

Abstract
Acquisition of resistance to tamoxifen is a critical therapeutic problem in breast cancer patients. Epithelial–mesenchymal transition (EMT), where cells undergo a developmental switch from a polarized epithelial phenotype to a highly motile mesenchymal phenotype, is associated with invasion and motility of cancer cells. Here, we found that tamoxifen‐resistant (TAMR)‐MCF‐7 cells had undergone EMT, as evidenced by mesenchymal‐like cell shape, downregulation of basal E‐cadherin expression, and overexpression of N‐cadherin and vimentin, as well as increased Snail transcriptional activity and protein expression. Given the roles of glycogen synthase kinase (GSK)‐3β and nuclear factor (NF)‐κB in Snail‐mediated E‐cadherin deregulation during EMT, we examined the role of these signaling pathways in the EMT of TAMR‐MCF‐7 cells. Both Ser9‐phosphorylated GSK‐3β (inactive form) and NF‐κB reporter activity were increased in TAMR‐MCF‐7 cells, as was activation of the phosphatase and tensin homolog depleted on chromosome ten (PTEN)–phosphoinositide 3 (PI3)‐kinase–Akt pathway. Pin1, a peptidyl‐prolyl isomerase, was overexpressed in TAMR‐MCF‐7 cells, and Snail transcription and the expression of EMT markers could be decreased by Pin1 siRNA treatment. These results imply that Pin1 overexpression in TAMR‐MCF‐7 cells is involved in the EMT process via PTEN–PI3‐kinase–Akt–GSK‐3β and/or GSK‐3β–NF‐κB‐dependent Snail activation, and suggest the potential involvement of Pin1 in EMT during breast cancer development. (Cancer Sci 2009; 100: 1834–1841)
Breast cancer is the most common malignancy in Western women. Hormone‐dependent control of growth is a characteristic of breast cancer. Ovarian steroid hormones, including estrogen, are essential for both mammary gland development and breast carcinoma formation.( 1 , 2 ) Hence, the administration of anti‐estrogens to reduce breast tumor growth has played a key role in the endocrine therapy of breast cancer. Tamoxifen (TAM), a non‐steroidal anti‐estrogen, is the most widely used anti‐estrogen in estrogen receptor‐positive breast cancer patients.( 3 ) Despite an initial response to TAM, the majority of patients will ultimately relapse and present with disease progression. Therefore, resistance to TAM is a major challenge in the management of breast cancer patients.( 4 , 5 ) To mimic this condition, we and others have established an MCF‐7‐derived TAM‐resistant cell line (TAMR‐MCF‐7 cells) by long‐term (>9 months) culture of MCF‐7 cells with 4‐hydroxytamoxifen.( 6 , 7 )
Epithelial tumors metastasize by initially invading the adjacent tissues, a process involving the loss of their cell–cell adhesions and the acquisition of migratory capabilities. These processes coincide with phenotype changes associated with epithelial–mesenchymal transitions (EMT), similar to those that take place during certain steps of embryonic development.( 8 ) EMT is involved in the efficient invasion and motility of cancer cells, because migration of single cancer cells is elicited by using either mesenchymal or amoeboid phenotypes.( 9 , 10 ) The invasive and metastatic phenotype is associated with downregulation of E‐cadherin expression.( 11 ) Several mechanisms have been implicated in the regulation of E‐cadherin expression during tumor progression, including genetic, epigenetic, and transcriptional changes.( 12 , 13 ) Snail transcription factor is a direct repressor of E‐cadherin expression in epithelial cells; the expression of Snail induces a full EMT and increases migration and invasion in different physiological and pathological situations.( 14 , 15 , 16 ) Moreover, Snail expression occurs in different invasive carcinoma and melanoma cell lines and, importantly, in invasive regions of squamous cell carcinomas and dedifferentiated ductal breast cancer carcinomas and hepatocarcinomas.( 16 , 17 ) Upon acquisition of tamoxifen resistance, breast cancer cells impart greater in vitro metastatic ability to these cells, as demonstrated by an increase in their motile and invasive behavior.( 18 ) Although Hiscox et al. showed that TAM resistance promotes EMT‐like behavior,( 19 ) the molecular mechanistic basis focusing on the cellular signaling pathway has not been fully studied.
