

Abstract
Recent understanding of the molecular events crucial in overcoming immunosuppressive tumor microenvironments and generating effective antitumor immunity provides us with the wreath opportunity to manipulate genes that have a key role in antitumor immune responses. Granulocyte‐macrophage colony stimulating factor (GM‐CSF) and interleukin‐12 (IL‐12) are two indispensable cytokines for activating dendritic cells and boosting the strong immune responses against cancer. In this review, we describe the antitumor mechanisms and clinical application of gene‐modified tumor cells and dendritic cells to secrete GM‐CSF or IL‐12, respectively, in various preclinical and clinical settings. The principles operative in these vaccination strategies may prove applicable to other immunotherapy strategies, especially in combination with other therapeutic modalities, such as chemotherapy and targeted therapy. (Cancer Sci 2009)
Cytokines function as “double‐edged sword” against cancers
The interplay of tumor cells and host immunity is increasingly recognized to play a decisive role throughout the multiple stages of carcinogenesis, and the manipulation of endogenous immune systems enables us to scrutinize effective anticancer therapeutics.( 1 , 2 ) Previous studies demonstrate that dense intratumoral lymphocyte infiltrates are associated with favorable clinico‐pathological outcomes in patients with various types of cancer. Indeed, cytotoxic, memory CD8+ T‐cell infiltrates within tumors are strongly correlated with reduced disease recurrence and prolonged survival following various therapeutic settings of malignant melanoma, colorectal carcinoma, esophageal carcinoma, liver cancer, and renal carcinoma.( 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 ) Together, these findings suggest a potential contribution for nascent host immune responses in modulating the clinical outcome of human malignant diseases.
Notwithstanding this protective role for endogenous immunity, most patients fail to achieve durable clinical benefits. Accumulating evidences demonstrate that host elements, especially various cytokines, are frequently influenced on the context of tumor microenvironments.( 11 , 12 ) Tumor cells and tumor‐infiltrating lymphocytes frequently adopt the dedicated strategy to evade the antitumor processes through the manipulation of multiple cytokine networks. Various immunomodulatory cytokines, such as Interleukin‐10 (IL‐10), Transforming growth factor β (TGFβ), Vascular endothelial growth factor (VEGF), etc. released from tumor cells and tumor‐infiltrating immune cells play a critical role in suppressing antitumor innate and adaptive responses.( 13 ) Multiple studies also demonstrate that diverse cancers frequently arise within a background of chronic inflammation, by which inflammatory cytokines such as Tumor necrosis factor α (TNFα) and IL‐1β, within the inflammatory microenvironment may enhance tumor growth, angiogenesis, and metastasis.( 11 , 14 ) Together, these observations underscore the complex regulatory mechanisms of host immunity by cytokines in cancer pathogenesis (Fig. 1).
Figure 1.
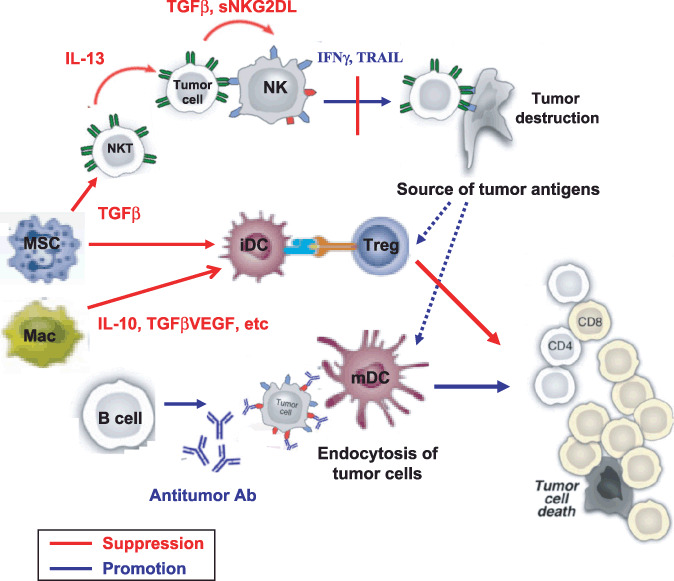
Various effector or regulatory cytokines produced from tumor cells or innate immune cells (Natural killer [NK], Natural killer T [NKT], macrophage, myeloid suppressor cells, etc.) have decisive roles in shaping the interplay between tumors and host immune systems. The cytokine milieu in tumor microenvironments regulates the function of dendritic cells, which in turn prime or suppress tumor‐specific T‐cell responses. IFNγ, Interferon γ; IL‐13, interleukin‐13; sNKG2D, soluble‐natural killer group‐2D; TGFβ, Transforming growth factor β; TRAIL, tumor necrosis factor receptor apoptosis‐inducing ligand; VEGF, Vascular endothelial growth factor.
Interleukin‐12 (IL‐12) and granulocyte‐macrophage colony stimulating factor (GM‐CSF) as critical mediators that generate antitumor immunogenicity
Deciphering the distinct molecular pathways through which cytokines recognize tumor at different stages and pathogenesis should contribute to improve the therapeutic quality of current immunotherapeutic strategies. Emerging evidences have identified the key molecular events that have a critical role in determining how host immunity recognizes nascent tumor microenvironments as antagonizing targets. Expression profiling analysis of colorectal carcinomas indicates that tumor samples rich in T helper type 1 (Th1) immune phenotypes are strongly indicative of favorable clinical outcomes, while inflammatory and immunosuppressive genes manifest a virulent disease with poor survival.( 15 ) These findings strongly suggested that in the clinical settings, the deviation toward Th1 phenotypes plays an important role in forming protective antitumor responses. Furthermore, enormous evidences have established the importance of dendritic cells (DCs) in surveying antitumor immunity by enhancing the cross‐presentation of immunogenic tumor antigens and linking innate and adaptive antitumor immune responses. Given the critical roles of DCs in determining the T helper type differentiation, targeting DCs at tumor microenvironments implicate the suitable therapeutic strategy to efficiently boost the Th1‐type immune responses and antitumor immunogenicity.( 16 , 17 ) The cytokines produced within the tumor microenvironment sharpen the quality and magnitude of the interplay between tumors and immune system.( 18 ) In particular, dendritic cells can secrete various cytokines that have a pivotal role in sensing the immune system against cancer. Indeed, the therapeutic strategies that manipulate tumor microenvironments by cytokine gene transfer attempt to render tumor‐associated lymphocytes to recover and activate antitumor immunity in preclinical and clinical settings (1, 2). In particular, GM‐CSF and IL‐12 are represented as the cytokines in stimulating DC cross‐presentation and promoting Th1 differentiation, respectively.( 17 , 18 )
Table 1.
