

Abstract
(Cancer Sci 2010; 101: 735–742)
Adenovirus‐mediated gene therapy shows promise for cancer therapy, but transgene expression of replication‐defective adenovirus may be low and transient in clinical settings. Recent reports have shown that the use of a conditionally replication‐competent adenovirus (CRAd) enhanced the gene transduction of a replication‐defective adenovirus vector. The control of tumor–stromal interactions has also been determined to be important in cancer therapy. In this study, we investigated the effect of the human telomerase reverse transcriptase (hTERT)‐CRAd, Ad5/3hTERTE1, which possesses the tumor‐specific hTERT promoter with the chimeric fiber 5/3, on the transgene expression and therapeutic efficacy of a replication‐defective adenovirus vector expressing NK4 under the control of the CMV promoter, Ad‐NK4. In addition, we established a new strategy to target both cancer cells and cancer–stromal interactions. Human pancreatic cancer cells were infected with Ad‐NK4 and either Ad5/3hTERTE1 (CRAd‐combination group) or Ad5/3hTERTLuc (control‐combination group). In the CRAd‐combination group, Ad‐NK4‐delivered transgene expression was increased, leading to an enhanced inhibitory effect on the invasion of cancer cells. In in vivo experiments, NK4 expression within tumors and its inhibitory effect on tumor growth, angiogenesis, and metastasis were enhanced in the CRAd‐combination group. These results suggest that hTERT‐CRAd enhances the transgene expression and therapeutic efficacies of Ad‐NK4, possibly through the in‐trans replication of Ad‐NK4 induced by adenovirus E1 derived from co‐infected hTERT‐CRAd. This approach may be a promising combination therapy against advanced pancreatic cancer.
Despite progress in surgical, chemotherapeutic, and radiation therapies, sufficient results for the treatment of advanced cancer have not been achieved. In particular, pancreatic cancer is a very aggressive malignant tumor, and has an extremely poor prognosis.( 1 , 2 , 3 ) Recently, many researchers have reported that tumor–stromal interactions are involved in the poor prognosis of malignant cancers through mechanisms of angiogenesis, metastasis, and recurrence.( 4 , 5 ) Therefore, cancer should be treated as a cancer‐associated mass that includes cancer cells and stromal cells, and a new strategy should be established to target not only cancer cells, but also tumor–stromal interactions.
Hepatocyte growth factor (HGF) is a potent mitogen for hepatocytes. The interaction of HGF and c‐Met receptor is involved in the malignant behavior of pancreatic cancer. Fibroblast‐derived HGF confers mitogenic potential by binding to the c‐Met receptor, which is frequently overexpressed in pancreatic cancer cells, and the phosphorylation of c‐Met receptor accelerates the migratory and invasive ability of pancreatic cancer cells.( 6 , 7 , 8 ) Therefore, for pancreatic cancer, an important strategy is to prevent the HGF–c‐Met association.
Adenovirus cancer therapy is a new strategy for advanced cancers that includes two approaches.( 9 , 10 ) The first approach is adenovirus gene therapy, which delivers target genes using a replication‐defective adenovirus vector. The second approach is the use of conditionally replicative oncolytic adenovirus therapy (CRAd) designed to infect target cells with replication‐competent adenovirus, which leads to the lysis of the target cells. Previously, we reported that gene therapy using a replication‐defective adenoviral vector expressing NK4 under the control of the CMV promoter (Ad‐NK4), which acts as an HGF antagonist and an angiogenesis inhibitor, inhibited the in vitro invasion, in vivo growth, and metastasis of human pancreatic cancer cells.( 11 , 12 , 13 ) Moreover, we reported that the human telomerase reverse transcriptase (hTERT) promoter‐dependent conditionally replicative adenovirus (hTERT‐CRAd), Ad5/3hTERTE1, which possesses the tumor‐specific hTERT promoter with the chimeric fiber 5/3, showed antitumor effects in non‐small‐cell lung cancer cell lines.( 14 )
These two adenovirus approaches have shown specific efficacies. However, their efficacies may be limited when used as a single agent,( 15 , 16 ) since the transgene expression and oncolytic effect delivered by these adenoviruses are low and transient in vivo.( 17 , 18 ) Several studies reported that transgene expression delivered by adenovirus vectors were enhanced when combined with CRAds( 19 , 20 , 21 , 22 ) including hTERT‐CRAd.( 23 , 24 ) Therefore, we hypothesized that NK4 expression delivered by Ad‐NK4 would be enhanced when combined with the hTERT‐CRAd, Ad5/3hTERTE1, and that this combination may be an effective cancer therapy that targets both cancer cells and tumor–stromal interactions in pancreatic cancer.
In this study, we investigated the effects of hTERT‐CRAd on the transgene expression of AdNK4 in cancer cells in vitro, and the effects on both transgene expression and therapeutic efficacies using in vivo models. We also used hTERT‐CRAd with the chimeric fiber 5/3, Ad5/3hTERTE1, because the expression levels of Coxsackie and adenovirus receptor (CAR), which is a receptor of adenovirus fiber 5,( 25 ) are often low in cancer and the chimeric fiber 5/3 can overcome this limitation.( 26 ) In addition, we previously reported that a dual‐knob mosaic adenovirus possessing serotype Ad5 and Ad3 knobs generated by co‐infection of E1‐containing cells of Ad5 and Ad5/3 chimeric virus exhibits expanded tropoism.( 27 ) Therefore, it may be useful to combine with different types of fibers as used in the present study.
Our results suggest that combination therapy with hTERT‐CRAd and AdNK4 may be an effective strategy against advanced cancers.
Materials and Methods
Cultured cells and reagents. We used the human pancreatic cancer cell line SUIT‐2 (generous gift from Dr H. Iguchi, National Shikoku Cancer Center, Matsuyama, Japan). Cells were cultured in DMEM supplemented with streptomycin (100 μg/mL), penicillin (100 U/mL), and 10% FBS at 37°C in a humidified atmosphere of 90% air.
Construction of recombinant adenoviruses. A recombinant adenoviral vector expressing human NK4, denoted Ad‐NK4, was constructed as described previously,( 28 , 29 , 30 ) and a control vector expressing the bacterial β‐galactosidase (β‐gal) gene (lacZ), denoted Ad‐LacZ, was constructed by the same procedure. We constructed an E1‐containing hTERT‐promoter‐dependent CRAd expressing the Ad5/3 chimeric fiber, which is serotype 5 fiber with a serotype 3 knob, denoted Ad5/3hTERTE1, and an E1‐deleted hTERT‐promoter‐dependent adenovirus containing both the luciferase gene driven by the hTERT promoter and the Ad5/3 chimeric fiber, denoted Ad5/3hTERTLuc, as described previously.( 14 ) The recombinant adenoviruses were propagated in HEK293 cells. The adenovirus titers in plaque‐forming units (pfu) were determined by plaque formation assays using HEK293 cells. The multiplicity of infection (MOI) was defined as the ratio of the total number of pfus used in a particular infection to the total number of cells infected.