Overexpression of the peptidyl‐prolyl isomerase Pin1 is a prognostic marker in several cancer types, including breast and prostate cancers.( 20 , 21 ) Activation of Pin1 affects oncogenic pathways and/or contributes to the transformation of breast epithelial cells. In the present study, we found that Pin1 is overexpressed in TAMR‐MCF‐7 cells and we used Pin1 siRNA to clarify its contribution to EMT. Given the important roles of the glycogen synthase kinase (GSK)‐3β–Snail and nuclear factor (NF)‐κB–Snail axis in E‐cadherin deregulation during EMT, we also investigated the role of GSK‐3β and NF‐κB–Snail signaling in EMT induction in TAMR‐MCF‐7 cells. Herein, we report for the first time Pin1 involvement in EMT induction of TAMR‐MCF‐7 cells through changes in the activities of the phosphatase and tensin homolog depleted on chromosome ten (PTEN)–PI3‐kinase–Akt and/or NF‐κB pathways.
Materials and Methods
Materials. Anti‐E‐cadherin, anti‐N‐cadherin, and anti‐snail antibodies were supplied by BD Transduction (San Jose, CA, USA) and Abcam (Cambridge Science Park, UK), respectively. Anti‐vimentin, anti‐Pin1, and anti‐GSK‐3β antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies for phosphorylated Akt (serine‐473), Akt, and serine‐9 phosphorylated GSK‐3β were purchased from Cell Signaling Technology (Beverly, MA, USA). Horseradish peroxidase‐conjugated donkey anti‐rabbit IgG, anti‐goat IgG, and alkaline phosphatase‐conjugated donkey anti‐mouse IgG were purchased from Jackson Immunoresearch Laboratories (West Grove, PA, USA). Anti‐actin antibody and most of the reagents used for molecular studies were obtained from Sigma (St Louis, MO, USA). siRNA targeting human Pin1 and control siRNA were purchased from Ambion (Austin, TX, USA). Snail‐Luc plasmids and GSK‐3β overexpression vector were kindly donated by Dr K. Y. Lee (Chonnam National University, Gwangju, Korea).
Cell culture. MCF‐7 cells were cultured at 37°C in 5% CO2/95% air in DMEM containing 10% FBS, 100 units/mL penicillin, and 100 mg/mL streptomycin. TAMR‐MCF‐7 cells were established using the methodology reported elsewhere.( 7 ) Briefly, MCF‐7 cells were washed with PBS, and the culture medium was changed to phenol‐red‐free DMEM containing 10% charcoal‐stripped, steroid‐depleted FBS (Hyclone, Logan, UT, USA) and 4‐hydroxytamoxifen (0.1 µM). The cells were continuously exposed to this treatment regimen for 2 weeks and the concentration of 4‐hydroxytamoxifen was gradually increased to 3 µM over a 9‐month period. Initially the cell growth rates were reduced. However, after exposure to the medium for 9 months, the rate of cell growth gradually increased, showing the establishment of a TAM‐resistant cell line.( 6 )
Immunoblot analysis. After washing with sterile PBS, the MCF‐7 or TAMR‐MCF‐7 cells were lysed in EBC lysis buffer containing 20 mM Tris‐HCl (pH 7.5), 1% Triton X‐100, 137 mM sodium chloride, 10% glycerol, 2 mM EDTA, 1 mM sodium orthovanadate, 25 mM β‐glycerophosphate, 2 mM sodium pyrophosphate, 1 mM phenylmethylsulfonylfluoride, and 1 µg/mL leupeptin. The cell lysates were centrifuged at 10 000 g for 10 min to remove the debris, and the proteins were fractionated using a 10% separating gel. The fractionated proteins were then transferred electrophoretically to nitrocellulose paper, and the proteins were immunoblotted. Horseradish peroxidase‐ or alkaline phosphatase‐conjugated anti‐IgG antibodies were used as the secondary antibodies. The nitrocellulose papers were developed using 5‐bromo‐4‐chloro‐3‐indolylphosphate and 4‐nitroblue tetrazolium or an ECL chemiluminescence system. For ECL chemiluminescence detection, the LAS3000‐mini system (Fujifilm, Tokyo, Japan) was used.