Summary of cytokine gene transfer cancer vaccines
| Gene | Cell | Activities | Clinical study |
|---|---|---|---|
| GM‐CSF | Tumor | Significant clinical activities | Yes |
| Flt‐3L | Tumor | Massive DC infiltration at tumors | No |
| IL‐1β | Tumor | Controversial results | No |
| IL‐2 | Tumor | Reduced tumor burden in mice | Yes |
| IL‐4 | Tumor | Th2‐dependent antitumor effects | No |
| IL‐7 | Tumor | Lymphocyte infiltration at tumor | No |
| IL‐12 | Fibroblast, DC | Significant clinical activities | Yes |
| IL‐18 | DC | Tumor growth inhibition in mice | No |
| IL‐21 | Tumor, DC | Enhanced cellular and humoral responses | No |
| IL‐23 | DC, DNA plasmid | Coordinated action with IL‐12 | No |
| TNFα | Tumor | Tumor inhibition with little toxicity | No |
| TRAIL | Tumor | Activation of innate immunity | No |
| IFNβ | Tumor, DNA plasmid | NK and CTL activation | No |
| LIGHT | Tumor | Eradication of established tumors | No |
| RANK/RANK‐L | DC | Enhanced T‐cell responses | No |
| Chemokine/chemokine receptor | DC, T‐cells | Lymphocyte infiltration at tumor | No |
| Dominant negative TGFβ | T‐cells | T‐cell resitance to immunosuppression | No |
DC, dentritic cells; Flt‐3L, fms‐like tyrosine kinase‐3 ligand; IFNβ, Interferon β; IL, interleukin; GM‐CSF, granulocyte‐macrophage colony stimulating factor; NT, Natural killer; RANK/RANK‐L, Receptor activator of nuclear factor‐κB/Receptor activator of nuclear factor‐κB‐ligand; TGFβ, Transforming growth factor β; Th2, T helper type 2; TNFα, Tumor necrosis factor α; TRAIL, tumor necrosis factor receptor apoptosis‐inducing ligand.
Table 2.
Summary of cytokine gene therapy clinical trials
| Gene | Vector/vaccine vehicle | Tumor type | Clinical phase | Reference | Clinical responses |
|---|---|---|---|---|---|
| GM‐CSF | Retrovirus/Tumor cells (autologous) | Melanoma | Phase I | 26, 34 | Local immune responses in 11/16 pt |
| Adenovirus/Tumor cells (autologous) | Melanoma | Phase I | 27 | Local immune responses in 19/26 pt, 10/26 survival 36M after therapy | |
| Adenovirus/Tumor cells (autologous) | Non‐small cell lung cancer | Phase I | 28, 29 | Local immune infiltration in 18/25 pt, SD in 4/25 pt | |
| Retrovirus/Tumor cells (autologous) | Hormone‐refractory prostate cancer | Phase I | 30 | Local immune responses, diverse humoral reactions | |
| Retrovirus/Tumor cells (autologous) | Renal cell carcinoma | Phase I | 31 | Cellular and humoral reactions in pt, long‐term survival with IL‐2 in 2/4 pt | |
| Retrovirus/Tumor cells (allogenic) | Pancreatic cancer | Phase II | 33, 93 | Immune reactions with prolonged survival in 3/14pt | |
| Adenovirus/Tumor cells (allogenic) | Hormone‐refractory prostate cancer | Phae II/II | 32 | PSA decline, prolonged survival compared to expected survival time | |
| Adenovirus/Tumor cells (allogenic) | Hormone‐refractory prostate cancer | Phase III | 36 | VITAL‐I (GVAX vs Docetaxel+PSL): Similar overall survival between two arms | |
| Phase III | 35 | VITAL‐II (GVAX+Docetaxel vs Docetaxel+PSL): Increased mortality in GVAX+Docetaxel | |||
| IL‐12 | Plasmid DNA | Melanoma | Phase I | 77, 78, 79 | Local immune response and decreased angiogenesis in responders; CR in 1/9, SD in 2/9 |
| Retrovirus vector/Fibroblast (autologous) | Melanoma | Phase I | 74, 75 | Tumor reduction in 4/9 pt | |
| Adenovirus‐transfer/DCs | GI tumor | Phase I | 84 | Local immune infiltration in 3/9 pt; SD in 2/9, PR in 1/9 | |
| IL‐2 | Adenovirus‐transfer/TIL | Melanoma | Phase I/II | 93, 94 | PR in 1/7 pt with local immune responses |
| Retrovirus vector (with HSV‐TK) | Glioblastoma | Phase I | 95 | Local cytokine cascade with 50% of response rate in a total of 12 pt | |
| cDNA | Prostate carcinoma | Phase I | 96 | Locval immune infiltrate | |
| Retrovirus (with MUC‐1 gene) | Prostate carcinoma | Phase I | 97 | PSA decline in 5/5 pt | |
| Canarypox virus | Melanoma | Phase I | 98 | PR in 3/8 pt with immune infiltration | |
| cDNA/Vero cells | Melanoma | Phase II | 99 | Cellular and humoral responses in 3/18 pt; SD in 50% | |
| cDNA/Tumor cells (autologous) | Lymphoma, CRC, RCC | Phase I | 100 | Clinical response in 1 pt with lymphoma | |
| Retrovirus/Tumor cells (autologous) | Melanoma | Phase I | 101 | Increased CTL response in 4/20 pt; SD in 3/20 pt | |
| IFNγ | Retrovirus/Tumor cells (autologous) | Melanoma | Phase I | 102 | SD with surgical removal in 1/5 pt |
CR, complete response; CRC, Colorectal carcinoma; DC, dentritic cells; GI, Gastrointestinal; GM‐CSF, granulocyte‐macrophage colony stimulating factor; HSV‐TK, herpes simplex virus‐tymisine kinase; IFNγ, Interferon γ; IL, interleukin; MUC‐1, Mucine‐1; PR, partial response; PSA, Prostate specific antigen; PSL, Predonisolone; RCC, renal cell carcinoma; SD, stable disease; TIL, tumor infiltrating lymphocytes.
Granulocyte‐macrophage colony stimulating factor (GM‐CSF) engenders protective immunity by stimulating the recruitment, maturation, and function of DCs,( 19 , 20 ) while IL‐12 released from DCs directly primes effector lymphocytes at local environments. These different modes of actions by these two cytokines were further clarified by comparing GM‐CSF and IL‐12 gene‐transferred DCs in inducing antitumor immune responses.( 21 , 22 ) In this regard, the gene transfer of GM‐CSF into dying tumor cells may be more suitable form to elicit local antitumor responses by differentiating DC in situ and providing immunogenic tumor antigens available from vaccinated tumor cells for cross‐presentation by paracrine fashion, whereas the injection of IL‐12 gene‐transferred DCs at tumors may be an appropriate strategy to maximally boost antitumor effector functions.
Overall, the therapeutic strategy to translate immunogene therapy using the two cytokines into clinical settings might be pivotal to improve the current cancer immunotherapy.
Granulocyte‐macrophage colony stimulating factor (GM‐CSF)‐secreting tumor cell vaccines
A comparative analysis of the relative abilities of multiple immunostimulatory molecules to enhance host responses, following gene transfer into tumor cells, identified GM‐CSF as the most potent of 10 gene products tested.( 23 ) Indeed, several preclinical studies revealed that DCs that are activated by GM‐CSF‐secreting tumor cells and sampled tumor antigens from apoptotic tumor cells in situ primed specific CD4+ and CD8+ T‐cells, CD1d‐restricted invariant Natural killer T (NKT) cells, and antibody producing B cells for effectuating tumor destruction.( 24 , 25 )
Based upon these preclinical studies, multiple phase I clinical trials of vaccination with irradiated tumor cells engineered to secrete GM‐CSF were undertaken in patients with diverse malignant disorders. In these studies, autologous tumor cells were engineered to secrete GM‐CSF by either retroviral or adenoviral mediated gene transfer in patients with metastatic melanoma,( 26 , 27 ) non‐small cell lung carcinoma,( 28 , 29 ) hormone therapy–refractory metastatic prostate cancer,( 30 ) or renal cell carcinoma,( 31 ) whereas standardized allogeneic tumor cell lines were stably transfected with expression plasmids encoding GM‐CSF.( 32 , 33 )
Overall, these trials demonstrated that vaccination consistently elicits local infiltrates composed of dendritic cells, granulocytes, macrophages, and T‐cells, and elicited durable cellular and humoral antitumor immune responses.( 34 ) The strong evidence for stimulating tumor immunity together with the lack of significant toxicity formed the basis for advancing this vaccination strategy to phase II and III testing in several malignancies.
