant CML stem cells with a niche may keep stem cells dormant and undifferentiated. Thus, inhibitors of the niche signal could activate cell cycling or induce differentiation, promoting TKI‐sensitivity. Together, factors capable of inhibiting CML stem cell/niche interaction are important candidates for CML therapies.
Acknowledgments
We thank Drs T. Suda, N. Komatsu, and S. Nakao for helpful discussions.
References
- 1. Fialkow PJ, Jacobson RJ, Papayannopoulou T. Chronic myelocytic leukemia: clonal origin in a stem cell common to the granulocyte, erythrocyte, platelet and monocyte/macrophage. Am J Med 1977; 63: 125–30. [DOI] [PubMed] [Google Scholar]
- 2. Jamieson CH, Ailles LE, Dylla SJ et al. Granulocyte‐macrophage progenitors as candidate leukemic stem cells in blast‐crisis CML. N Engl J Med 2004; 351: 657–67. [DOI] [PubMed] [Google Scholar]
- 3. Ren R. Mechanisms of BCR‐ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer 2005; 5: 172–83. [DOI] [PubMed] [Google Scholar]
- 4. Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science 1990; 247: 824–30. [DOI] [PubMed] [Google Scholar]
- 5. Kelliher MA, McLaughlin J, Witte ON, Rosenberg N. Induction of a chronic myelogenous leukemia‐like syndrome in mice with v‐abl and BCR/ABL. Proc Natl Acad Sci U S A 1990; 87: 6649–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Elefanty AG, Cory S. Hematologic disease induced in BALB/c mice by a bcr‐abl retrovirus is influenced by the infection conditions. Mol Cell Biol 1992; 12: 1755–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gishizky ML, Johnson‐White J, Witte ON. Efficient transplantation of BCR‐ABL‐induced chronic myelogenous leukemia‐like syndrome in mice. Proc Natl Acad Sci U S A 1993; 90: 3755–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pear WS, Miller JP, Xu L et al. Efficient and rapid induction of a chronic myelogenous leukemia‐like myeloproliferative disease in mice receiving P210 bcr/abl‐transduced bone marrow. Blood 1998; 92: 3780–92. [PubMed] [Google Scholar]
- 9. Huntly BJ, Shigematsu H, Deguchi K et al. MOZ‐TIF2, but not BCR‐ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell 2004; 6: 587–96. [DOI] [PubMed] [Google Scholar]
- 10. Hu Y, Swerdlow S, Duffy TM et al. Targeting multiple kinase pathways in leukemic progenitors and stem cells is essential for improved treatment of Ph+ leukemia in mice. Proc Natl Acad Sci U S A 2006; 103: 16870–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhao C, Blum J, Chen A et al. Loss of β‐catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell 2007; 12: 528–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ito K, Bernardi R, Morotti A et al. PML targeting eradicates quiescent leukaemia‐initiating cells. Nature 2008; 453: 1072–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhao C, Chen A, Jamieson CH et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 2009; 458: 776–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Naka K, Hoshii T, Muraguchi T et al. TGF‐β‐FOXO signalling maintains leukaemia‐initiating cells in chronic myeloid leukaemia. Nature 2010; 463: 676–80. [DOI] [PubMed] [Google Scholar]
- 15. Holyoake T, Jiang X, Eaves C, Eaves A. Isolation of a highly quiescent subpopulation of primitive leukemic cells in chronic myeloid leukemia. Blood 1999; 94: 2056–64. [PubMed] [Google Scholar]