Treatment with adenoviruses. Cells were seeded on plates and cultured in DMEM supplemented with 10% FBS for 24 h. The cells were then infected simultaneously with Ad‐NK4 and Ad5/3hTERTE1 or Ad5/3hTERTLuc at various MOIs for 1 h, followed by replacement of the culture media with fresh DMEM supplemented with 10% FBS.
Quantitative analysis of β‐gal mRNA levels by one‐step real‐time RT‐PCR. Total RNA was extracted from cultured cells using a High Pure RNA Isolation Kit (Roche Diagnostics, Mannheim, Germany) with DNaseI (Roche Diagnostics) treatment, according to the manufacturer’s instructions. We designed specific primers as follows: β‐gal, forward primer, 5′‐cacggcacatac‐acttgctg‐3′ and reverse primer, 5′‐atcgccatttgaccactacc‐3′; 18S rRNA, forward primer, 5′‐gtaacccgttgaaccccatt‐3′ and reverse primer, 5′‐ccatccaatcggtagtagccg‐3′. We performed BLAST searches to ensure the specificities of these primers. One‐step quantitative RT‐PCR (qRT‐PCR) was performed using a QuantiTect SYBR Green Reverse Transcription‐PCR kit (Qiagen, Tokyo, Japan) with Chrom4 Real‐Time PCR Detection System (Bio‐Rad Laboratories, Hercules, CA, USA) as described previously.( 31 ) Each sample was run in triplicate and the expression of each gene was presented as the ratio between the expression of each target gene mRNA and that of 18S rRNA.
Assessment of transgene distributions by evaluation of β‐gal expression. After treatment of SUIT‐2 cells with Ad‐lacZ and Ad5/3hTERTE1 or Ad5/3hTERTLuc, the treated cells were rinsed twice with PBS and fixed with 0.25% glutaraldehyde in PBS for 15 min at 4°C. β‐Gal activity was detected by immersing the cells in 5‐bromo‐4‐chloro‐3‐indolyl‐β‐galactopyranoside (X‐gal) staining solution (5 mm K4FeCN, 5 mm K3FeCN, 2 mm MgCl2, 1 mg/mL X‐gal) for 12 h at 37°C.
Extraction of proteins from Ad‐NK4‐infected cells. SUIT‐2 cells were infected with Ad‐NK4 and Ad5/3hTERTE1 or Ad5/3hTERTLuc, as described above. After these treatments, the cells were lysed in 500 μL of ice‐cold lysis buffer (20 mm Tris‐HCl [pH 7.5], 150 mm NaCl, 10 mm EDTA, 5 μg/mL leupeptin, 1 mm phenylmethyl sulfonyl fluoride, and 0.5% (v/v) Triton X‐100). Cell debris was removed by centrifugation at 14 000 g for 20 min at 4°C and the supernatants were collected. The protein concentrations of the supernatants were measured by the absorbance at 280 nm using an ND‐1000 Spectrophotometer (NanoDrop Technologies, Rockland, DE, USA).
Measurement of NK4 expression levels. The conditioned media and proteins extracted from cells infected with Ad‐NK4 were measured using a human HGF ELISA Kit (IMMUNIS HGF EIA; Institute of Immunology, Tokyo, Japan), according to the manufacturer’s recommendations.
Quantitative analysis of Ad‐NK4 DNA after co‐infection. We infected SUIT‐2 cells simultaneously with Ad‐NK4 at 10 MOI and Ad5/3hTERTLuc or Ad5/3hTERTE1 at 0.01 MOI, and measured the Ad‐NK4‐specific genome by quantitative PCR. Total DNA was extracted with DNeasy Blood & Tissue Kit (Qiagen). We designed Ad‐NK4 specific primers as follows: NK4, forward primer, 5′‐gcaattaaaacatgcgctga‐3′ and reverse primer, 5′‐attgacagtgcccctgtagc‐3′. Quantitative PCR was performed using a SYBR Premix Ex Taq (Takara Bio, Shiga, Japan) with a Chrom4 Real‐Time PCR Detection System (Bio‐Rad Laboratories). Each sample was run in triplicate and the expression of each gene was presented as the ratio between the expression of each NK4 gene DNA and that of 18S DNA.
Cell proliferation assay after infection. Cell proliferation was evaluated by the fluorescence intensity of propidium iodide (PI), as described previously.( 32 ) Cells were plated at 2 × 104 cells/well in 24‐well tissue culture plates (Becton Dickinson Labware, Bedford, MA, USA) and cultured overnight. After determination of the initial cell numbers, the cells were infected with Ad‐NK4 at 10 MOIs and Ad5/3hTERTE1 or Ad5/3hTERTLuc at various MOIs. PI (30 μm) and digitonin (600 μm) were added to each well to label all nuclei with PI. The fluorescence intensity, corresponding to the total cells, was measured using a CytoFluor II multi‐well plate reader (PerSeptive Biosystems, Framingham, MA, USA) with 530‐nm excitation and 645‐nm emission filters. All experiments were performed in triplicate wells and repeated at least three times.
Invasion assay. SUIT‐2 cells were infected with Ad‐NK4 at an MOI of 10 and Ad5/3hTERTE1 or Ad5/3hTERTLuc at MOIs of 0.01. After infection, the conditioned culture media were collected on days 1 and 3. Invasion of tumor cells was measured as the number of cells invading through Matrigel‐coated transwell inserts (Becton Dickinson, Franklin Lakes, NJ, USA), as described previously.( 33 ) Briefly, fresh untreated SUIT‐2 cells were seeded in 24‐well plates at a density of 5 × 104 cells/cm2 in 100 μL of DMEM mixed with 150 μL of conditioned culture media in the inner chamber, and cultured with 150 μL of conditioned culture media in the outer chamber. After 48 h of incubation in the presence of 3 ng/mL HGF, cells that had invaded to the lower surface of the Matrigel‐coated membrane were fixed with 70% ethanol, stained with hematoxylin–eosin (H&E), and counted in five randomly selected fields under a light microscope (ECLIPSE TE2000; Nikon, Tokyo, Japan).