However, the results of a recently completed randomized phase III trial of immunization with irradiated, GM‐CSF‐secreting allogeneic prostate carcinoma cells in patients with hormone refractory metastatic disease has unveiled the difficulty of achieving sufficient clinical benefit in comparison with standard anticancer regimens. The GM‐CSF‐secreting tumor cell vaccines in combination with Docetaxel caused increased mortality due to disease progression compared to decetaxel regimens, while GM‐CSF‐secreting vaccines elicit survival benefit compatible to decetaxel and prednisolone, the standard chemotherapeutic regimens.( 35 , 36 ) Although these randomized trials showed that there is little evidence that this vaccination strategy against hormone therapy–refractory prostate cancer increases survival compared to conventional cytotoxic chemotherapy, further trials have been underway to assess clinical efficacy against several malignancies, such as pancreatic carcinoma, malignant melanoma, and non‐small cell lung cancer.
Overall, the rigorous assessments would be required to clarify which therapeutic strategies (autologous vs allogenioc, combination with another immunotherapy, etc.) should be suitable for effectuating clinical activities of GM‐CSF‐secreting tumor cell vaccines in future clinical trials.
Mechanisms that attenuate antitumor efficacy of GM‐CSF‐based vaccines
Given the disappointing results of phase III clinical studies, it is important to clarify the molecular mechanisms that restrict clinical efficacy of GM‐CSF‐secreting vaccines. Recent reports explored the mechanisms underlying several pathways that restrain the efficacy of GM‐CSF‐secreting tumor cell vaccines to further improve the antitumor activities.( 37 ) Paradoxically, sustained GM‐CSF production within the tumor microenvironment is associated with disease progression in some experimental models. In both murine models and patients with advanced melanoma, GM‐CSF may recruit immature CD11b+Gr‐1+ myeloid suppressor cells into the sites of tumor, where they may trigger antitumor T‐cell dysfunction.( 38 , 39 , 40 ) These contradictory activities of GM‐CSF might depend upon additional factors operative in the tumor microenvironment. To determine more about the physiologic functions of GM‐CSF, we generated and characterized mice deficient in the cytokine. These animals developed the progressive accumulation of surfactant proteins in the pulmonary air spaces, a disorder reminiscent of idiopathic pulmonary alveolar proteinosis in humans.( 41 , 42 ) In addition to the pulmonary abnormalities, GM‐CSF‐deficient mice also display systemic chronic inflammatory manifestations as represented by systemic lupus erythematosis‐like disorder, immune complex–mediated glomerulonephritis, and the induction of autoimmune diabetes.( 43 , 44 ) These pathologic findings unveiled a key role for GM‐CSF in the maintenance of immune tolerance against self‐antigens. To clarify these mechanisms, we characterized the function of antigen presenting cells in the GM‐CSF deficient mice. The most apparent manifestation of phagocytosis of GM‐CSF‐deficient macrophages and dendritic cells was the marked impairment of phagocytosis of apoptotic cells. The efficient clearance of apoptotic cells by phagocytes is critical for maintaining immune homeostasis.( 43 , 45 ) Multiple phagocyte receptors and secreted proteins effectuate the clearance of dying cells by dendritic cells and macrophages. To explore the pathways that were responsible for mediating apoptotic cell uptake in a GM‐CSF‐dependent pathway, we determined the expression profiles of several candidate molecules in GM‐CSF‐deficient antigen‐presenting cells (APCs). Of the molecules tested, milk fat globule epidermal growth factor protein‐8 (MFG‐E8) displayed the greatest reduction in GM‐CSF‐deficient macrophages and dendritic cells compared to wild‐type controls.( 45 )
Milk fat globule epidermal growth factor protein‐8 (MFG‐E8) was initially identified as a component of milk fat globules,( 46 ) but subsequent work revealed that macrophages and dendritic cells also secrete the protein. Oxidized phosphatidylserine exposed on the apoptotic cell surfaces delivers a major signal for facilitating phagocyte engulfment.( 47 , 48 ) MFG‐E8 binds phosphatidylserine and promotes apoptotic cell ingestion through engaging αvβ3–5 integrins on phagocytes.( 49 ) The essential roles of these molecules in apoptotic cell uptake and immune tolerance is validated by the development of autoimmunity and persistent inflammation in mice deficient in these pathways.( 50 )
The genetic reconstitution of MFG‐E8 in GM‐CSF‐deficient dendritic cells restored the efficient clearance of apoptotic cells, confirming the requirement for MFG‐E8 in GM‐CSF‐dependent phagocyte activities. Furthermore, the clearance of dying cells endowed dendritic cells that ingested apoptotic cells to promote the differentiation of CD4+ T‐cells into FoxP3+ regulatory T‐cells (Treg) and restrict the generation of Th1 and Th17 CD4+ cells.( 45 , 51 )
The abilities of MFG‐E8 to stimulate Treg expansion render this opsonin an important target for cancer therapy. Indeed, the induction of MFG‐E8 by GM‐CSF might create a negative feedback loop that attenuates the potency of tumor cell vaccination: the coexpression of MFG‐E8 abrogated the protective immunity of GVAX against subcutaneous B16 murine melanoma tumors through the expansion of tumor infiltrating Tregs, which blocked the expansion and activity of cytotoxic T‐cells. The vaccination with GVAX combined with pharmacological blockade of MFG‐E8 accomplished complete regressions of established B16 melanomas, with the inhibited Tregs and the expanded CD8+ cytotoxic T‐cells.( 45 ) These findings raise the possibility that MFG‐E8 blockade might enhance the clinical activity of GM‐CSF‐secreting tumor cell vaccines (Fig. 2). Other mechanisms to attenuate therapeutic efficacy of GM‐CSF‐based cancer vaccines have been identified in our studies, such as the cytotoxic T lymphocyte associated antigen‐4 (CTLA‐4)‐mediated negative costimulation( 52 ) and the suppression of NKG2D‐dependent innate and adaptive antitumor cytotoxicity.( 53 )
Figure 2.
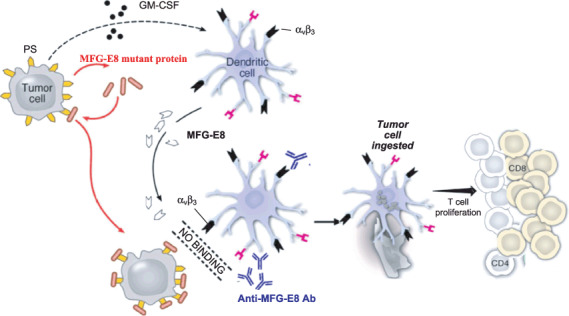
Milk fat globule epidermal growth factor protein‐8 (MFG‐E8) blocking antibodies or tumor cells engineered to produce dominant negative MFG‐E8 mutant efficiently blocks the binding of MFG‐E8 with its receptor αvβ3 or αvβ5 integrin on tumor cells or dendritic cells, thereby blocking the immunosuppressive effects of apoptotic cells. Thus, granulocyte‐macrophage colony stimulating factor (GM‐CSF)‐secreting tumor cell vaccine stimulates a potent antitumor response through an MFG‐E8‐independent pathway.