Reporter gene assay. Promoter activity was determined using a dual‐luciferase reporter assay system (Promega, Madison, WI, USA). Briefly, cells (3 × 105 cells/well) were replated in 12‐well plates overnight and transiently transfected with the Snail‐Luc plasmids containing –1.5 kb or –2.5 kb human snail promoter and phRL‐SV plasmid (hRenilla luciferase expression for normalization) (Promega) or the NF‐κB minimal reporter plasmid and phRL‐SV plasmid using Hilymax reagent (Dojindo Molecular Technologies, Gaithersburg, MD, USA). The cells were then incubated in the culture medium without serum for 18 h, and the firefly and Renilla luciferase activities in the cell lysates were measured using a luminometer (LB941; Berthold Technologies, Bad Wildbad, Germany). The relative luciferase activities were calculated by normalizing the promoter‐driven firefly luciferase activity versus Renilla luciferase.
Establishment of stable Pin1‐overexpressing MCF‐7 cells. MCF‐7 cells stably overexpressing Pin1 (Pin1‐MCF‐7 cells) were established using a murine stem cell virus (MSCV)‐GFP retrovirus system. Briefly, Pin1 cDNA was subcloned into the MSCV‐GFP retroviral vector, and phoenix cells (a packaging cell line) were transfected with either the MSCV‐GFP (control) or the MSCV‐Pin1‐GFP (Pin1 overexpression) plasmid. Supernatants containing amphotrophic replication‐incompetent retroviruses were collected and stored at –80°C until required. MCF‐7 cells at 30% confluence were multiply infected (12 times) with retrovirus particles. Intensities of infection were monitored by GFP fluorescence and western blot analysis using a Pin1 antibody.
Statistical analysis. Paired Student's t‐test was used to examine the significant intergroup differences. Statistical significance was accepted at either P < 0.05 or P < 0.01.
Results
Snail‐mediated EMT induction in TAMR‐MCF‐7 cells. TAMR‐MCF‐7 cells, which have acquired resistance to TAM, display enhanced invasive capacity in vitro ( 18 ) and a morphology distinct from the parent MCF‐7 cells. Whereas MCF‐7 cells show highly organized cell–cell adhesion and cell contact, TAMR‐MCF‐7 cells had a refractive and elongated appearance, with cell scattering and loss of cell–cell contacts (Fig. 1a). The cobblestone‐like morphology of MCF‐7 cells at confluence was changed in TAMR‐MCF‐7 cells to a spindle‐like fibroblastic morphology (Fig. 1a). Loss of the epithelial adherence junction protein, E‐cadherin, and upregulation of mesenchymal proteins such as N‐cadherin and vimentin are biochemical markers of EMT. In fact, the loss of E‐cadherin is a common marker of EMT in mammary epithelial cells and is also associated with an aggressive phenotype with poor prognosis during mammary carcinogenesis.( 22 ) The levels of total cellular E‐cadherin were significantly reduced in TAMR‐MCF‐7 compared to MCF‐7 cells (Fig. 1b). Conversely, the expression levels of N‐cadherin and vimentin were elevated in TAMR‐MCF‐7 cells (Fig. 1b). Therefore, both the morphological and molecular changes in the TAMR‐MCF‐7 cells demonstrated that these cells had undergone EMT.
Figure 1.
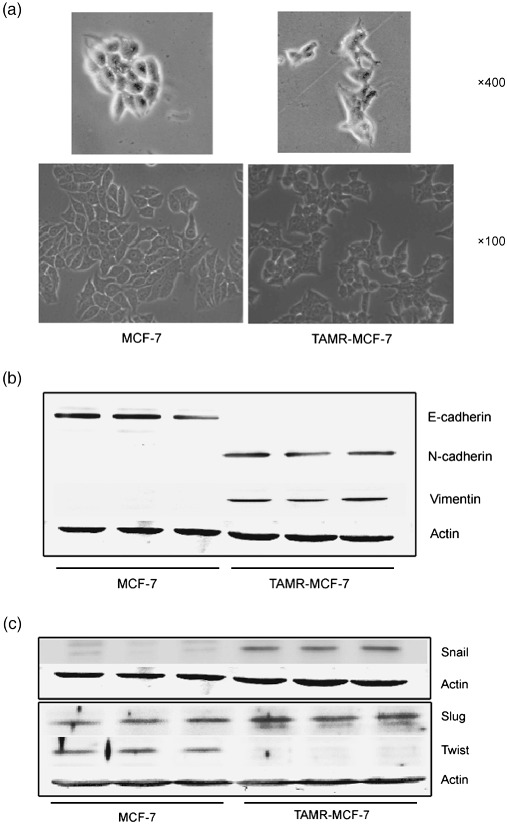
Snail‐mediated epithelial–mesenchymal transition (EMT) induction in tamoxifen‐resistant (TAMR)‐MCF‐7 cells. (a) Cell morphology of MCF‐7 and TAMR‐MCF‐7 cells. (b) Immunoblot analysis of E‐cadherin, N‐cadherin, and vimentin. Equal loading of proteins was verified by actin immunoblot. (c) Immunoblot analysis of Snail, Slug, and Twist. Representative immunoblots show the protein levels of Snail, Slug, and Twist in both MCF‐7 and TAMR‐MCF‐7 cells deprived of serum for 18 h.
