

Abstract
Despite high response rates and clinical benefits, androgen ablation often fails to cure advanced or relapsed prostate cancer because castration‐resistant prostate cancer (CRPC) cells inevitably emerge. CRPC cells not only grow under castration, but also behave more aggressively, indicating that a number of malignant signaling pathways are activated in CRPC cells as well as androgen receptor signaling. Based on information from the gene expression profiles of clinical CRPC cells, we here identified one overexpressed gene, serine/threonine/tyrosine kinase 1 (STYK1), encoding a potential kinase, as a molecular target for CRPC. RNA and immunohistochemical analyses validated the overexpression of STYK1 in prostate cancer cells, and its expression was distinct in CRPC cells. Knockdown of STYK1 by siRNA resulted in drastic suppression of prostate cancer cell growth and, concordantly, enforced expression of STYK1 promoted cell proliferation, whereas ectopic expression of a kinase‐dead mutant STYK1 did not. An in vitro kinase assay using recombinant STYK1 demonstrated that STYK1 could have some potential as a kinase, although its specific substrates are unknown. These findings suggest that STYK1 could be a possible molecular target for CRPC, and small molecules specifically inhibiting STYK1 kinase could be a possible approach for the development of novel CRPC therapies. (Cancer Sci 2009)
Prostate cancer (PC) is the most common malignancy in men and the second leading cause of cancer‐related death in the USA and Europe.( 1 ) Detection of PC at an early stage by serum test for prostate‐specific antigen and subsequent surgery and radiation therapy can cure most of the localized disease, but nearly 30% of patients suffer relapse.( 2 , 3 , 4 ) Despite high response rates and clinical benefits, androgen ablation therapy (castration) does not cure advanced or relapsed PC because castration‐resistant prostate cancer (CRPC) cells inevitably emerge, leading PC patients to death. These CRPC cells not only grow under castration conditions (low serum level of androgen), but also behave more aggressively as cancer cells, which indicates that a number of growth‐promoting or malignant signaling pathways that bypass the androgen receptor (AR) pathway are activated in CRPC as well as the AR signaling pathway.( 5 , 6 , 7 , 8 , 9 ) At present there are very limited treatments available for these CRPC. Hence, development of new therapies for CRPC on the basis of the molecular mechanisms of prostate carcinogenesis and castration‐resistant progression is eagerly required.
There is a growing body of evidence supporting the idea that AR signaling pathways are still active in CRPC cells, and the mechanism of CRPC progression could be divided into two pathways: one still involving AR and the other independent of AR. The CRPC phenotype where PC cells can survive under castration is mainly dependent on AR reactivation,( 9 ) but activation of multiple AR‐independent pathways in CRPC cells is likely to contribute to their more malignant or aggressive phenotype. These two mechanisms are not mutually exclusive and often coexist in CRPC cells. One of the possible hypotheses about AR activation under castration is AR phosphorylation by protein kinases. ( 8, 9 ) For example, HER2 is overexpressed in some CRPC cells and HER2 could increase AR activity through the activation of mitogen‐activated protein kinase and Akt. ( 9, 10, 11 ) On the other hand, the PTEN–phosphatidylinositol 3‐kinase (PI3K)–Akt pathway is likely to be one of the most critical pathways that can explain the malignant or aggressive phenotype of CRPC and these phosphorylations and kinases can promote PC cell growth by enhanced AR activation and other signaling pathways.
It is apparent that protein phosphorylation and kinases play important roles in a variety of cancer cell activities including transformation, proliferation, survival, and metastasis. Among the human kinase genome (kinome) including approximately 518 kinases,( 12 ) mutations and deregulation of more than 100 kinases were observed in cancer and some of them can play causal roles in oncogenesis and modify the phenotype of cancer.( 13 ) These enzymes have become one of the most intensively pursued classes of drug target with more than 30 distinct kinase targets being developed to the level of a phase I clinical trial for cancer treatment,( 14 ) and activated or deregulated kinases in CRPC are the most attractive molecular targets for developing novel therapies for PC. To identify novel molecular targets for CRPC, we previously carried out genome‐wide expression profile analysis of CRPC using cDNA microarray in combination with microdissection. ( 15 ) Among dozens of genes that were commonly transactivated in clinical CRPC cells, we here focused on serine/threonine/tyrosine kinase 1 ( STYK1 ), which was overexpressed in CRPC cells. STYK1, a human putative protein kinase, is approximately 30% similar to the mouse fibroblast growth factor (FGF) and platelet‐derived growth factor (PDGF) receptor superfamily,( 16 , 17 ) and is predicted to have a transmembrane domain and protein kinase domain, belonging to a receptor protein tyrosine kinase family. A recent study suggested that it could be involved in oncogenesis and metastasis,( 17 ) and STYK1 mRNA was also upregulated in breast cancer and lung cancer.( 18 , 19 ) However, the details of its functions in cancer and its potential as a kinase are still unclear and should be clarified.