Furthermore, the cytokines (IL‐12, IL‐6, etc.), chemokines (Thymus and activation‐regulated chemokines [TARC], Regulated on activation, normal T‐cell expressed and secreted‐APCs Antigen‐presenting cells [RANTES]), and costimulatory molecules (B7‐1) have been reported to enhance the therapeutic efficacy of GM‐CSF‐secreting tumor cell vaccines, suggesting that combination with other immunotherapeutic strategies may overcome the limited clinical efficacy of GVAX.( 54 , 55 , 56 , 57 , 58 , 59 , 60 )
Together, these results clarify the key regulatory circuits that attenuate the clinical activity of GM‐CSF‐based vaccines and aid in the definition of new strategies to enhance their activities.
Interleukin‐12 (IL‐12)‐based cancer immunotherapy
Interleukin‐12 (IL‐12) has been identified as a master regulator of innate and adaptive immune responses against various pathogens and tumors.( 61 , 62 ) IL‐12 is mainly produced from myeloid DCs, and exerts protective immunity primarily by promoting Th1 differentiation of CD4+ T‐cells, stimulating macrophage and NK cell cytotoxicity, and recruiting CTL into tumors through the induction of various chemokine repertoires. IL‐12 also initiates antiangiogenic programs through the inhibition of proangiogenic factors VEGF and matrix metalloproteinase 2/9 (MMP2/9) and direct antiangiogenic activity of Interferon γ (IFNγ).( 63 ) The antitumor activities of IL‐12 have been extensively evaluated and showed extensive antitumor responses in murine models including various types of tumors.( 64 , 65 , 66 ) In phase I clinical settings, IL‐12, in a form of recombinant protein, has been initially administered in patients with advanced malignancies including metastatic melanoma, renal cell carcinoma, and head and neck suquamous cell carcinoma, to investigate its safety and antitumor activities. With exception of the results obtained in cutaneous T‐cell lymphoma and Hodgkin's disease,( 67 , 68 ) the clinical efficacy remains minimal, with an objective response rate ranging 0–11%. Intolerable toxicities followed by systemic IL‐12 infusion were observed which mainly consisted of bone marrow complications, such as agranulocytosis and hemolytic anemia, which appear to be associated with systemic IFNγ levels.( 69 , 70 , 71 , 72 , 73 )
To achieve sufficient amounts of IL‐12 at tumors and draining lymph nodes and to ameliorate the systemic toxicity, clinical studies were conducted to evaluate antitumor activities of fibroblast or tumor cells engineered to transfer IL‐12 gene‐encoded vector including plasmid DNA or recombinant viruses in patients with solid malignancies.( 74 , 75 , 76 , 77 , 78 , 79 ) These trials demonstrated that vaccination frequently triggered clinical responses, represented as transit tumor shrinkage, as well as local infiltrates of immune cells, and elicited durable cellular antitumor immune responses. Furthermore, the significant toxicities were not observed in all cases. The strong immunostimulatory effects of IL‐12 may serve as an adjuvant in combination with various immunization strategies, and several clinical studies supported this idea.( 80 ) Therefore, the strategies designed to combine antigen‐specific approaches (peptide, tumor cell vaccines) with IL‐12 gene therapy may further effectuate clinical responses and trigger durable antitumor immune responses. Overall, notwithstanding the limiting antitumor clinical efficacy, IL‐12‐based vaccines still provide various perspectives for exploring the clinical development of effective cancer vaccines. Furthermore, deciphering the molecular machinery that restrains the antitumor immunity of IL‐12 may provide new strategies to improve the clinical efficiency of IL‐12 gene therapy.
Interleukin‐12 (IL‐12) gene‐transduced DC vaccines
Ex vivo transfer of genes encoding tumor antigens, cytokines, or other immunostimulatory elements into DCs has been shown to effectively prime antitumor T‐cells very effectively in tumor‐draining lymph nodes, and induce long‐term immunity against tumors, in various murine models.( 17 , 81 ) Among the numerous candidates that are important in enhancing DC immunogenicity at local tumor microenvironments, IL‐12 gene transfer may be a suitable strategy in triggering innate and adaptive immune responses as well as antiangiogenic activities, while maintaining the endocytic capacity for optimally selecting suitable tumor antigens released from dying tumor cells in situ. IL‐12 also endowed DCs to acquire the migratory capacities toward draining lymph node and priming antitumor CTL through enhanced cross‐presentation of residual DCs, further amplifying strong antitumor immune reactivity ( Fig. 3 ). Furthermore, the localized expression of IL‐12 more closely resembles the physiological setting and lessens toxicity concerns, as observed in earlier clinical studies. In the preclinical studies, intratumoral injection of DCs engineered to secrete IL‐12 was found to express MHC class I/II and costimulatory molecules at compatible levels to immature phenotypes, but still provoked extensive antitumor responses with marked reductions in tumor burden and prolonged survival in murine models.( 22 , 81 , 82 ) These results further support the idea that the unique characters of IL‐12 gene transfer approaches that selectively enhance DC‐mediated T‐cell polarization may be combined with the other immunostimulatory approaches to accelerate immune‐mediated rejection of tumors in future clinical settings.
Figure 3.
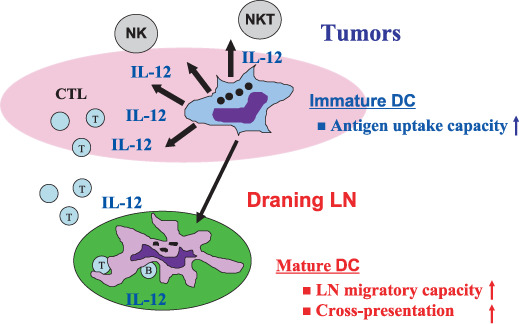
Interleukin (IL)‐12 gene transfer into dendritic cells (DCs) manifest antitumor activities by triggering innate and T helper type 1 (Th1) immune responses as well as antiangiogenic activities, but retain endocytic capacity to ingest tumor antigens released from dying tumor cells in situ, in tumors. The IL‐12‐transferred DCs migrate toward the draining lymphnodes and strongly prime antitumor cytotoxic T lymphocyte reactivity to further amplify antitumor responses. LN, lymph node; NK, Natural killer; NKT, Natural killer T.
Based upon the above results, phase I clinical trials of vaccination with monocyte‐derived DCs engineered to secrete IL‐12 were undertaken in patients with diverse gastrointestinal malignancies. In these studies, DCs were engineered to secrete IL‐12 by replication‐defective adenoviral‐mediated gene transfer. The vaccination consistently elicits transit clinical activities, and elicited durable cellular and humoral antitumor immune responses in some patients.( 83 ) Further clinical studies should validate the feasibility of IL‐12 gene‐modified DC vaccines in antitumor mechanisms and clinical utilities in the near future.