In vivo analysis of NK4 expression levels in subcutaneously implanted tumors. Six‐week‐old female nude mice were injected in the back subcutaneously with 5 × 106 SUIT‐2 cells (200 μL). Fifteen mice were used in each experimental group. After 7 days (day 0), the control‐combination group mice were administered 2 × 108 pfu of Ad‐NK4 (50 μL) and 2 × 108 pfu of Ad5/3hTERTE1 (50 μL), and the CRAd‐combination group mice were administered 2 × 108 pfu of Ad‐NK4 (50 μL) and 2 × 108 pfu of Ad5/3hTERTLuc (50 μL) peritumorally on day 1. Five mice in each group were sacrificed and their tumors were excised for protein extraction on days 5, 8, and 10. The tumors were homogenized in ice‐cold lysis buffer, and NK4 was measured by ELISA, as described above.
In vivo efficacy of combination therapy in subcutaneously implanted tumors. Six‐week‐old female nude mice were injected in the back subcutaneously with 2 × 106 SUIT‐2 cells (200 μL). After 7 days (day 0), each group were administered 2 × 108 pfu/50 μL of adenovirus particles (control group, Ad‐LacZ+Ad5/3hTERTLuc; Ad‐NK4 group, Ad‐NK4 + Ad5/3hTERTLuc; CRAd group, Ad‐LacZ+ Ad5/3hTERTEl; CRAd‐combination group, Ad‐NK4 + Ad5/3hTERTEl) by weekly peritumoral injections on days 1, 8, and 15. In the tumor growth experiment, seven mice were used in each group. The size of the tumors was measured weekly, using digital vernier calipers (Mitutoyo, Kawasaki, Japan), and the volume of the tumors was calculated according to the following: tumor volume = ab 2/2, where a is the longest diameter of the tumor, and b is the shortest diameter. In the tumor angiogenesis experiment, another five mice for each group (control group, Ad‐NK4 group, CRAd‐combination group) were treated as the same way, and on day 21 they were sacrificed and their tumors were excised for immunohistochemical staining.
Immunohistochemical staining. To stain microvessels, the peroxidase‐conjugated avidin–biotin complex method was used with a Vectastain Elite ABC Kit (Vector Laboratories, Burlingame, CA, USA). Mouse monoclonal CD31 antibody JC/70A (NeoMarkers, Fremont, CA, USA) was used at a dilution of 1:50 followed by application ofbiotinylated antimouse IgG (1:100; Vector Laboratories). Microvessel density (MVD) was assessed in tumor areas showing high‐density staining. The number of vessels was counted in 10 fields/section at ×200 magnification (0.739 mm2/field), and the mean counts were recorded.
In vivo efficacy of combination therapy in intrasplenic implantation of SUIT‐2 cells. 2 × 106 SUIT‐2 cells (100 μL) were implanted by open injection into the spleen of 6‐week‐old female nude mice under anesthesia. Seven mice in each group were injected with 2 × 108 pfu/50 μL of adenovirus particles (control group, Ad‐NK4 group, CRAd group, CRAd‐combination group) into the spleen on day 3. Mice were sacrificed on day 21. Livers from animals were sectioned completely and metastatic tumors were evaluated by histological examination.
Animal procedures. All animal procedures were approved by the Ethics Committee of Kyushu University.
Statistical analysis. Statistical significance was evaluated by the nonparametric Mann–Whitney U‐test. Statistical significance was defined as values of P < 0.05 based on a two‐tailed test.
Results
Ad5/3hTERTE1 enhances transgene expression delivered by replication defective adenovirus vectors. To investigate the effects of hTERT‐promoter‐dependent CRAd on the transgene expression delivered by replication‐defective adenoviral vectors, we measured the expression levels of β‐gal delivered by Ad‐lacZ combined with either Ad5/3hTERTE1 or Ad5/3hTERTLuc. SUIT‐2 cells were infected with Ad‐lacZ at an MOI of 25 and Ad5/3hTERTE1 (CRAd‐combination group) or Ad5/3hTERTLuc (control‐combination group) at an MOI of 0.01. After infection, we evaluated the β‐gal mRNA levels by qRT‐PCR and the β‐gal activity by X‐gal staining. Compared with the control‐combination group cells, the β‐gal mRNA expression levels increased in the CRAd‐combination group cells in a time‐dependent manner (Fig. 1A, P < 0.01, days 1, 2, 3), and the numbers of cells with positive X‐gal staining (blue‐stained cells) also increased in the CRAd‐combination group cells (Fig. 1B, day 3). These results indicate that Ad5/3hTERTE1 enhances the transgene expression of β‐gal delivered by Ad‐lacZ.
Figure 1.
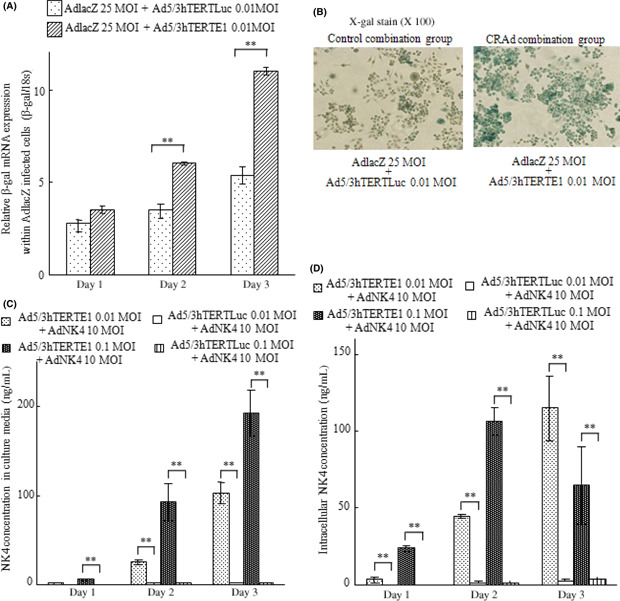
Ad5/3hTERTE1 enhances transgene expression delivered by replication‐ defective adenovirus vectors. (A,B) Control‐combination group SUIT‐2 cells were infected with Ad‐lacZ (25 MOI) and Ad5/3hTERTLuc (0.01 MOI), and CRAd‐combination group SUIT‐2 cells were infected with Ad‐lacZ (25 MOI) and Ad5/3hTERTLuc (0.01 MOI). (A) Total RNA samples were extracted on the indicated days. The expression levels of β‐gal mRNA were measured by qRT‐PCR and normalized by the corresponding expression level of 18S rRNA. Bars represent relative expression levels of mRNA. (B) Photomicrographs of SUIT‐2 cells from the control‐combination and CRAd‐combination groups. β‐Gal activity was assessed by X‐gal staining (magnification, ×100). (C,D) Control‐combination group SUIT‐2 cells were treated with Ad‐NK4 (10 MOI) and Ad5/3hTERTLuc (0.01 MOI or 0.1 MOI), and CRAd‐combination group SUIT‐2 cells were treated with Ad‐NK4 (10 MOI) and Ad5/3hTERTE1 (0.01 MOI or 0.1 MOI). The NK4 expression levels were measured in the culture media (C) and within infected cells (D) on days 1, 2, and 3. Bars represent NK4 concentrations. Each value represents the mean ± SD of three independent samples. **P < 0.01.