In the present study, we validated STYK1 overexpression in CRPC and some proportions of castration‐naïve prostate cancer (CNPC). We also demonstrated its positive involvement in the proliferation of PC cells, and STYK1 is likely to have some kinase activity that could involve PC cell growth. Our data could provide new insights into the molecular mechanisms of PC progression and some clues to develop new therapeutic strategies targeting STYK1 kinase for PC.
Materials and Methods
Cell lines and clinical samples. COS7 cells and the human PC cell lines LNCaP, 22Rv1, PC‐3, and DU145 were obtained from the American Type Culture Collection (Rockville, MD, USA). C4‐2B, a castration‐resistant derivative from LNCaP, was purchased from ViroMed Laboratories (Minnetonka, MN, USA). All of the cell lines were cultured as monolayers in appropriate medium supplemented with 10% FBS. Cells were maintained at 37°C in an atmosphere of humidified air with 5% CO2. Non‐treated PC tissues, that is, CNPC tissues, were obtained from the patients who underwent prostatectomy at Kochi University Medical School, and CRPC tissues were obtained by autopsy and transurethral prostatectomy at Kochi University Medical School, Iwate Medical Collage, Okayama University Medical School, and Kyoto Prefectural University of Medicine, under the appropriate informed consents. Clinical CRPC was defined by elevation of serum prostate‐specific antigen levels at three consecutive times and/or enlargement of tumor in spite of androgen ablation therapy.
Semi‐quantitative RT‐PCR. Microdissection of CRPC cells, CNPC cells, and normal prostate epithelium, and RNA purification and T7‐based amplification were described previously.( 20 ) The primer sequences for RT‐PCR were 5′‐TTGGCTTGACTCAGGATTTA‐3′ and 5′‐ATGCTATCACCTCCCCTGTG‐3′ for β‐actin (ACTB), and 5′‐GGACATGGATTCTTGATCTTCCT‐3′ and 5′‐ATGTGGTTCCAGAGGAAACTAGC‐3′ for STYK1. The RT‐PCR exponential phase was determined to allow semiquantitative comparisons among cDNA developed from identical reactions. Each PCR regime involved a 95°C, 5‐min initial denaturation step followed by 20 cycles (for ACTB) or 30 cycles (for STYK1) at 95°C for 30 s, 55°C for 30 s, and 72°C for 30 s, on a Gene Amp PCR system 9600 (PE Applied Biosystems, Foster, CA, USA).
Northern blot analysis. Total RNA was extracted from PC cell lines as described above. After treatment with DNaseI (Qiagen, Crawley, UK), mRNA was purified with mRNA Purification Kit (GE Healthcare, Piscataway, NJ, USA), according to the manufacturer’s protocols. A 1‐μg aliquot of each mRNA from the PC cell lines, as well as those isolated from normal human adult heart, lung, liver, kidney, brain, and prostate (BD Biosciences, Palo Alto, CA, USA), were separated on 1% denaturing agarose gels and transferred onto nylon membranes. The 247‐bp probe specific to STYK1 was prepared by PCR using the primers described above. Hybridization with a random‐primed, 32P‐dCTP‐labeled probe was carried out according to the instructions for the Megaprime DNA labeling system (GE Healthcare). Prehybridization, hybridization, and washing were carried out according to the supplier’s recommendations. The blots were autoradiographed with intensifying screens at −80°C for 7 days.
Western blot analysis. To detect exogenous STYK1 protein, cells were lysed with RIPA buffer (50 mM Tris‐HCl [pH 8.0], 150 mM NaCl, 1% Nonidet P‐40, 0.5% deoxycholate Na, 0.1% SDS) containing Protease Inhibitor Cocktail Set III (Calbiochem, La Jolla, CA, USA). Protein samples were separated by SDS‐PAGE and electroblotted onto Hybond‐ECL nitrocellulose membranes (GE Healthcare). Blots were incubated with a mouse monoclonal anti‐Myc antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) or a mouse monoclonal β‐actin (ACTB) antibody (Sigma‐Aldrich, St Louis, MO, USA). Protein bands were visualized by enhanced chemiluminescence western blotting detection reagents (GE Healthcare).
Generation of anti‐STYK1 polyclonal antibody. The cDNA fragment encoding the partial‐length STYK1 protein (codons 50–150, Genbank Accession no. NP_060893) was generated using PrimeSTAR polymerase (Takara, Kyoto, Japan) using the primers 5′‐AGAGAACAAAGAACTCAACAGC‐3′ and 5′‐TCATAAAGCCTTGAGAATAACAC‐3′, and cloned into pGEX‐6P‐1 (GE Healthcare). The recombinant STYK1 protein fused with GST was expressed in Escherichia coli BL21 codon plus (Stratagene, La Jolla, CA, USA), and purified with GST beads under native conditions according to the supplier’s protocol. The purified GST‐fused STYK1 was cleaved by PreScission Protease (GE Healthcare), and the recombinant STYK1 protein was purified. The purified recombinant STYK1 protein was immunized into rabbits, and the immune sera was purified on affinity columns packed with Affi‐Gel 10 activated affinity media (Bio‐Rad, Hercules, CA, USA) conjugated to recombinant STYK1 protein. Western blot analysis using lysates from STYK1‐transfected cells confirmed that this antibody could recognize STYK1 specifically.