Interleukin‐23 (IL‐23)‐based immunogene therapy
Interleukin‐23 (IL‐23) shares the p40 chain with IL‐12, but such subunit associates with a p19 chain.( 84 ) Similar to IL‐12, IL‐23 is produced predominantly by macrophages and dendritic cells. These two cytokines seem to have similar roles in the priming and activating of T‐cell responses, but with quite different roles in polarizing helper functions and influencing inflammation and cancer.
Interleukin‐23 (IL‐23) plays an important role in the survival and activation of newly identified Th17 cells.( 85 ) IL‐23 has been recently identified as an essential mediator in promoting tumorigenesis and suppressing Th1 responses within the background of chronic inflammation.( 86 ) However, it remains obscure how therapeutic manipulation of IL‐23 through the recombinant protein or gene transfer has any impact on host immune responses against tumors. Emerging evidences have uncovered that IL‐23 may provoke antitumor immunity and elicit the reduction of tumor burden in various murine models.( 87 , 88 ) As the mechanism by which IL‐23 enhances the antitumor immunity, we uncovered that systemic IL‐23 gene transfer induced marked shrinkage of established tumors, and Th1 responses mediated by endogenous IL‐12 play an indispensable roles in their antitumor effects.( 87 ) Overall, caution would be justified in future clinical development of IL‐23‐based cancer immunotherapy until further preclinical studies clarify the detailed mechanisms of how IL‐23 modifies the interplay between tumor cells and lymphocytes within tumor microenvironments.
Future perspectives
The recent breakthrough clarifying the molecular machinery of antitumor immunity provides us a wealth opportunity to realize successful tumor immunotherapy in clinical settings. In this regard, other immunostimulatory cytokines, such as IL‐15, IL‐18, IL‐21, etc., elicit antitumor responses through the activation of innate and adaptive immune cells mediated by distinct pathways.( 89 , 90 ) The targeting of negative (cytotoxic T lymphocyte antigen‐4 [CTLA‐4], programmed death‐1 [PD‐1], B and T lymphocyte attenuator [BTLA], etc.) as well as positive (CD137, CD28, etc.) checkpoint machinery also coordinately stimulate durable antitumor immunity with cytokine gene therapy.( 91 ) Recent studies revealed the molecular mechanisms by which conventional cytotoxic chemotherapy triggers tumor immunogenicity and provokes antitumor immune responses.( 92 ) The clinical application of targets that augment anticancer immune responses by cytotoxic therapy may serve as an ideal combination with immunogene therapy. Together, the targeting of these pathways may provide another complementary strategy to reconstitute immune homeostasis and promote immune‐mediated tumor destruction.
Conclusion
We describe the landscape of the current status and future perspectives for two immunogene therapies, GM‐CSF gene‐transduced tumor cells and IL‐12 gene‐transduced DC vaccines. Although these strategies have provoked significant antitumor reactivity in several cases, their limited clinical efficacy warrants further investigation in preclinical and clinical settings. Technological advances and increasing understanding of immunoregulatory mechanisms by which host responses are compromised within tumor microenvironments may lead to combined immunomodulatory gene approaches with improved efficacy for the treatment of cancer patients.
References
- 1. Pardoll D. Does the immune system see tumors as foreign or self? Annu Rev Immunol 2003; 21: 807–39. [DOI] [PubMed] [Google Scholar]
- 2. Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med 2002; 10: 909–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Clark W, Elder D, Guerry D et al . Model predicting survival in stage I melanoma based on tumor progression. J Natl Cancer Inst 1989; 81: 1893–904. [DOI] [PubMed] [Google Scholar]
- 4. Pages F, Berger A, Camus M et al . Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med 2005; 353: 2654–66. [DOI] [PubMed] [Google Scholar]
- 5. Gao Q, Qiu SJ, Fan J et al . Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J Clin Oncol 2007; 25: 2586–93. [DOI] [PubMed] [Google Scholar]
- 6. Nakano O, Sato M, Naito Y et al . Proliferative activity of intratumoral CD8 (+) T‐lymphocytes as a prognostic factor in human renal cell carcinoma: clinicopathologic demonstration of antitumor immunity. Cancer Res 2001; 61: 5132–6. [PubMed] [Google Scholar]
- 7. Tahara H, Shiozaki H, Kobayashi K et al . Phenotypic characterisrics of tumor‐infiltrating lymphocytes in human oesophageal cancer tissues defined by quantitative two‐colour analysis with flow‐cytometry. Virchows Arch a Pathol Anat 1990; 416: 329–34. [DOI] [PubMed] [Google Scholar]
- 8. Dave SS, Wright G, Tan B et al . Prediction of survival in follicular lymphoma based on molecular features of tumor‐infiltrating immune cells. N Engl J Med 2004; 351: 2159–69. [DOI] [PubMed] [Google Scholar]
- 9. Zhang L, Conejo‐Garcia J, Katsaros D et al . Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Eng J Med 2003; 348: 203–13. [DOI] [PubMed] [Google Scholar]
- 10. Sato E, Olson SH, Ahn J et al . Intraepithelial CD8+ tumor‐infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A 2005; 102: 18538–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Coussens L, Werb Z. Inflammation and cancer. Nature 2002; 420: 860–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gabrilovich D. Mechanisms and functional significance of tumour‐induced dendritic‐cell defects. Nat Rev Immunol 2004; 4: 941–52. [DOI] [PubMed] [Google Scholar]
- 13. Karin M, Lawrence T, Nizet V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell 2006; 124: 823–35. [DOI] [PubMed] [Google Scholar]
- 14. De Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer 2006; 6: 24–37. [DOI] [PubMed] [Google Scholar]
- 15. Galon J, Costes A, Sanchez‐Cabo F et al . Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006; 313: 1960–4. [DOI] [PubMed] [Google Scholar]
- 16. Huang AY, Golumbek P, Ahmadzadeh M, Jaffee E, Pardoll D, Levitsky H. Role of bone marrow‐derived cells in presenting MHC class I‐restricted tumor antigens. Science 1994; 264: 961–5. [DOI] [PubMed] [Google Scholar]
- 17. Engleman EG. Dendritic cell‐based cancer immunotherapy. Semin Oncol 2003; 30: 23–9. [DOI] [PubMed] [Google Scholar]
- 18. Dranoff G. Cytokines in cancer pathogenesis and cancer therapy. Nat Rev Cancer 2004; 4: 11–22. [DOI] [PubMed] [Google Scholar]
- 19. Mach N, Gillessen S, Wilson SB, Sheehan C, Mihm M, Dranoff G. Differences in dendritic cells stimulated in vivo by tumors engineered to secrete granulocyte‐macrophage colony‐stimulating factor or flt3‐ligand. Cancer Res 2000; 60: 3239–46. [PubMed] [Google Scholar]