Next, we investigated the effects of Ad5/3hTERTE1 on the expression levels of NK4 delivered by Ad‐NK4. SUIT‐2 cells were infected with Ad‐NK4 at an MOI of 10 and either Ad5/3hTERTE1 or Ad5/3hTERTLuc at an MOI of 0.01 or 0.1. We measured the NK4 expression levels in culture media and among intracellular proteins extracted from the infected cells on days 1, 2, and 3. Similar to the case for Ad‐lacZ, the CRAd‐combination group significantly increased the NK4 expression levels in the culture media in a dose‐ and time‐dependent manner (Fig. 1C, P < 0.01 for 0.01 and 0.1 MOI, days 1, 2, 3). In addition, in the CRAd‐combination group, the intracellular NK4 expression levels increased significantly in a dose‐dependent manner (Fig. 1D, P < 0.01 for 0.01 and 0.1 MOI, days 1, 2). The results indicate that hTERT‐dependent CRAd enhances the intracellular transgene expression delivered by replication‐defective adenovirus vectors, as well as the extracellular transgene levels. In addition, this enhancement effect was also observed in breast cancer cell line MCF7, colon cancer cell line HCT116, and gastric cancer cell line AGS (Fig. S1).
hTERT‐CRAd, which contains E1 genomes, allows in‐trans replication of Ad‐NK4 DNA, leading to the coproduction of Ad‐NK4 particles. Adenovirus E1 includes E1A, which is essential for replication, and E1B, which is important for replication productivity.( 34 , 35 ) E1‐deleted non‐replicating adenovirus can replicate in E1‐containing cells such as human HEK293 or cells infected with E1‐containing adenovirus.( 21 , 36 ) Therefore, we investigated the effect of adenovirus E1 supplied from hTERT‐promoter‐dependent CRAd on viral replication of Ad‐NK4. At first, we confirmed that the NK4 expression levels of E1 (+) SUIT‐2 cells were significantly higher than those of E1 (−) SUIT‐2 cells, and that this increase was initiated on day 1 (Fig. S2). Next, we infected SUIT‐2 cells simultaneously with Ad‐NK4 at 10 MOI and either Ad5/3hTERTLuc (control‐combination group) or Ad5/3hTERTE1 (CRAd‐combination group) at 0.01 MOI, and measured the Ad‐NK4‐specific genome within the first 48 h after co‐infection by quantitative PCR. As shown in Figure 2(A), NK4 DNA levels began to increase gradually from 1 h, and then increased intensively at 24 h after co‐infection. This result suggests that hTERT‐CRAd allows in‐trans replication of Ad‐NK4 DNA immediately after co‐infection. Then, we assessed whether replicated Ad‐NK4 was released into the culture media as Ad‐NK4 particles, possibly leading to the bystander effect on neighboring cells. We infected SUIT‐2 cells with Ad‐NK4 at 10 MOI and either Ad5/3hTERTE1 or Ad5/3hTERTLuc at 0.01 MOI or 0.1 MOI. At 48 h after infection, we collected supernatants from cells infected with Ad‐NK4 and Ad5/3hTERTE1 (CRAd‐conditioned media) and Ad‐NK4 and Ad5/3hTERTLuc (control‐conditioned media). Next, new parental SUIT‐2 cells were treated with the CRAd‐conditioned media or control‐conditioned media for 1 h. At 48 h after treatment, we collected the supernatants and measured the NK4 protein levels. As shown in Figure 2(B), we did not detect NK4 protein in the supernatant treated with control‐conditioned media, but detected high expression levels of NK4 in supernatant treated with CRAd‐conditioned media. This result indicates that Ad‐NK4 particles were coproduced and released into the supernatant by at least 2 days after co‐infection with the hTERT‐CRAd.
Figure 2.

Human telomerase reverse transcriptase (hTERT) promoter‐dependent conditionally replicative adenovirus (hTERT‐CRAd), which contains E1 genomes, allowed in‐trans replication of Ad‐NK4 DNA, leading to the co‐production of Ad‐NK4 particles. (A) Quantitative analysis of Ad‐NK4 DNA after co‐infection. SUIT‐2 cells were infected with Ad‐NK4 at 10 MOI and either Ad5/3hTERTLuc or Ad5/3hTERTE1 at 0.01 MOI simultaneously. Total DNA was extracted at the indicated time after co‐infection. Ad‐NK4 DNA was measured by qPCR and normalized by the corresponding levels of 18S DNA. Each value represents the mean ± SD of three independent samples. *P < 0.05, **P < 0.01. (B) Co‐produced Ad‐NK4 particles were released into the culture media and allowed the bystander effect. SUIT‐2 cells were infected with Ad‐NK4 (10 MOI) and either Ad5/3hTERTLuc (0.01 MOI or 0.1 MOI) or Ad5/3hTERTE1 (0.01 MOI or 0.1 MOI). After 48 h, each media was collected. Then, fresh SUIT‐2 cells were infected with each collected media, and after 48 h, the levels of NK4 expression in the new collected supernatants were measured using ELISA. Each value represents the mean ± SD of three independent samples. **P < 0.01.
Ad5/3hTERTE1 enhances the inhibitory effects of Ad‐NK4 on cancer cell invasion. NK4, which inhibits biological events driven by HGF–Met signaling, inhibits invasion, but has no effect on the proliferation and survival of pancreatic cancer cells.( 30 , 37 ) To investigate whether NK4 expression enhanced by the combination of Ad5/3hTERTE1 had biological effects on cancer cell invasion, we simultaneously infected SUIT‐2 cells with Ad‐NK4 (10 MOI) and either Ad5/3hTERTE1 (0.01 MOI) or Ad5/3hTERTLuc (0.01 MOI). After replacement with fresh media to remove adenoviral particles, the cells were cultured and the culture media was collected on days 1 and 3 to use for invasion assays. As shown in Figure 3(A), combination with Ad5/3hTERTLuc or Ad5/3hTERTE1 at 0.01 MOI did not have any cytotoxic effect on SUIT‐2 cells on days 1–3, suggesting that the culture media derived from cells treated with this combination therapy on days 1–3 does not contain adenovirus particles. Based on these findings, in this experiment, we used Ad5/3hTERTE1 at the dose of 0.01 MOI and evaluated the number of invasive cells at 48 h after treatment to avoid the effect of newly released virus particles. On day 1, there was no difference in the effect on invasion between the treatment with the culture media derived from the Ad‐NK4‐ and Ad5/3hTERTE1‐infected cells and the treatment with control media derived from the Ad‐NK4‐ and Ad5/3hTERTLuc‐infected cells. However, the media from combination on day 3 inhibited invasion of cancer cells remarkably compared with the control media (Fig. 3B). These findings suggest that hTERT‐CRAd‐increased NK4 transgene also had the biological effects of NK4 on cancer cell invasion.