Cell fractionation. Cells were suspended in homogenate buffer (10 mM Tris‐HCl [pH 7.4], 250 mM sucrose, 1 mM EDTA) containing Protease Inhibitor Cocktail Set III. After suspension, cell homogenates were ultracentrifuged (Optima TL Ultracentrifuge; Beckman Instruments, Fullerton, CA, USA) at 600g for 10 min (for the nuclear fraction) and 130 000g for 30 min (pellet for the microsomal fraction, supernatant for the cytoplasmic fraction) at 4°C, and collected as the pellet of each step. The pellets were suspended in lysis buffer (50 mM Tris‐HCl [pH7.4], 1 mM EDTA, 20% glycerol, protease inhibitor cocktail) for western blot analysis.
Immunofluorescence analysis. COS7 cells transfected with plasmids expressing Myc‐tagged STYK1 (pcDNA3.1/STYK1‐Myc‐His) were seeded on a glass coverslip. The cells were fixed with ice‐cold methanol : acetone (1:1) for 20 min at −20°C and subsequently washed with PBS. After blocking with PBS containing 3% BSA, the cells were incubated with rabbit polyclonal anti‐STYK1 and mouse monoclonal anti‐Myc (Santa Cruz Biotechnology) diluted in PBS containing 3% BSA for 1 h at room temperature. After being washed with PBS, the cells were stained with FITC‐conjugated antimouse secondary antibody (Santa Cruz Biotechnology) and antirabbit secondary antibody conjugated to Alexa Fluor 594 (Molecular Probes, Eugene, OR, USA) for 1 h at room temperature. After another wash with PBS, each specimen was mounted with Vectashield (Vector Laboratories, Burlingame, CA, USA) containing 4′,6′‐diamidine‐2′‐phenylindolendihydrochrolide (DAPI) and visualized with Spectral Confocal Scanning Systems (Leica, Bensheim, Germany).
Immunohistochemical staining. Paraffin‐embedded tissue sections were deparaffinized, subjected to microwave treatment at 360 W for 1 min four times in antigen‐retrieval solution, high pH (Dako, Carpinteria, CA, USA), and then treated with peroxidase blocking reagent (Dako) followed by protein block reagent (Dako). Immunohistochemical study was carried out using the Ventana automated IHC systems (Discovery, Ventana Medical Systems, Tucson, AZ, USA). Sections were incubated with a 1 : 10 diluted solution of the rabbit polyclonal anti‐STYK1 antibody overnight at 4°C. The protocol is based on an indirect biotin–avidin system using a biotinylated universal secondary antibody and diaminobenzidine substrate with hematoxylin counterstaining.
shRNA‐expressing vectors and transfection. To knockdown endogenous STYK1 expression in PC cells, we used psiU6BX3.0 vector for expression of shRNA against a target gene as described previously.( 21 ) The target sequences of the shRNA to STYK1 (Genebank Accession no. NM_018423) were as follows: si1, 5′‐GGTGGTACCTGAACTGTAT‐3′; si2, 5′‐CAGAGAATGGTCTTTCCCA‐3′; si3, 5′‐GGTGGAGGAGTCATTTCAT‐3′; and scramble‐si (SCR), 5′‐GCGCGCTTTGTAGGATTCG‐3′ as a negative control. Two PC cell lines, 22Rv1 and LNCaP, both of which expressed STYK1, were seeded onto six‐well plates, and transfected with 8 μg of each of the shRNA‐expressing vectors to STYK1 by using FuGENE6 (Roche, Indianapolis, IN, USA) according to the manufacturer’s instructions. The cells were selected using 0.4 mg/mL (for 22Rv1) or 0.8 mg/mL (for LNCaP) geneticin (Sigma‐Aldrich) for 9 days, and then harvested to analyze the knockdown effect on STYK1 expression. For colony formation assay, transfectants expressing shRNA were grown for 24 days in media containing geneticin. After fixation with methanol, the transfected cells were stained with crystal violet solution to assess colony formation. Cell viability was quantified using Cell Counting Kit‐8 (Dojindo, Kumamoto, Japan). After 24 days incubation in the geneticin‐containing medium, the solution was added at a final concentration of 10%. Following incubation at 37°C for 2 h, absorbance at 450 nm was measured with Microplate Reader 550 (Bio‐Rad).