Snail, a zinc finger transcription repressor, inhibits E‐cadherin transcription and its increase is closely related with EMT during cancer cell growth and invasiveness.( 23 , 24 ) Snail is absent in epithelial cells but expressed in tumors, and its expression correlates with tumor grade.( 25 ) In comparison to MCF‐7 cells, Snail levels were increased in TAMR‐MCF‐7 cells (Fig. 1c). We also determined the expression levels of Slug and Twist. Slug expression was not distinctly enhanced in TAMR‐MCF‐7 cells (Fig. 1c). Twist expression in TAMR‐MCF‐7 cells was rather lower than that in control MCF‐7 cells (Fig. 1c). Hence, Snail upregulation could be considered as a critical event to induce EMT in TAMR‐MCF‐7 cells.
Role of GSK‐3β inactivation in Snail upregulation. In order to clarify whether Snail upregulation in TAMR‐MCF‐7 cells is associated with transcriptional activation of the snail gene, we determined snail promoter activities using –1.5 kb or –2.5 kb Snail‐Luc plasmids. Both of the reporter activities were significantly increased in TAMR‐MCF‐7 cells (Fig. 2a).
Figure 2.
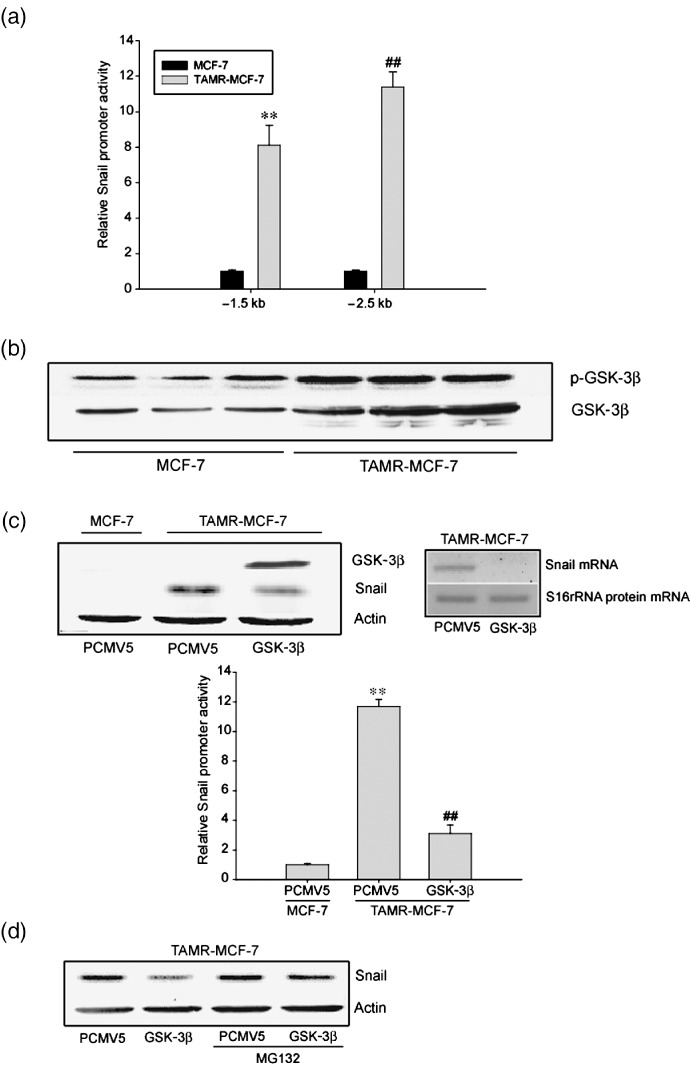
Role of glycogen synthase kinase (GSK)‐3β inactivation in Snail upregulation. (a) Basal reporter activity of Snail promoters (Snail‐Luc) in MCF‐7 and tamoxifen‐resistant (TAMR)‐MCF‐7 cells. Each cell type was transiently transfected with –1.5kb or –2.5kb Snail‐Luc plasmid. Dual luciferase reporter assays were carried out on the lysed cells cotransfected with Snail‐Luc plasmids (firefly luciferase) and phRL‐SV (hRenilla luciferase) (a ratio of 100:1) 18 h after transfection. Reporter gene activation was calculated as a relative ratio of firefly luciferase to hRenilla luciferase activity. Data represent means ± SD with four different samples (significant vs MCF‐7 cells, **P < 0.01, ## P < 0.01). (b) Immunoblot analysis of GSK‐3β and phosphorylated (p)‐GSK‐3β. Representative immunoblots show each protein in both MCF‐7 and TAMR‐MCF‐7 cells serum‐deprived for 18 h. (c) Inhibition of Snail transactivation by GSK‐3β overexpression vector. Left upper panel: Levels of GSK‐3β and Snail were determined by immunoblotting in TAMR‐MCF‐7 cells transfected with GSK‐3β overexpression vector (1 