Figure 3.
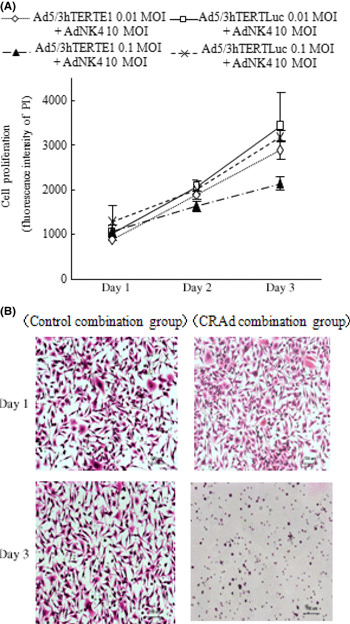
Ad5/3hTERTE1 enhanced the inhibitory effects of Ad‐NK4 on cancer cell invasion. (A) SUIT‐2 cells were infected with Ad‐NK4 (10 MOI) and either Ad5/3hTERTE1 or Ad5/3hTERTLuc (0.01 MOI or 0.1 MOI). Cell proliferation was measured by the fluorescence intensity of propidium iodide (PI), which correlates with the cell number, on the indicated days. All experiments were performed in triplicate wells and repeated at least three times, and each value represents the mean ± SD of three independent samples. (B) Control‐combination group SUIT‐2 cells were treated with Ad‐NK4 (10 MOI) and Ad5/3hTERTLuc (0.01 MOI), and the culture media were collected. Fresh untreated SUIT‐2 cells were seeded in DMEM mixed with conditioned culture media in the inner chamber and cultured with conditioned culture media in the outer chamber. After 48 h of culture in the presence of 3 ng/mL hepatocyte growth factor (HGF), cells that had invaded the lower surface of the Matrigel‐coated membrane were fixed with 70% ethanol, and stained with H&E. Photomicrographs of SUIT‐2 cells that invaded to the lower surface of the Matrigel‐coated membrane are shown.
Ad5/3hTERTE1 sustains the expression levels of NK4 delivered by Ad‐NK4 in tumors in vivo. We have shown that Ad5/3hTERTE1 enhances the transgene expression of NK4 delivered by Ad‐NK4, leading to the enhancement of inhibitory effects of Ad‐NK4 on cancer cell invasion in vitro. Therefore, we investigated whether the same enhanced effect was observed in in vivo settings.
We administered Ad‐NK4 and either Ad5/3hTERTE1 (CRAd‐combination group) or Ad5/3hTERTLuc (control‐combination group) peritumorally on day 1. The subcutaneously implanted tumors were excised and examined for their NK4 expression levels on days 5, 8, and 10. As shown in Figure 4(A), the levels of NK4 expression in the tumors gradually decreased in the control‐combination group, whereas the levels of NK4 expression in the CRAd‐combination group were sustained for 10 days at the level attained on day 3 (P < 0.01, day 10).
Figure 4.
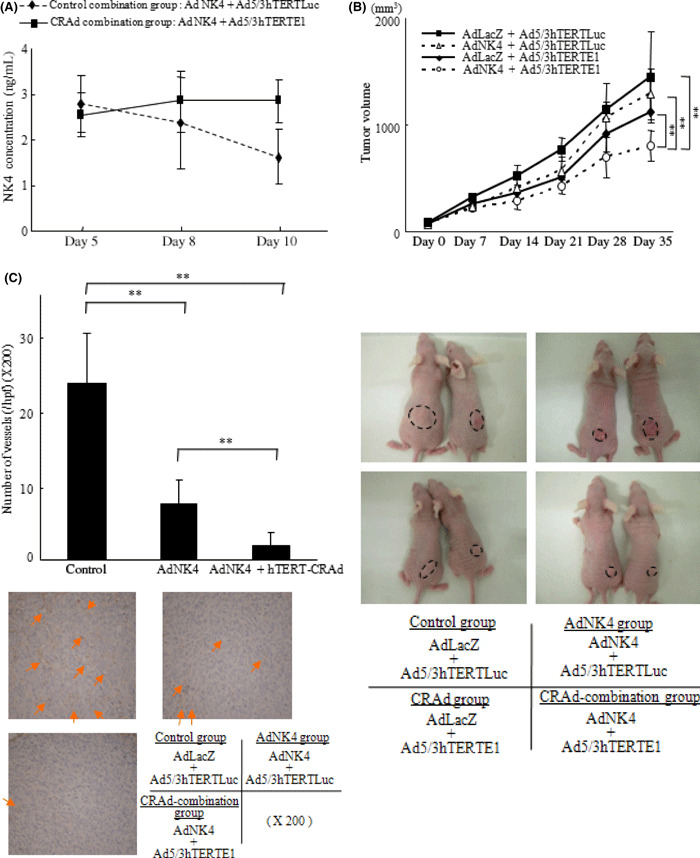
Combination with Ad5/3hTERTE1 sustained the expression levels of NK4 delivered by Ad‐NK4 in tumors and enhanced the therapeutic efficacy in vivo. Six‐week‐old female nude mice were used in the in vivo study. (A) Mice were injected subcutaneously with SUIT‐2 cells. The control‐combination group mice were administered Ad‐NK4 and Ad5/3hTERTLuc peritumorally, and the CRAd‐combination group mice were administered Ad‐NK4 and Ad5/3hTERTE1 on day 1. Five mice in each group were sacrificed and their tumors were excised for protein extraction on days 5, 8, and 10. NK4 was measured by ELISA. Each value represents the mean ± SD of three independent samples. **P < 0.01. (B) Seven mice in each group were subcutaneously implanted with SUIT‐2 cells in the back. Each group was administered weekly peritumoral injections on days 1, 8, and 15 as follows: control group, Ad‐LacZ+Ad5/3hTERTLuc; Ad‐NK4 group, AdAd‐NK4 + Ad5/3hTERTLuc; CRAd group, Ad‐LacZ + Ad5/3hTERTEl; and CRAd‐combination group, Ad‐NK4 + Ad5/3hTERTEl. The size of the tumors was measured weekly, and the volume of the tumors was calculated. (C) Five mice in each group (control group, Ad‐LacZ + Ad5/3hTERTLuc; Ad‐NK4 group, AdAd‐NK4 + Ad5/3hTERTLuc; and CRAd‐combination group, Ad‐NK4 + Ad5/3hTERTEl) were treated the same way, and on day 21, tumors were excised for immunohistochemical staining. Microvessels were determined by anti‐CD31 antibody and the microvessel density (MVD) was counted. Arrows indicate microvessels. Data are expressed as mean ± SD. **P < 0.01.