In vitro kinase assay. Partial length of STYK1 cDNA (codons 146–1268) was prepared by PCR amplification, and the PCR product was inserted into pGEX‐6P‐1 vector for expressing GST‐tagged protein (GE Healthcare). Recombinant STYK1 was purified with glutathione sepharose‐4B and PreScission protease (GE Healthcare), according to the manufacturer’s protocols. Five μg of recombinant STYK1 protein was incubated in 30 μL kinase assay buffer (25 mM Tris‐HCl [pH 8.0], 5 mM MgCl2, 0.2 mM EDTA, 4 mM DTT, and 100 μM ATP) and then supplemented with 1.85 MBqγ32P‐ATP (GE Healthcare). As the candidate substrates of the in vitro kinase assay, 2 μg histone H1 (Roche), histone H3 (Roche), myelin basic protein (MBP: upstate, Lake Placid, NY, USA), or 22Rv1 cell lysate were prepared in the reaction solutions. After 30 min incubation at 30°C, the reactions were terminated by addition of SDS sample buffer. After boiling, the protein samples were electrophoresed on 5–20% gradient gel (Bio‐Rad), and then autoradiographed.
Expression vectors and cell growth assay. The full‐length cDNA of STYK1 was cloned into pIRES/myc‐his (Takara) for wild‐type STYK1 (pIRES/STYK1‐myc‐his). We also carried out site‐directed mutagenesis PCR to generate a kinase‐dead (KD) mutant in which Lys147 was substituted to arginine (K147R). The primer set used for K147R was 5′‐GAGTGTTATTCTCAGGGCTTTAAAAGAAC‐3′ and 5′‐GTTCTTTTAAAGCCCTGAGAATAACACTC‐3′ (underlining indicates nucleotides that were replaced from the wild‐type). For in vitro growth assay, 1 × 105 cells of 22Rv1 were seeded into each of the wells of a six‐well culture plate and incubated in DMEM containing 5% FBS. The cells were transfected with pIRES for mock, pIRES/STYK1 wild‐type, or pIRES/STYK1 KD mutant (K147R). After 48 h incubation, the cell numbers were measured using Cell‐counting Kit‐8.
Results
STYK1 was overexpressed in CRPC cells. Among the dozens of up‐regulated genes in CRPC cells identified through our expression profile analysis of clinical CRPC cells,( 15 ) we here focused on STYK1 for further expression and functional analysis. Semiquantitative RT‐PCR analysis using RNA from microdissected CRPC cells, CNPC cells, and normal prostate epithelial cells (NP mix) demonstrated that STYK1 expression was significantly upregulated in CRPC cells compared with CNPC cells and normal prostate epithelial cells (Fig. 1a left), and RT‐PCR showed that all of the PC cell lines we examined also showed STYK1 expression more or less (Fig. 1a right). Further comparisons of STYK1 expression patterns in PC cells and vital organs by northern blot analysis and RT‐PCR analysis clearly demonstrated STYK1 expression in all of the PC cells as well as normal prostate (Fig. 1b). Northern blot analysis on human normal adult organs showed STYK1 expression in the prostate, but hardly detected expression in any other adult organs (Fig.1c).
Figure 1.
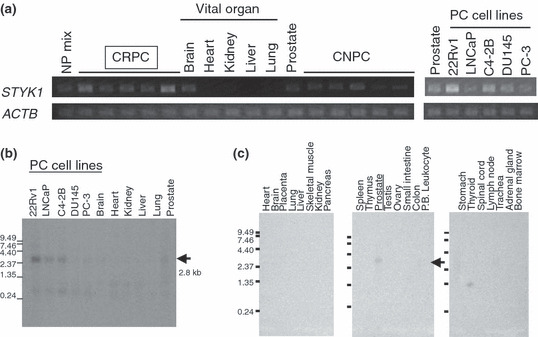
Overexpression of STYK1 in castration‐resistant prostate cancer (CRPC) cells. (a) Semiquantitative RT‐PCR validated that STYK1 expression was up‐regulated in the microdissected CRPC cells compared with castration‐naïve prostate cancer (CNPC) cells, normal prostate epithelial cells (NP mix) that were also microdissected, and several vital organs (brain, heart, kidney, liver, lung, and prostate). All five prostate cancer (PC) cell lines we examined also expressed STYK1 more or less. The expression of β‐actin (ACTB) served as a quantitative control. (b) Northern blot analysis showed the strong expression of STYK1, indicated by a 2.8‐kb band, in PC cell lines, whereas faint expression was observed in adult normal prostate, and there was no expression in vital organs including heart, lung, liver, and kidney. (c) Northern blot analysis of multiple tissues showed faint expression of STYK1 only in normal prostate among various normal human adult organs. P.B. Leukocyte, peripheral blood leukocyte.
Subcellular localization of STYK1 protein. STYK1 protein had one putative transmembrane domain (PTLLVTIFLILLGVI) and there was some possibility that STYK1 could be localized to the plasma membrane as a receptor protein tyrosine kinase. To investigate the subcellular localization of STYK1 protein, we carried out immunofluorescence analysis using the STYK1‐transfected cells. In COS7 cells expressing Myc‐tagged STYK1 (pcDNA3.1/STYK1‐Myc‐His), exogenous STYK1 protein was observed in the cytoplasm, not in the plasma membrane (Fig. 2a). We also carried out western blot analysis using the fractionated lysates of the STYK1‐transfected cells. As a result, STYK1 protein was detected mainly in the microsomal fraction (Fig. 2b). STYK1 is likely to be localized to an organelle, such as the endoplasmic reticulum.