- 16. Holyoake TL, Jiang X, Jorgensen HG et al. Primitive quiescent leukemic cells from patients with chronic myeloid leukemia spontaneously initiate factor‐independent growth in vitro in association with up‐regulation of expression of interleukin‐3. Blood 2001; 97: 720–8. [DOI] [PubMed] [Google Scholar]
- 17. Bernardi R, Pandolfi PP. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol 2007; 8: 1006–16. [DOI] [PubMed] [Google Scholar]
- 18. Reya T, Duncan AW, Ailles L et al. A role for Wnt signalling in self‐renewal of haematopoietic stem cells. Nature 2003; 423: 409–14. [DOI] [PubMed] [Google Scholar]
- 19. Scheller M, Huelsken J, Rosenbauer F et al. Hematopoietic stem cell and multilineage defects generated by constitutive β‐catenin activation. Nat Immun 2006; 7: 1037–47. [DOI] [PubMed] [Google Scholar]
- 20. Rohatgi R, Scott MP. Patching the gaps in Hedgehog signalling. Nat Cell Biol 2007; 9: 1005–9. [DOI] [PubMed] [Google Scholar]
- 21. Dierks C, Beigi R, Guo GR et al. Expansion of Bcr‐Abl‐positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell 2008; 14: 238–49. [DOI] [PubMed] [Google Scholar]
- 22. Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 2005; 24: 7410–25. [DOI] [PubMed] [Google Scholar]
- 23. Tothova Z, Kollipara R, Huntly BJ et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 2007; 128: 325–39. [DOI] [PubMed] [Google Scholar]
- 24. Miyamoto K, Araki KY, Naka K et al. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell 2007; 1: 101–12. [DOI] [PubMed] [Google Scholar]
- 25. Yalcin S, Zhang X, Luciano JP et al. Foxo3 is essential for the regulation of ataxia telangiectasia mutated and oxidative stress‐mediated homeostasis of hematopoietic stem cells. J Biol Chem 2008; 283: 25692–705. [DOI] [PubMed] [Google Scholar]
- 26. Komatsu N, Watanabe T, Uchida M et al. A member of forkhead transcription factor FKHRL1 is a downstream effector of STI571‐induced cell cycle arrest in BCR‐ABL‐expressing cells. J Biol Chem 2003; 278: 6411–19. [DOI] [PubMed] [Google Scholar]
- 27. Ghaffari S, Jagani Z, Kitidis C et al. Cytokines and BCR‐ABL mediate suppression of TRAIL‐induced apoptosis through inhibition of forkhead FOXO3a transcription factor. Proc Natl Acad Sci U S A 2003; 100: 6523–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Essafi A, Fernandez de Mattos S, Hassen YA et al. Direct transcriptional regulation of Bim by FoxO3a mediates STI571‐induced apoptosis in Bcr‐Abl‐expressing cells. Oncogene 2005; 24: 2317–29. [DOI] [PubMed] [Google Scholar]
- 29. Druker BJ, Guilhot F, O’Brien SG et al. Five‐year follow‐up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med 2006; 355: 2408–17. [DOI] [PubMed] [Google Scholar]
- 30. Bhatia R, Holtz M, Niu N et al. Persistence of malignant hematopoietic progenitors in chronic myelogenous leukemia patients in complete cytogenetic remission following imatinib mesylate treatment. Blood 2003; 101: 4701–7. [DOI] [PubMed] [Google Scholar]
- 31. Cortes J, O’Brien S, Kantarjian H. Discontinuation of imatinib therapy after achieving a molecular response. Blood 2004; 104: 2204–5. [DOI] [PubMed] [Google Scholar]
- 32. Goldman J, Gordon M. Why do chronic myelogenous leukemia stem cells survive allogeneic stem cell transplantation or imatinib: does it really matter? Leuk Lymphoma 2006; 47: 1–7. [DOI] [PubMed] [Google Scholar]
- 33. Savona M, Talpaz M. Getting to the stem of chronic myeloid leukaemia. Nat Rev Cancer 2008; 8: 341–50. [DOI] [PubMed] [Google Scholar]