Combination with Ad5/3hTERTE1 enhances the therapeutic efficacies of Ad‐NK4 in tumors in vivo. To investigate the antitumor effects of the combination therapy in vivo, we used a subcutaneously implanted pancreatic cancer mouse model and assessed the effect on tumor growth and angiogenesis within tumors. As shown in Figure 4(B,C), mice treated with Ad‐NK4 + Ad5/3hTERTE1 (CRAd‐combination group) significantly suppressed tumor growth and angiogenesis (P < 0.01). In addition, in an intrasplenic‐implanted pancreatic cancer model, metastatic tumors in the liver were identified at the following rates: control group, 3/7; Ad‐NK4 group, 1/7; CRAd group, 1/7; and CRAd‐combination group, 0/7. These results also indicate that combination with hTERT‐CRAd enhances the therapeutic effects of Ad‐NK4 in vivo, as well as in vitro.
Discussion
The results of the present study revealed that the combination with Ad5/3hTERTE1 enhances NK4 expression delivered by Ad‐NK4 and the inhibitory effect on in vitro cancer invasion. In addition, our in vivo studies demonstrated that this combination therapy has remarkable antitumor effects on tumor growth and angiogenesis within tumors in subcutaneously implanted models and on liver metastasis in the intrasplenic‐injected models. These results are consistent with previous reports describing that transgene expression delivered by adenovirus vectors is enhanced when combined with replication‐competent adenovirus.( 19 , 20 , 21 , 22 ) Several studies have reported that combination with hTERT‐CRAd also enhances the functions of replication‐defective adenovirus vectors.( 23 , 24 ) In the present in vitro analysis, the CRAd‐combination group significantly increased NK4 transgene expression levels both in culture media and within infected cells, and this enhanced effect was due to the in‐trans replication of Ad‐NK4by adenovirus E1 derived from co‐infected hTERT‐CRAd. Our data suggest that replication of Ad‐NK4 DNA began immediately after co‐infection with hTERT‐CRAd. We also observed that intracellular NK4 expression in infected cells began to increase at day 1 after treatment. These findings are consistent with the ‘in‐trans replication’ mechanism described previously.( 21 , 36 ) We also observed that the replicated Ad‐NK4 particles were released when the infected host cells were killed by hTERT‐CRAd, possibly leading to the bystander effect on neighboring cells.
Adenovirus cancer therapy has some problems that need to be overcome before clinical use. In the clinical use of replication‐defective adenovirus vectors, their gene expression is too low and transient, and sometimes the intratumoral dispersion of adenovirus vectors after intratumoral injection is limited to the injection site. The present study revealed that CRAd enhanced and sustained transgene expression levels of replication‐defective adenovirus vectors and enhanced their biological effects. In addition, it has been reported that CRAd improved the penetration of adenoviruses and target proteins within tumors by creating spaces through the cell‐killing effect of CRAd.( 38 ) However, most CRAds are unable to carry a therapeutic gene because of size constraints, and, even if they can carry a therapeutic gene, transgene expression may be short because CRAds kill the host cells before synthesizing target proteins adequately. Also, cancers consist of genetically heterogeneous cells leading to frequent resistance to a single use of CRAd( 39 , 40 ) because CRAds replication is restricted to cells with specific characteristics, such as mutant‐p53 and telomerase.( 41 , 42 , 43 , 44 , 45 ) Therefore, it is reasonable to administer both adenovirus vectors carrying therapeutic genes and CRAd separately at the same time, especially when targeting both cancer cells and tumor–stromal interactions for cancer therapy.
In conclusion, in this study, we found that combination with the hTERT‐CRAd, Ad5/3hTERTE1, enhanced the transgene expression and therapeutic effects of adenovirus vector Ad‐NK4. This strategy is promising in the targeting of both cancer cells and tumor–stromal interactions in pancreatic cancer.
Supporting information
Fig. S1. Ad5/3hTERTE1 enhances transgene expression delivered by replication‐defective adenovirus vectors in various cancer cell lines. We used three cancer cell lines: (a) breast cancer cell line MCF7, (b) colon cancer cell line HCT116, and (c) gastric cancer cell line AGS. Control‐combination group cells were treated with Ad‐NK4 (10 MOI) and Ad5/3hTERTLuc (0.01 MOI or 0.1 MOI), and CRAd‐combination group cells were treated with Ad‐NK4 (10 MOI) and Ad5/3hTERTE1 (0.01 MOI or 0.1 MOI). The NK4 expression levels were measured in the culture media on days 1, 2, and 3. Bars represent NK4 concentrations. Each value represents the mean ± SD of three independent samples. *P < 0.05, **P < 0.01.
Fig. S2. Ad‐NK4 was transduced at higher levels in E1‐transfected SUIT‐2 cells than in E1‐non‐transfected SUIT‐2 cells. (a) SUIT‐2 cells were transfected with E1‐expressing plasmid (pShuttle TERT E1) or E1‐non‐expressiong plasmid (pShuttle TERT Luc). After transfection, total RNA samples were extracted on the indicated days. The expression levels of E1 mRNA (a) were measured by qRT‐PCR and normalized by the expression levels of 18S rRNA. Bars represent relative expression levels of mRNA. (b) E1‐transfected or E1‐non‐tranfected SUIT‐2 cells were infected with Ad‐NK4 (5 MOI). After infection, the culture media were collected on days 1, 2, and 3, and NK4 protein expression levels were measured by ELISA. Each value represents the mean ± SD of three independent samples. **P < 0.01.