Figure 2.
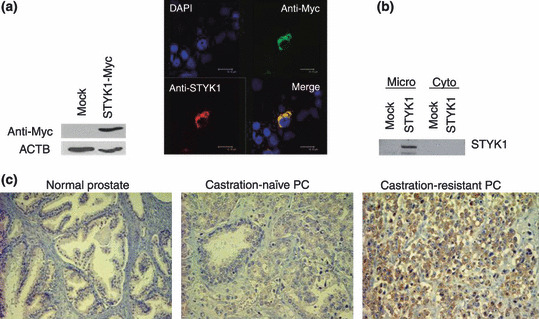
Subcellular localization of STYK1 and expression in prostate cancer (PC) tissues. (a) Immunocytochemical analysis showed that exogeneous STYK1 protein was localized to the cytoplasm. Green, anti‐Myc antibody; red, anti‐STYK1 antibody; blue, DAPI staining. Exogenous STYK1 expression was validated by western blot analysis using anti‐Myc antibody (left). (b) Western blot analysis with anti‐Myc antibody on the cellular fractions of STYK1‐transfected cells revealed that exogenous STYK1 was localized mainly to the microsome. Micro, microsome fraction; Cyto, cytoplasmic fraction. (c) Immunohistochemical study on clinical PC tissues using anti‐STYK1 antibody. Intense staining was observed in castration‐resistant prostate cancer (CRPC) tissue (right), whereas castration‐naïve prostate cancer (CNPC) tissue (middle) and normal prostate tissue (left) showed weak or no staining. In total, all six CRPC showed strong staining, whereas 34 out of 52 CNPC (65%) showed strong positive staining. The remaining CNPC and normal prostate epithelial cells showed very weak or no expression of STYK1.
Immunohistochemical analysis of clinical PC tissues. To further investigate STYK1 protein expression in PC tissue sections, we carried out immunohistochemical staining using 58 clinical PC tissues with rabbit polyclonal anti‐STYK1 antibody. To check the specificity of our anti‐STYK1 antibody, we made paraffin‐embedded blocks of four PC cell lines and confirmed that the staining signals in immunohistochemistry were concordant with RNA expression of STYK1 in these PC cell lines (Fig. S1). Immunohistochemical analysis demonstrated strong positive staining for STYK1 in six of the CRPC we examined, and 34 out of 52 CNPC (65%) showed strong positive staining for STYK1 (Fig. 2c). The staining signals were observed in the cytoplasm of PC cells. The remaining CNPC and normal prostate epithelial cells showed very weak or no expression of STYK1. These staining patterns in CNPC were not correlated with Gleason score.
Knockdown of STYK1 by shRNA attenuated PC cell growth. To investigate the biological significance of STYK1 overexpression in PC cells and examine its potential as a molecular target for PC treatment, we constructed several shRNA‐expression vectors specific to STYK1 (si1, si2, si3, and SCR) and transfected each of them into two PC cell lines (22Rv1 and LNCaP) that endogenously expressed STYK1. Semiquantitative RT‐PCR showed that si1 and si3 significantly knocked down endogenous STYK1 expression, whereas si2 and the control SCR did not (Fig. 3a,d). After 24 days of selection in culture medium containing geneticin, we carried out MTT assays (Fig. 3b,e) and colony formation assays (Fig. 3c,f), and found that introduction of si1 and si3 shRNA in 22Rv1 and LNCaP cells drastically attenuated their growth or viability, whereas that of other shRNA did not affect STYK1 expression and PC cell growth. These findings indicated that STYK1 expression could play some essential roles in PC viability and STYK1 could have some potential as a molecular target for PC.
Figure 3.
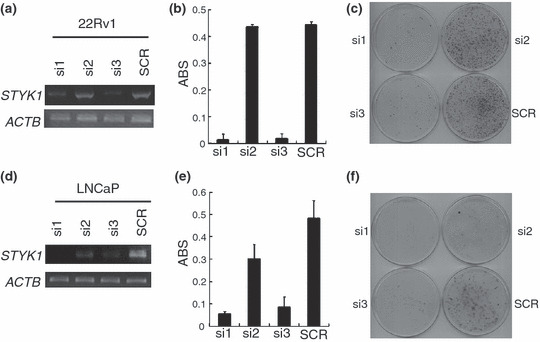
Knockdown effect of shRNA on STYK1 attenuated prostate cancer (PC) cell viability. Three STYK1 shRNA‐expression vectors (si1, si2, and si3) and a scramble shRNA‐expression vector (SCR) as a negative control were transfected into (a–c) 22Rv1 and (d–f) LNCaP cells that showed STYK1 overexpression. The knockdown effect on STYK1 expression was validated by (a,d) RT‐PCR with β‐actin (ACTB) expression as a quantitative control. Transfection with si1 and si3 revealed strong knockdown effects on STYK1 expression, whereas transfection with si2 and SCR did not show any effect on STYK1 expression. Transfection with si1 and si3 resulted in drastic reductions of (b,e) the numbers of viable cells measured by MTT assay (P < 0.002 by Student’s t‐test) and (c,f) the number of colony formation compared with the cells transfected with si2 and SCR, which did not show a knockdown effect on STYK1. ABS on the y‐axis at MTT assay (b,e) means absorbance at 490 nm, and at 630 nm as reference, measured with a microplate reader.