- 34. Graham SM, Jorgensen HG, Allan E et al. Primitive, quiescent, Philadelphia‐positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood 2002; 99: 319–25. [DOI] [PubMed] [Google Scholar]
- 35. Copland M, Hamilton A, Elrick LJ et al. Dasatinib (BMS‐354825) targets an earlier progenitor population than imatinib in primary CML but does not eliminate the quiescent fraction. Blood 2006; 107: 4532–9. [DOI] [PubMed] [Google Scholar]
- 36. Jorgensen HG, Allan EK, Jordanides NE et al. Nilotinib exerts equipotent antiproliferative effects to imatinib and does not induce apoptosis in CD34+ CML cells. Blood 2007; 109: 4016–19. [DOI] [PubMed] [Google Scholar]
- 37. Konig H, Copland M, Chu S et al. Effects of dasatinib on SRC kinase activity and downstream intracellular signaling in primitive chronic myelogenous leukemia hematopoietic cells. Cancer Res 2008; 68: 9624–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Konig H, Holtz M, Modi H et al. Enhanced BCR‐ABL kinase inhibition does not result in increased inhibition of downstream signaling pathways or increased growth suppression in CML progenitors. Leukemia 2008; 22: 748–55. [DOI] [PubMed] [Google Scholar]
- 39. Konig H, Holyoake TL, Bhatia R. Effective and selective inhibition of chronic myeloid leukemia primitive hematopoietic progenitors by the dual Src/Abl kinase inhibitor SKI‐606. Blood 2008; 111: 2329–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Michor F, Hughes TP, Iwasa Y et al. Dynamics of chronic myeloid leukaemia. Nature 2005; 435: 1267–70. [DOI] [PubMed] [Google Scholar]
- 41. Roeder I, Horn M, Glauche I et al. Dynamic modeling of imatinib‐treated chronic myeloid leukemia: functional insights and clinical implications. Nat Med 2006; 12: 1181–4. [DOI] [PubMed] [Google Scholar]
- 42. Kavalerchik E, Goff D, Jamieson CH. Chronic myeloid leukemia stem cells. J Clin Oncol 2008; 26: 2911–15. [DOI] [PubMed] [Google Scholar]
- 43. Yamazaki S, Iwama A, Takayanagi SI et al. TGF‐β as a candidate bone marrow niche signal to induce hematopoietic stem cell hibernation. Blood 2009; 113: 1250–6. [DOI] [PubMed] [Google Scholar]
- 44. Moller GM, Frost V, Melo JV, Chantry A. Upregulation of the TGFβ signalling pathway by Bcr‐Abl: implications for haemopoietic cell growth and chronic myeloid leukaemia. FEBS Lett 2007; 581: 1329–34. [DOI] [PubMed] [Google Scholar]
- 45. Chen Y, Hu Y, Zhang H et al. Loss of the Alox5 gene impairs leukemia stem cells and prevents chronic myeloid leukemia. Nat Genet 2009; 41: 783–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Radmark O, Werz O, Steinhilber D, Samuelsson B. 5‐Lipoxygenase: regulation of expression and enzyme activity. Trends Biochem Sci 2007; 32: 332–41. [DOI] [PubMed] [Google Scholar]
- 47. Wymann MP, Schneiter R. Lipid signalling in disease. Nat Rev Mol Cell Biol 2008; 9: 162–76. [DOI] [PubMed] [Google Scholar]
- 48. Rousselot P, Huguet F, Rea D et al. Imatinib mesylate discontinuation in patients with chronic myelogenous leukemia in complete molecular remission for more than 2 years. Blood 2007; 109: 58–60. [DOI] [PubMed] [Google Scholar]
- 49. Essers MA, Offner S, Blanco‐Bose WE et al. IFNα activates dormant haematopoietic stem cells in vivo. Nature 2009; 458: 904–8. [DOI] [PubMed] [Google Scholar]
- 50. Sato T, Onai N, Yoshihara H et al. Interferon regulatory factor‐2 protects quiescent hematopoietic stem cells from type I interferon‐dependent exhaustion. Nat Med 2009; 15: 696–700. [DOI] [PubMed] [Google Scholar]