Appendix S1. Materials and methods.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Acknowledgments
The authors are grateful to Mrs Shoko Sadatomi for her outstanding technical support and to Dr Akifumi Hayashi for his helpful pathological evaluation. This study was supported in part by a Grant‐in‐Aid from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- 1. Li J, Saif MW. Advancements in the management of pancreatic cancer. JOP 2009; 10: 109–17. [PubMed] [Google Scholar]
- 2. Mancuso A, Calabro F, Sternberg CN. Current therapies and advances in the treatment of pancreatic cancer. Crit Rev Oncol Hematol 2006; 58: 231–41. [DOI] [PubMed] [Google Scholar]
- 3. Warshaw AL, Fernandez‐del Castillo C. Pancreatic carcinoma. N Engl J Med 1992; 326: 455–65. [DOI] [PubMed] [Google Scholar]
- 4. Chu GC, Kimmelman AC, Hezel AF, DePinho RA. Stromal biology of pancreatic cancer. J Cell Biochem 2007; 101: 887–907. [DOI] [PubMed] [Google Scholar]
- 5. Mahadevan D, Von Hoff DD. Tumor‐stroma interactions in pancreatic ductal adenocarcinoma. Mol Cancer Ther 2007; 6: 1186–97. [DOI] [PubMed] [Google Scholar]
- 6. Nakamura T, Matsumoto K, Kiritoshi A, Tano Y. Induction of hepatocyte growth factor in fibroblasts by tumor‐derived factors affects invasive growth of tumor cells: in vitro analysis of tumor‐stromal interactions. Cancer Res 1997; 57: 3305–13. [PubMed] [Google Scholar]
- 7. Seslar SP, Nakamura T, Byers SW. Regulation of fibroblast hepatocyte growth factor/scatter factor expression by human breast carcinoma cell lines and peptide growth factors. Cancer Res 1993; 53: 1233–8. [PubMed] [Google Scholar]
- 8. Di Renzo MF, Poulsom R, Olivero M, Comoglio PM, Lemoine NR. Expression of the Met/hepatocyte growth factor receptor in human pancreatic cancer. Cancer Res 1995; 55: 1129–38. [PubMed] [Google Scholar]
- 9. Green NK, Seymour LW. Adenoviral vectors: systemic delivery and tumor targeting. Cancer Gene Ther 2002; 9: 1036–42. [DOI] [PubMed] [Google Scholar]
- 10. Alemany R, Balague C, Curiel DT. Replicative adenoviruses for cancer therapy. Nat Biotechnol 2000; 18: 723–7. [DOI] [PubMed] [Google Scholar]
- 11. Maehara N, Matsumoto K, Kuba K, Mizumoto K, Tanaka M, Nakamura T. NK4, a four‐kringle antagonist of HGF, inhibits spreading and invasion of human pancreatic cancer cells. Br J Cancer 2001; 84: 864–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Maehara N, Nagai E, Mizumoto K et al. Gene transduction of NK4, HGF antagonist, inhibits in vitro invasion and in vivo growth of human pancreatic cancer. Clin Exp Metastasis 2002; 19: 417–26. [DOI] [PubMed] [Google Scholar]
- 13. Murakami M, Nagai E, Mizumoto K et al. Suppression of metastasis of human pancreatic cancer to the liver by transportal injection of recombinant adenoviral NK4 in nude mice. Int J Cancer 2005; 117: 160–5. [DOI] [PubMed] [Google Scholar]
- 14. Uchino J, Takayama K, Harada A et al. Infectivity enhanced, hTERT promoter‐based conditionally replicative adenoviruses are useful for SCLC treatment. Cancer Gene Ther 2005; 12: 737–48. [DOI] [PubMed] [Google Scholar]
- 15. Ram Z, Culver KW, Oshiro EM et al. Therapy of malignant brain tumors by intratumoral implantation of retroviral vector‐producing cells. Nat Med 1997; 3: 1354–61. [DOI] [PubMed] [Google Scholar]
- 16. Mulvihill S, Warren R, Venook A et al. Safety and feasibility of injection with an E1B‐55 kDa gene‐deleted, replication‐selective adenovirus (ONYX‐015) into primary carcinomas of the pancreas: a phase I trial. Gene Ther 2001; 8: 308–15. [DOI] [PubMed] [Google Scholar]
- 17. Sangro B, Mazzolini G, Ruiz J et al. Phase I trial of intratumoral injection of an adenovirus encoding interleukin‐12 for advanced digestive tumors. J Clin Oncol 2004; 22: 1389–97. [DOI] [PubMed] [Google Scholar]
- 18. Crystal RG. Transfer of genes to humans: early lessons and obstacles to success. Science 1995; 270: 404–10. [DOI] [PubMed] [Google Scholar]
- 19. Lee CT, Lee YJ, Kwon SY et al. In vivo imaging of adenovirus transduction and enhanced therapeutic efficacy of combination therapy with conditionally replicating adenovirus and adenovirus‐p27. Cancer Res 2006; 66: 372–7. [DOI] [PubMed] [Google Scholar]
- 20. Wolkersdorfer GW, Morris JC, Ehninger G, Ramsey WJ. Trans‐complementing adenoviral vectors for oncolytic therapy of malignant melanoma. J Gene Med 2004; 6: 652–62. [DOI] [PubMed] [Google Scholar]
- 21. Habib NA, Mitry R, Seth P et al. Adenovirus replication‐competent vectors (KD1, KD3) complement the cytotoxicity and transgene expression from replication‐defective vectors (Ad‐GFP, Ad‐Luc). Cancer Gene Ther 2002; 9: 651–4. [DOI] [PubMed] [Google Scholar]
- 22. Lee CT, Park KH, Yanagisawa K et al. Combination therapy with conditionally replicating adenovirus and replication defective adenovirus. Cancer Res 2004; 64: 6660–5. [DOI] [PubMed] [Google Scholar]
- 23. Lanson NA Jr, Friedlander PL, Schwarzenberger P, Kolls JK, Wang G. Replication of an adenoviral vector controlled by the human telomerase reverse transcriptase promoter causes tumor‐selective tumor lysis. Cancer Res 2003; 63: 7936–41. [PubMed] [Google Scholar]
- 24. Hioki M, Kagawa S, Fujiwara T et al. Combination of oncolytic adenovirotherapy and Bax gene therapy in human cancer xenografted models. Potential merits and hurdles for combination therapy. Int J Cancer 2008; 122: 2628–33. [DOI] [PubMed] [Google Scholar]
- 25. Bewley MC, Springer K, Zhang YB, Freimuth P, Flanagan JM. Structural analysis of the mechanism of adenovirus binding to its human cellular receptor, CAR. Science 1999; 286: 1579–83. [DOI] [PubMed] [Google Scholar]
- 26. Kanerva A, Mikheeva GV, Krasnykh V et al. Targeting adenovirus to the serotype 3 receptor increases gene transfer efficiency to ovarian cancer cells. Clin Cancer Res 2002; 8: 275–80. [PubMed] [Google Scholar]