Protein kinase activity and growth‐promoting effect of STYK1. In silico analysis of the STYK1 amino acid sequence indicated the presence of a protein kinase domain in STYK1. Generally, a kinase has three important motifs, VAIK, HRD, and DFG motifs, within the catalytic domain, and they are thought to be critical for the catalytic function of a kinase.( 12 , 22 ) STYK1 has well conserved VAIK and HRD motifs; however, it lacks an activation loop (DFG motif), which is important in regulating kinase activity (Fig. 4a). Protein kinases without conserved DFG motifs were almost inactive in phosphorylating substrates. Hence, the kinase catalytic activity of STYK1 is questionable, and to investigate its actual kinase activity, we generated the recombinant protein of partial STYK1 (Fig. 4b) and carried out an in vitro kinase assay using this recombinant protein. As substrates of STYK1 have not been identified so far, we used some common phosphorylated proteins (histones and MBP) and the crude cell lysate of PC cell lines as substrates for the in vitro kinase assay. As shown in Figure 4(c), the in vitro kinase assay detected a faint 15‐kDa phosphorylated band in the crude cell lysate from 22Rv1, but not in histones and MBP. This 15‐kDa protein is likely to be a good candidate substrate of STYK1 kinase in the PC cells, and this in vitro finding can support some potential kinase activity of STYK1.
Figure 4.
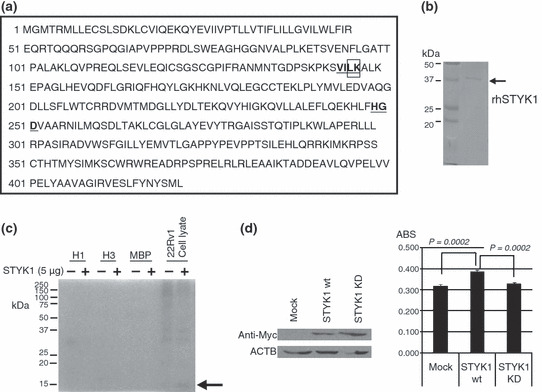
In vitro kinase assay of STYK1 and the growth‐promoting effect of STYK1 overexpression. (a) The amino acid sequence of human STYK1 has VAIK and HRD motifs (underlined), but no DFG motif is found. Lys147 in the VAIK motif was substituted for Arg in the STYK1 kinase‐dead mutant (K147R; STYK1 KD), indicated by the box. (b) Recombinant protein of partial STYK1 (∼37 kDa, without the transmembrane region) was purified for the in vitro kinase assay. Purified STYK1 was subjected to SDS‐PAGE and Coomassie Brilliant Blue (CBB) staining. (c) Histone H1, histone H3, myelin basic protein (MBP), and 22Rv1 whole cell lysate were incubated with γ32P‐ATP with or without recombinant STYK1 protein. A phosphorylated band of ∼15 kDa was detected in the autoradiography only in the incubation of 22Rv1 whole cell lysate with STYK1 recombinant protein (arrow). (d) Wild‐type STYK1 (STYK1 wt) or STYK1 KD was overexpressed in 22Rv1 cells. Western blot analysis using Myc‐tag antibody confirmed their expression in 22Rv1 cells. MTT assay 48 h after the transfection demonstrated that the transfection of STYK1 wt significantly promoted PC cell proliferation compared with mock transfection (Student’s t‐test, P = 0.0002). On the other hand, the transfection of STYK1 KD did not promote PC cell growth and STYK1 KD lost its growth‐promoting effect on PC cells (Student’s t‐test, P = 0.0002).
To further investigate whether the possible kinase activity of STYK1 could involve PC cell proliferation, we carried out the cell growth assay by transfecting STYK1‐expression vectors into PC cells. We also generated a KD mutant of STYK1 (STYK1 KD), in which its conserved lysine residue at codon 147 within the predicted ATP‐binding site was replaced with arginine (K147R; Fig. 4a boxed). Each of the STYK1 expression vectors was transfected into 22Rv1 cells and western blotting confirmed their expression in 22Rv1 cells (Fig. 4d). MTT assay 48 h after the transfection demonstrated that the overexpression of wild‐type STYK1 significantly promoted PC cell proliferation, compared with mock transfection. On the other hand, overexpression of STYK1 KD did not promote PC cell growth and this KD STYK1 lost its growth‐promoting effect on PC cells (Fig.4d). These findings supported that STYK1 overexpression could promote PC cell growth as a kinase.