- 20. Hung K, Hayashi R, Lafond‐Walker A, Lowenstein C, Pardoll H, Levitsky H. The central role of CD4+ T cells in the antitumor immune response. J Exp Med 1998; 188: 2357–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Curiel‐Lewandrowski C, Mahnke K, Labeur M et al . Transfection of immature murine bone marrow‐derived dendritic cells with the granulocyte‐macrophage colony‐stimulating factor gene potently enhances their in vivo antigen‐presenting capacity. J Immunol 1999; 163: 174–83. [PubMed] [Google Scholar]
- 22. Nishioka Y, Hirao M, Robbins PD, Lotze MT, Tahara H. Induction of systemic and therapeutic antitumor immunity using intratumoral injection of dendritic cells genetically modified to express interleukin 12. Cancer Res 1999; 55: 4035–404. [PubMed] [Google Scholar]
- 23. Dranoff G, Jaffee E, Lazenby A et al . Vaccination with irradiated tumor cells engineered to secrete murine granulocyte‐macrophage colony‐stimulating factor stimulates potent, specific, and long‐lasting anti‐tumor immunity. Proc Natl Acad Sci USA 1993; 90: 3539–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gillessen S, Naumov YN, Nieuwenhuis EE et al . CD1d‐restricted T cells regulate dendritic cell function and antitumor immunity in a granulocyte‐macrophage colony‐stimulating factor‐dependent fashion. Proc Natl Acad Sci USA 2003; 100: 8874–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Simons JW, Jaffee EM, Weber CE et al . Bioactivity of autologous irradiated renal cell carcinoma vaccines generated by ex vivo granulocyte‐macrophage colony‐stimulating factor gene transfer. Cancer Res 1997; 57: 1537–46. [PMC free article] [PubMed] [Google Scholar]
- 26. Soiffer R, Hodi FS, Haluska F et al . Vaccination with irradiated, autologous melanoma cells engineered to secrete granulocyte‐macrophage colony‐stimulating factor by adenoviral‐mediated gene transfer augments antitumor immunity y in patients with metastatic melanoma. J Clin Oncol 2003; 21: 3343–50. [DOI] [PubMed] [Google Scholar]
- 27. Soiffer R, Lynch T, Mihm M et al . Vaccination with irradiated, autologous melanoma cells engineered to secrete human granulocyte‐macrophage colony stimulating factor generates potent anti‐tumor immunity in patients with metastatic melanoma. Proc Natl Acad Sci USA 1998; 95: 13141–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Salgia R, Lynch T, Skarin A et al . Vaccination with irradiated autologous tumor cells engineered to secrete granulocyte‐macrophage colony‐stimulating factor augments antitumor immunity in some patients with metastatic non‐small‐cell lung carcinoma. J Clin Oncol 2003; 21: 624–30. [DOI] [PubMed] [Google Scholar]
- 29. Nemunaitis J, Sterman D, Jablons D et al . Granulocyte‐macrophage colony‐stimulating factor gene‐modified autologous tumor vaccines in non‐small‐cell lung cancer. J Natl Cancer Inst 2004; 96: 326–31. [DOI] [PubMed] [Google Scholar]
- 30. Simons J, Mikhak B, Chang J‐F et al . Induction of immunity to prostate cancer antigens: results of a clinical trial of vaccination with irradiated autologous prostate tumor cells engineered to secrete granulocyte‐macrophage colony‐stimulating factor using ex vivo gene transfer. Cancer Res 1999; 59: 5160–8. [PubMed] [Google Scholar]
- 31. Tani K, Azuma M, Nakazaki Y et al . Phase I study of autologous tumor vaccines transduced with the GM‐CSF gene in four patients with stage IV renal cell cancer in Japan: clinical and immunological findings. Mol Ther 2004; 10: 799–816. [DOI] [PubMed] [Google Scholar]
- 32. Small EJ, Sacks N, Nemunaitis J et al . Granulocyte macrophage colony‐stimulating factor – secreting allogeneic cellular immunotherapy for hormone‐refractory prostate cancer. Clin Cancer Res 2007; 13: 3883–91. [DOI] [PubMed] [Google Scholar]
- 33. Jaffee E, Hruban R, Biedrzycki B et al . Novel allogeneic granulocyte‐macrophage colony‐stimulating factor‐secreting tumor vaccine for pancreatic cancer: a phase I trial of safety and immune activation. J Clin Oncol 2001; 19: 145–56. [DOI] [PubMed] [Google Scholar]
- 34. Luiten RM, Kueter EW, Mooi W et al . Immunogenicity, including vitiligo, and feasibility of vaccination with autologous GM‐CSF‐transduced tumor cells in metastatic melanoma patients. J Clin Oncol 2005; 23: 8978–91. [DOI] [PubMed] [Google Scholar]
- 35. Higano C, Saad F, Somer B et al . A phase III trial of GVAX immunotherapy for prostate cancer versus docetaxel plus prednisone in asymptomatic, castration‐resistant prostate cancer (CRPC). Proceedings of the 2009 Genitourinary Cancer Symposium (Abstract No. LBA‐150); American Society of Clinical Oncology (ASCO), Orlando, FL, USA. 26–28 Feb, 2009.
- 36. Small E, Demkow T, Gerritsen WR et al . A phase III trial of GVAX immunotherapy for prostate cancer in combination with docetaxel versus docetaxel plus prednisone in symptomatic, castration‐resistant prostate cancer (CRPC). Proceedings of the 2009 Genitourinary Cancer Symposium (Abstract No. 4); American Society of Clinical Oncology (ASCO), Orlando, FL, USA. 26–28 Feb, 2009.
- 37. Sotomayor EM, Fu YX, Lopez‐Cepero M et al . Role of tumor‐derived cytokines on the immune system of mice bearing a mammary adenocarcinoma. II. Down‐regulation of macrophage‐mediated cytotoxicity by tumor‐derived granulocyte‐macrophage colony‐stimulating factor. J Immunol 1991; 147: 2816–23. [PubMed] [Google Scholar]
- 38. Serafini P, Carbley R, Noonan KA, Tan G, Bronte V, Borrello I. High‐dose granulocyte‐macrophage colony‐stimulating factor‐producing vaccines impair the immune response through the recruitment of myeloid suppressor cells. Cancer Res 2004; 64: 6337–43. [DOI] [PubMed] [Google Scholar]
- 39. Filipazzi P, Valenti R, Huber V et al . Identification of a new subset of myeloid suppressor cells in peripheral blood of melanoma patients with modulation by a granulocyte‐macrophage colony‐stimulation factor‐based antitumor vaccine. J Clin Oncol 2007; 25: 2546–53. [DOI] [PubMed] [Google Scholar]
- 40. Jinushi M, Hodi FS, Dranoff G. Enhancing the clinical activity of granulocyte‐macrophage colony‐stimulating factor‐secreting tumor cell vaccines. Immunol Rev 2008; 222: 287–98. [DOI] [PubMed] [Google Scholar]
- 41. Dranoff G, Crawford AD, Sadelain M et al . Involvement of granulocyte‐macrophage colony‐stimulating factor in pulmonary homeostasis. Science 1994; 264: 713–6. [DOI] [PubMed] [Google Scholar]