- 27. Takayama K, Reynolds PN, Short JJ et al. A mosaic adenovirus possessing serotype Ad5 and serotype Ad3 knobs exhibits expanded tropism. Virology 2003; 309: 282–93. [DOI] [PubMed] [Google Scholar]
- 28. Maemondo M, Narumi K, Saijo Y et al. Targeting angiogenesis and HGF function using an adenoviral vector expressing the HGF antagonist NK4 for cancer therapy. Mol Ther 2002; 5: 177–85. [DOI] [PubMed] [Google Scholar]
- 29. Korst RJ, Bewig B, Crystal RG. In vitro and in vivo transfer and expression of human surfactant SP‐A‐ and SP‐B‐associated protein cDNAs mediated by replication‐deficient, recombinant adenoviral vectors. Hum Gene Ther 1995; 6: 277–87. [DOI] [PubMed] [Google Scholar]
- 30. Date K, Matsumoto K, Shimura H, Tanaka M, Nakamura T. HGF/NK4 is a specific antagonist for pleiotrophic actions of hepatocyte growth factor. FEBS Lett 1997; 420: 1–6. [DOI] [PubMed] [Google Scholar]
- 31. Ohuchida K, Mizumoto K, Ogura Y et al. Quantitative assessment of telomerase activity and human telomerase reverse transcriptase messenger RNA levels in pancreatic juice samples for the diagnosis of pancreatic cancer. Clin Cancer Res 2005; 11: 2285–92. [DOI] [PubMed] [Google Scholar]
- 32. Zhang L, Mizumoto K, Sato N et al. Quantitative determination of apoptotic death in cultured human pancreatic cancer cells by propidium iodide and digitonin. Cancer Lett 1999; 142: 129–37. [DOI] [PubMed] [Google Scholar]
- 33. Sato N, Maehara N, Mizumoto K et al. Telomerase activity of cultured human pancreatic carcinoma cell lines correlates with their potential for migration and invasion. Cancer 2001; 91: 496–504. [DOI] [PubMed] [Google Scholar]
- 34. Flint J, Shenk T. Adenovirus E1A protein paradigm viral transactivator. Annu Rev Genet 1989; 23: 141–61. [DOI] [PubMed] [Google Scholar]
- 35. Graham FL, Van Der Eb AJ, Heijneker HL. Size and location of the transforming region in human adenovirus type 5 DNA. Nature 1974; 251: 687–91. [DOI] [PubMed] [Google Scholar]
- 36. Fechner H, Wang X, Wang H et al. Trans‐complementation of vector replication versus Coxsackie‐adenovirus‐receptor overexpression to improve transgene expression in poorly permissive cancer cells. Gene Ther 2000; 7: 1954–68. [DOI] [PubMed] [Google Scholar]
- 37. Date K, Matsumoto K, Kuba K, Shimura H, Tanaka M, Nakamura T. Inhibition of tumor growth and invasion by a four‐kringle antagonist (HGF/NK4) for hepatocyte growth factor. Oncogene 1998; 17: 3045–54. [DOI] [PubMed] [Google Scholar]
- 38. Nagano S, Perentes JY, Jain RK, Boucher Y. Cancer cell death enhances the penetration and efficacy of oncolytic herpes simplex virus in tumors. Cancer Res 2008; 68: 3795–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Reid T, Warren R, Kirn D. Intravascular adenoviral agents in cancer patients: lessons from clinical trials. Cancer Gene Ther 2002; 9: 979–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ries S, Korn WM. ONYX‐015: mechanisms of action and clinical potential of a replication‐selective adenovirus. Br J Cancer 2002; 86: 5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bischoff JR, Kirn DH, Williams A et al. An adenovirus mutant that replicates selectively in p53‐deficient human tumor cells. Science 1996; 274: 373–6. [DOI] [PubMed] [Google Scholar]
- 42. Kirn D. Oncolytic virotherapy for cancer with the adenovirus dl1520 (Onyx‐015): results of phase I and II trials. Expert Opin Biol Ther 2001; 1: 525–38. [DOI] [PubMed] [Google Scholar]
- 43. Huang TG, Savontaus MJ, Shinozaki K, Sauter BV, Woo SL. Telomerase‐dependent oncolytic adenovirus for cancer treatment. Gene Ther 2003; 10: 1241–7. [DOI] [PubMed] [Google Scholar]
- 44. Huang Q, Zhang X, Wang H et al. A novel conditionally replicative adenovirus vector targeting telomerase‐positive tumor cells. Clin Cancer Res 2004; 10: 1439–45. [DOI] [PubMed] [Google Scholar]
- 45. Wirth T, Zender L, Schulte B et al. A telomerase‐dependent conditionally replicating adenovirus for selective treatment of cancer. Cancer Res 2003; 63: 3181–8. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Ad5/3hTERTE1 enhances transgene expression delivered by replication‐defective adenovirus vectors in various cancer cell lines. We used three cancer cell lines: (a) breast cancer cell line MCF7, (b) colon cancer cell line HCT116, and (c) gastric cancer cell line AGS. Control‐combination group cells were treated with Ad‐NK4 (10 MOI) and Ad5/3hTERTLuc (0.01 MOI or 0.1 MOI), and CRAd‐combination group cells were treated with Ad‐NK4 (10 MOI) and Ad5/3hTERTE1 (0.01 MOI or 0.1 MOI). The NK4 expression levels were measured in the culture media on days 1, 2, and 3. Bars represent NK4 concentrations. Each value represents the mean ± SD of three independent samples. *P < 0.05, **P < 0.01.
Fig. S2. Ad‐NK4 was transduced at higher levels in E1‐transfected SUIT‐2 cells than in E1‐non‐transfected SUIT‐2 cells. (a) SUIT‐2 cells were transfected with E1‐expressing plasmid (pShuttle TERT E1) or E1‐non‐expressiong plasmid (pShuttle TERT Luc). After transfection, total RNA samples were extracted on the indicated days. The expression levels of E1 mRNA (a) were measured by qRT‐PCR and normalized by the expression levels of 18S rRNA. Bars represent relative expression levels of mRNA. (b) E1‐transfected or E1‐non‐tranfected SUIT‐2 cells were infected with Ad‐NK4 (5 MOI). After infection, the culture media were collected on days 1, 2, and 3, and NK4 protein expression levels were measured by ELISA. Each value represents the mean ± SD of three independent samples. **P < 0.01.
Appendix S1. Materials and methods.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item