Discussion
The emergence of CRPC is one of the most critical problems in PC clinics, and effective therapeutics are not available for advanced stages of CRPC after the failure of androgen ablation therapy. Very recently, the combination therapy of docetaxel and prednisone was established as the new standard protocol for CRPC patients,( 9 , 23 , 24 ) but its therapeutic effect on CRPC is still limited, and hence, novel molecular targeting therapies for CRPC are urgently required. In the present study we identified overexpression of STYK1 encoding a possible kinase in CRPC and, as shown in this study, due to its restrictive expression in adult normal organs and its critical roles in PC cells, STYK1 kinase could be a promising target for a novel therapeutic approach with a minimal risk of adverse effects.
The in silico analysis on the primary structure of human STYK1 indicated that STYK1 has a putative kinase domain but its kinase activity was predicted to be very weak or absent due to the lack of a DFG motif in subdomain VII in the kinase domain, in which the aspartic acid binds the Mg2+ ions that coordinate the γ‐phosphate in the ATP‐binding cleft.( 12 , 14 , 22 ) In fact, some atypical receptor protein kinases, such as CCK‐4, H‐Ryk, VRK3, and DNT, with substitutions in the DFG motif were reported to be defective in their kinase activity, and they may interact with kinase‐active partners and activate other downstream signaling proteins.( 22 , 25 , 26 ) Nevertheless, our in vitro kinase assay using STYK1 recombinant protein and the growth assay by STYK1 overexpression suggested some possibility that STYK1 could function as a kinase to promote PC cell growth, although its specific substrates are still not identified. According to the human kinome mapping (http://www.cellsignal.com/reference/kinase/kinome.html), STYK1 does not belong to any major group of kinases( 12 ) and it is likely to be a unique kinase that possibly has novel functions or might be involved in a novel signaling pathway in cancer cells. The identification of its specific substrate in cancer cells is the next important step to clarify the functional and biological significance of STYK1 overexpression in CRPC and other cancers. Phosphorylation of protein is an important signaling mechanism in eukaryotic cells, and oncogenic activation of tyrosine kinases is a common feature in many types of cancer and novel anticancer drugs, such as imatinib and gefitinib, have been introduced to target these kinases( 12 , 27 ) and their great effects on cancer have been established. The identification of the substrates of STYK1 kinase and a small molecule specifically inhibiting its kinase activity should provide us with a novel therapeutic approach against CRPC.
Supporting information
Fig. S1. Immunohistochemical analysis by anti‐STYK1 polyclonal antibody (�10) on paraffin‐embedded blocks of four prostate cancer cell lines. 22Rv1 and C4‐2B cells showed strong signals, whereas LNCaP and PC‐3 cells showed very weak signals, which was concordant with their RNA expression of STYK1, which was analyzed by RT‐PCR.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
{kind=link}
Acknowledgments
We would like to thank Ms Mami U for her technical assistance. This work was supported by Grant‐in‐Aid for Scientific Research #18590323 (H. Nakagawa) and Research for the Future Program Grant #00L01402 (Y. Nakamura) from the Japan Society for the Promotion of Science.
References
- 1. Gronberg H. Prostate cancer epidemiology. Lancet 2003; 361: 859–64. [DOI] [PubMed] [Google Scholar]
- 2. Han M, Partin AW, Piantadosi S, Epstein JI, Walsh PC. Era specific biochemical recurrence‐free survival following radical prostatectomy for clinically localized prostate cancer. J Urol 2001; 166: 416–19. [PubMed] [Google Scholar]
- 3. Roberts SG, Blute ML, Bergstralh EJ, Slezak JM, Zincke H. PSA doubling time as a predictor of clinical progression after biochemical failure following radical prostatectomy for prostate cancer. Mayo Clinic Proc 2001; 76: 576–81. [DOI] [PubMed] [Google Scholar]
- 4. Roberts WW, Bergstralh EJ, Blute ML et al. Contemporary identification of patients at high risk of early prostate cancer recurrence after radical retropubic prostatectomy. Urology 2001; 57: 1033–7. [DOI] [PubMed] [Google Scholar]
- 5. Grossmann ME, Huang HJ, Tindall DJ. Androgen receptor signaling in androgen‐refractory prostate cancer. J Natl Cancer Inst 2001; 93: 1687–97. [DOI] [PubMed] [Google Scholar]
- 6. Craft N, Shostak Y, Carey M, Sawyers CL. A mechanism for hormone‐independent prostate cancer through modulation of androgen receptor signaling by the HER‐2/neu tyrosine kinase. Nat Med 1999; 5: 280–5. [DOI] [PubMed] [Google Scholar]