- 42. Stanley E, Lieschke GJ, Grail D et al . Granulocyte/macrophage colony‐stimulating factor‐deficient mice show no major perturbation of hematopoiesis but develop a characteristic pulmonary pathology. Proc Natl Acad Sci USA 1994; 91: 5592–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Enzler T, Gillessen S, Manis JP et al . Deficiencies of GM‐CSF and interferon‐gamma link inflammation and cancer. J Exp Med 2003; 197: 1213–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Enzler T, Gillessen S, Dougan M et al . Functional deficiencies of granulocyte‐macrophage colony stimulating factor and interleukin‐3 contribute to insulitis and destruction of beta cells. Blood 2007; 110: 954–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jinushi M, Nakazaki Y, Dougan M, Carrasco DR, Mihm M, Dranoff G. MFG‐E8 mediated uptake of apoptotic cells by APCs links the pro‐ and anti‐inflammatory activities of GM‐CSF. J Clin Invest 2007; 117: 1902–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stubbs JD, Lekutis C, Singer KL et al . cDNA cloning of a mouse mammary epithelial cell surface protein reveals the existence of epidermal growth factor‐like domains linked to factor VIII‐like sequences. Proc Natl Acad Sci U S A 1990; 87: 8417–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fadok VA, Bratton DL, Henson PM. Phagocyte receptors for apoptotic cells: recognition, uptake, and consequences. J Clin Invest 2001; 108: 957–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol 2002; 2: 965–75. [DOI] [PubMed] [Google Scholar]
- 49. Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S. Identification of a factor that links apoptotic cells to phagocytes. Nature 2002; 417: 182–7. [DOI] [PubMed] [Google Scholar]
- 50. Hanayama R, Tanaka M, Miyasaka K et al . Autoimmune disease and impaired uptake of apoptotic cells in MFG‐E8‐deficient mice. Science 2004; 304: 1147–50. [DOI] [PubMed] [Google Scholar]
- 51. Jinushi M, Nakazaki Y, Carrasco DR et al . Milk fat globule EGF‐8 promotes melanoma progression through coordinated Akt and twist signaling in the tumor microenvironment. Cancer Res 2008; 68: 8889–98. [DOI] [PubMed] [Google Scholar]
- 52. Peggs KS, Segal NH, Allison JP. Targeting immunosupportive cancer therapies: accentuate the positive, eliminate the negative. Cancer Cell 2007; 12: 192–9. [DOI] [PubMed] [Google Scholar]
- 53. Jinushi M, Hodi FS, Dranoff G. Therapy‐induced antibodies to MHC class I chain‐related protein A antagonize immune suppression and stimulate antitumor cytotoxicity. Proc Natl Acad Sci U S A 2006; 103: 9190–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chang CJ, Chen YH, Huang KW et al . Combined GM‐CSF and IL‐12 gene therapy synergistically suppresses the growth of orthotopic liver tumors. Hepatology 2007; 45: 746–54. [DOI] [PubMed] [Google Scholar]
- 55. Wang Z, Qiu SJ, Ye SL, Tang ZY, Xiao X. Combined IL‐12 and GM‐CSF gene therapy for murine hepatocellular carcinoma. Cancer Gene Ther 2001; 8: 751–8. [DOI] [PubMed] [Google Scholar]
- 56. Kinoshita Y, Kono T, Yasumoto R et al . Antitumor effect on murine renal cell carcinoma by autologous tumor vaccines genetically modified with granulocyte‐macrophage colony‐stimulating factor and interleukin‐6 cells. J Immunother 2001; 24: 205–11. [PubMed] [Google Scholar]
- 57. Inoue H, Iga M, Xin M et al . TARC and RANTES enhance antitumor immunity induced by the GM‐CSF‐transduced tumor vaccine in a mouse tumor model. Cancer Immunol Immunother 2008; 57: 1399–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sumimoto H, Tani K, Nakazaki Y et al . Superiority of interleukin‐12‐transduced murine lung cancer cells to GM‐CSF or B7‐1 (CD80) transfectants for therapeutic antitumor immunity in syngeneic immunocompetent mice. Cancer Gene Ther 1998; 5: 29–37. [PubMed] [Google Scholar]
- 59. Nakazaki Y, Tani K, Lin ZT et al . Vaccine effect of granulocyte‐macrophage colony‐stimulating factor or CD80 gene‐transduced murine hematopoietic tumor cells and their cooperative enhancement of antitumor immunity. Gene Ther 1998; 10: 1355–62. [DOI] [PubMed] [Google Scholar]
- 60. Ren SP, Wu CT, Huang WR et al . Adenoviral‐mediated transfer of human wild‐type p53, GM‐CSF and B7–1 genes results in growth suppression and autologous anti‐tumor cytotoxicity of multiple myeloma cells in vitro . Cancer Immunol Immunother 2006; 55: 375–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Trinchieri G. Interleukin‐12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol 2003; 3: 133–46. [DOI] [PubMed] [Google Scholar]
- 62. Colombo MP, Trinchieri G. Interleukin‐12 in anti‐tumor immunity and immunotherapy. Cytokine Growth Factor Rev 2002; 13: 155–68. [DOI] [PubMed] [Google Scholar]
- 63. Voest EE, Kenyon BM, O’Reilly MS, Truitt G, D’Amato RJ, Folkman J. Inhibition of angiogenesis in vivo by interleukin 12. J Natl Cancer Inst 1995; 87: 581–6. [DOI] [PubMed] [Google Scholar]
- 64. Brunda MJ, Luistro L, Warrier RR et al . Antitumor and antimetastatic activity of interleukin 12 against murine tumors. J Exp Med 1993; 178: 1223–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tahara H, Ze H, Storkus WJ et al . Fibroblasts genetically engineered to secrete interleukin‐12 can suppress tumor growth in vivo and induce antitumor immunity to a murine melanoma. Cancer Res 1994; 54: 182–9. [PubMed] [Google Scholar]
- 66. Nastala CL, Edington HD, McKinney TG et al . Recombinant IL‐12 administration induces tumor regression in association with IFN‐γ production. J Immunol 1994; 153: 1697–706. [PubMed] [Google Scholar]
- 67. Rook AH, Wood GS, Yoo EK et al . Interleukin‐12 therapy of cutaneous T‐cell lymphoma induces lesion regression and cytotoxic T‐cell responses. Blood 1999; 94: 902–8. [PubMed] [Google Scholar]
- 68. Younes A, Pro B, Robertson MJ et al . Phase II clinical trial of interleukin‐12 in patients with relapsed and refractory non‐Hodgkin's lymphoma and Hodgkin's disease. Clin Cancer Res 2004; 10: 5432–8. [DOI] [PubMed] [Google Scholar]
- 69. Gollob JA, Mier JW, Veenstra K et al . Phase I trial of twice‐weekly intravenous interleukin 12 in patients with metastatic renal cell cancer or malignant melanoma: ability to maintain IFN‐γ induction is associated with clinical response. Clin Cancer Res 2000; 6: 1678–92. [PubMed] [Google Scholar]
- 70. Van Herpen CM, Looman M, Zonneveld M et al . Intratumoral administration of recombinant human interleukin 12 in head and neck squamous cell carcinoma patients elicits a T‐helper 1 profile in the locoregional lymph nodes. Clin Cancer Res 2004; 10: 2626–35. [DOI] [PubMed] [Google Scholar]
- 71. Atkins MB, Robertson MJ, Gordon M et al . Phase I evaluation of intravenous recombinant human interleukin 12 in patients with advanced malignancies. Clin Cancer Res 1997; 3: 409–1. [PubMed] [Google Scholar]