- 7. Bernard D, Pourtier‐Manzanedo A, Gil J, Beach DH. Myc confers androgen‐independent prostate cancer cell growth. J Clin Invest 2003; 112: 1724–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Feldman BJ, Feldman D. The development of androgen‐independent prostate cancer. Nat Rev Cancer 2001; 1: 34–45. [DOI] [PubMed] [Google Scholar]
- 9. Scher HI, Sawyers CL. Biology of progressive, castration‐resistant prostate cancer: directed therapies targeting the androgen‐receptor signaling axis. J Clin Oncol 2005; 23: 8253–61. [DOI] [PubMed] [Google Scholar]
- 10. Signoretti S, Montironi R, Manola J et al. Her‐2‐neu expression and progression toward androgen independence in human prostate cancer. J Natl Cancer Inst 2000; 92: 1918–25. [DOI] [PubMed] [Google Scholar]
- 11. Osman I, Scher HI, Drobnjak M et al. HER‐2/neu (p185neu) protein expression in the natural or treated history of prostate cancer. Clin Cancer Res 2001; 7: 2643–7. [PubMed] [Google Scholar]
- 12. Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science 2002; 298: 1912–34. [DOI] [PubMed] [Google Scholar]
- 13. Davies H, Hunter C, Smith R et al. Somatic mutations of the protein kinase gene family in human lung cancer. Cancer Res 2005; 65: 7591–5. [DOI] [PubMed] [Google Scholar]
- 14. Zhang JM, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer 2009; 9: 28–39. [DOI] [PubMed] [Google Scholar]
- 15. Tamura K, Furihata M, Tsunoda T et al. Molecular features of hormone‐refractory prostate cancer cells by genome‐wide gene expression profiles. Cancer Res 2007; 67: 5117–25. [DOI] [PubMed] [Google Scholar]
- 16. Ye X, Ji CN, Huang QS et al. Isolation and characterization of a human putative receptor protein kinase cDNA STYK1. Mol Biol Rep 2003; 30: 91–6. [DOI] [PubMed] [Google Scholar]
- 17. Liu L, Yu XZ, Li TS et al. A novel protein tyrosine kinase NOK that shares homology with platelet‐derived growth factor/fibroblast growth factor receptors induces tumorigenesis and metastasis in nude mice. Cancer Res 2004; 64: 3491–9. [DOI] [PubMed] [Google Scholar]
- 18. Kimbro KS, Duschene K, Willard M, Moore JA, Freeman S. A novel gene STYK1/NOK is upregulated in estrogen receptor‐alpha negative estrogen receptor‐beta positive breast cancer cells following estrogen treatment. Mol Biol Rep 2008; 35: 23–7. [DOI] [PubMed] [Google Scholar]
- 19. Amachika T, Kobayashi D, Moriai R, Tsuji N, Watanabe N. Diagnostic relevance of overexpressed mRNA of novel oncogene with kinase‐domain (NOK) in lung cancers. Lung Cancer 2007; 56: 337–40. [DOI] [PubMed] [Google Scholar]
- 20. Ashida S, Nakagawa H, Katagiri T et al. Molecular features of the transition from prostatic intraepithelial neoplasia (PIN) to prostate cancer: genome‐wide gene‐expression profiles of prostate cancers and PINs. Cancer Res 2004; 64: 5963–72. [DOI] [PubMed] [Google Scholar]
- 21. Anazawa Y, Nakagawa H, Furihara M et al. PCOTH, a novel gene overexpressed in prostate cancers, promotes prostate cancer cell growth through phosphorylation of oncoprotein TAF‐Ibeta/SET. Cancer Res 2005; 65: 4578–86. [DOI] [PubMed] [Google Scholar]
- 22. Boudeau J, Miranda‐Saavedra D, Barton GJ, Alessi DR. Emerging roles of pseudokinases. Trends Cell Biol 2006; 16: 443–52. [DOI] [PubMed] [Google Scholar]
- 23. Tannock IF, De Wit R, Berry WR et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. New Engl J Med 2004; 351: 1502–12. [DOI] [PubMed] [Google Scholar]
- 24. Petrylak DP, Tangen CM, Hussain MHA et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. New Engl J Med 2004; 351: 1513–20. [DOI] [PubMed] [Google Scholar]
- 25. Katso RM, Russell RB, Ganesan TS. Functional analysis of H‐Ryk, an atypical member of the receptor tyrosine kinase family. Mol Cell Biol 1999; 19: 6427–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Scheeff ED, Eswaran J, Bunkoczi G, Knapp S, Manning G. Structure of the pseudokinase VRK3 reveals a degraded catalytic site, a highly conserved kinase fold, and a putative regulatory binding site. Structure 2009; 17: 128–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dancey J, Sausville EA. Issues and progress with protein kinase inhibitors for cancer treatment. Nat Rev Drug Discov 2003; 2: 296–313. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Immunohistochemical analysis by anti‐STYK1 polyclonal antibody (�10) on paraffin‐embedded blocks of four prostate cancer cell lines. 22Rv1 and C4‐2B cells showed strong signals, whereas LNCaP and PC‐3 cells showed very weak signals, which was concordant with their RNA expression of STYK1, which was analyzed by RT‐PCR.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item