- 72. Robertson MJ, Cameron C, Atkins MB et al . Immunological effects of interleukin 12 administered by bolus intravenous injection to patients with cancer. Clin Cancer Res 1999; 5: 9–16. [PubMed] [Google Scholar]
- 73. Motzer RJ, Rakhit A, Schwartz LH et al . Phase I trial of subcutaneous recombinant human interleukin‐12 in patients with advanced renal cell carcinoma. Clin Cancer Res 1998; 4: 1183–91. [PubMed] [Google Scholar]
- 74. Kang WK, Park C, Yoon HL et al . Interleukin 12 gene therapy of cancer by peritumoral injection of transduced autologous fibroblasts: outcome of a phase I study. Hum Gene Ther 2001; 12: 671–84. [DOI] [PubMed] [Google Scholar]
- 75. Sun Y, Jurgovsky K, Moller P. Vaccination with IL‐12 gene‐modified autologous melanoma cells: preclinical results and a first clinical phase I study. Gene Ther 1998; 5: 481–90. [DOI] [PubMed] [Google Scholar]
- 76. Sangro B, Melero I, Qian C, Prieto J. Gene therapy of cancer based on interleukin 12. Curr Gene Ther 2005; 5: 573–81. [DOI] [PubMed] [Google Scholar]
- 77. Heinzerling L, Burg G, Dummer R et al . Intratumoral injection of DNA encoding human interleukin‐12 into patients with metastatic melanoma: clinical efficacy. Hum Gene Ther 2005; 16: 35–48. [DOI] [PubMed] [Google Scholar]
- 78. Triozzi PI, Strong TV, Bucy RP et al . Intratumoral administration of a recombinant Canarypox virus expressing interleukin 12 in patients with metastatic melanoma. Hum Gene Ther 2005; 16: 91–100. [DOI] [PubMed] [Google Scholar]
- 79. Daud AI, DeConti RC, Andrews S et al . Phase I trial of interleukin‐12 plasmid electroporation in patients with metastatic melanoma. J Clin Oncol 2008; 26: 5896–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lee P, Wang F, Kuniyoshi J et al . Effects of interleukin‐12 on the immune response to a multipeptide vaccine for resected metastatic melanoma. J Clin Oncol 2001; 19: 3836–47. [DOI] [PubMed] [Google Scholar]
- 81. Ribas A, Butterfield LH, Glaspy JA, Economou LS. Cancer immunotherapy using gene‐modified dendritic cells. Curr Gene Ther 2002; 2: 57–78. [DOI] [PubMed] [Google Scholar]
- 82. Tatsumi T, Takehara T, Yamaguchi S et al . Injection of IL‐12‐transduced dendritic cells into mouse liver tumor lesions activates both innate and acquired immunity. Gene Ther 2007; 14: 863–71. [DOI] [PubMed] [Google Scholar]
- 83. Mazzolini G, Alfaro C, Sangro B et al . Intratumoral injection of dendritic cells engineered to secrete interleukin‐12 by recombinant adenovirus in patients with metastatic gastrointestinal carcinomas. J Clin Oncol 2005; 23: 999–1010. [DOI] [PubMed] [Google Scholar]
- 84. Oppmann B, Lesley R, Blom B et al . Novel p19 protein engages IL‐12p40 to form a cytokine, IL‐23, with biological activities similar as well as distinct from IL‐12. Immunity 2000; 13: 715–25. [DOI] [PubMed] [Google Scholar]
- 85. Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity 2006; 24: 677–88. [DOI] [PubMed] [Google Scholar]
- 86. Langowski JL, Zhang X, Wu L et al . IL‐23 promotes tumour incidence and growth. Nature 2006; 442: 461–5. [DOI] [PubMed] [Google Scholar]
- 87. Lo CH, Lee SC, Wu PY et al . Antitumor and antimetastatic activity of IL‐23. J Immnuol 2003; 171: 600–7. [DOI] [PubMed] [Google Scholar]
- 88. Kaiga T, Sato M, Kaneda H, Iwakura Y, Takayama T, Tahara H. Systemic administration of IL‐23 induces potent antitumor immunity primarily mediated through Th1‐type response in association with the endogenously expressed IL‐12. J Immunol 2007; 178: 7571–80. [DOI] [PubMed] [Google Scholar]
- 89. Waldmann TA. The biology of interleukin‐2 and interleukin‐15: implications for cancer therapy and vaccine design. Nat Rev Immunol 2006; 6: 595–601. [DOI] [PubMed] [Google Scholar]
- 90. Spolski R, Leonard WJ. Interleukin‐21: basic biology and implications for cancer and autoimmunity. Annu Rev Immnuol 2008; 26: 57–79. [DOI] [PubMed] [Google Scholar]
- 91. Melero I, Hervas‐Stubbs S, Glennie M, Pardoll DM, Chen L. Immunostimulatory monoclonal antibodies for cancer therapy. Nat Rev Cancer 2007; 7: 95–106. [DOI] [PubMed] [Google Scholar]
- 92. Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immnuol 2008; 8: 59–73. [DOI] [PubMed] [Google Scholar]
- 93. Heemskerk B, Liu K, Dudley ME et al . Adoptive cell therapy for patients with melanoma, using tumor‐infiltrating lymphocytes genetically engineered to secrete interleukin‐2. Hum Gene Ther 2008; 5: 496–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Hofbauer GF, Baur T, Bonnet MC et al . Clinical phase I intratumoral administration of two recombinant ALVAC canarypox viruses expressing human granulocyte‐macrophage colony‐stimulating factor or interleukin‐2: the transgene determines the composition of the inflammatory infiltrate. Melanoma Res 2008; 18: 104–11. [DOI] [PubMed] [Google Scholar]
- 95. Colombo F, Barzon L, Franchin E et al . Combined HSV‐TK/IL‐2 gene therapy in patients with recurrent glioblastoma multiforme: biological and clinical results. Cancer Gene Ther 2005; 12: 835–48. [DOI] [PubMed] [Google Scholar]
- 96. Trudel S, Trachtenberg J, Toi A et al . A phase I trial of adenovector‐mediated delivery of interleukin‐2 (AdIL‐2) in high‐risk localized prostate cancer. Cancer Gene Ther 2003; 10: 755–63. [DOI] [PubMed] [Google Scholar]
- 97. Rochlitz C, Dreno B, Jantscheff P et al . Immunotherapy of metastatic melanoma by intratumoral injections of Vero cells producing human IL‐2: phase II randomized study comparing two dose levels. Cancer Gene Ther 2002; 9: 289–95. [DOI] [PubMed] [Google Scholar]
- 98. Belldegrun A, Tso CL, Zisman A et al . Interleukin 2 gene therapy for prostate cancer: phase I clinical trial and basic biology. Hum Gene Ther 2001; 12: 883–92. [DOI] [PubMed] [Google Scholar]
- 99. Scholl SM, Balloul JM, Le Goc G et al . Recombinant vaccinia virus encoding human MUC1 and IL2 as immunotherapy in patients with breast cancer. J Immunother 2000; 23: 570–80. [DOI] [PubMed] [Google Scholar]
- 100. Schmidt‐Wolf IG, Finke S, Trojaneck B et al . Phase I clinical study applying autologous immunological effector cells transfected with the interleukin‐2 gene in patients with metastatic renal cancer, colorectal cancer and lymphoma. Br J Cancer 1999; 81: 1009–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Palmer K, Moore J, Everard M et al . Gene therapy with autologous, interleukin 2‐secreting tumor cells in patients with malignant melanoma. Hum Gene Ther 1999; 10: 1261–8. [DOI] [PubMed] [Google Scholar]
- 102. Nemunaitis J, Bohart C, Fong T et al . Phase I trial of retroviral vector‐mediated interferon (IFN)‐gamma gene transfer into autologous tumor cells in patients with metastatic melanoma. Cancer Gene Ther 1998; 5: 292–300. [PubMed] [Google Scholar]