µg/well) or mock vector PCMV5 (1 µg/well). Right upper panel: Snail mRNA expression was determined by RT‐PCR analysis in TAMR‐MCF‐7 cells transfected with GSK‐3β overexpression vector or PCMV5 (1 µg/well). Lower panel: MCF‐7 and TAMR‐MCF‐7 cells were cotransfected with Snail –2.5kb reporter plasmid in combination with GSK‐3β overexpression vector or PCMV5 (1 µg/well). Data represents means ± SD with four different samples (significant vs the control, **P < 0.01; significant vs PCMV5‐transfected group, ## P < 0.01; control level = 1). (d) Effect of MG‐132, a proteasome inhibitor, on the expression of Snail. GSK‐3β overexpression vector‐ or PCMV5 (1 µg/well)‐transfected TAMR‐MCF‐7 cells were incubated with or without MG‐132 (1 µM) for 24 h and total cell lysates were subjected to immunoblotting.
GSK‐3β is a ubiquitously expressed serine/threonine kinase initially identified as a kinase phosphorylating glycogen synthase. Snail protein levels and its repressor functions are regulated by GSK‐3β during EMT.( 26 , 27 ) In resting cells, GSK‐3β is constitutively active as a mitogenic signaling pathway; Akt inactivates the enzyme through Ser‐9 phosphorylation.( 28 , 29 ) The levels of phosphorylated (inactive) GSK‐3β in TAMR‐MCF‐7 cells were significantly higher than in MCF‐7 cells (Fig. 2b). To test whether GSK‐3β inactivation regulates Snail in TAMR cells, we determined the promoter activity and protein/mRNA levels of Snail in TAMR‐MCF‐7 cells cotransfected with a wild‐type GSK‐3β expression plasmid. GSK‐3β overexpression significantly decreased Snail reporter activity (Fig. 2c, bottom) and protein/mRNA expression (Fig. 2c, upper), which supports the notion that GSK‐3β inactivation causes snail upregulation in TAMR‐MCF‐7 cells. Proteasomal degradation of Snail can function as a regulatory step of the protein stability. After MG‐132 treatment, GSK‐3β‐mediated Snail reduction was partly reversed, indicating that GSK‐3β‐dependent proteasomal degradation of Snail is also downregulated in TAMR‐MCF‐7 cells (Fig. 2d).
GSK‐3β‐dependent snail increase is associated with the deregulated PTEN–PI3‐kinase–Akt signaling in TAMR‐MCF7 cells. The PTEN–PI3‐kinase–Akt pathway is involved in carcinogenesis.( 30 , 31 ) Oncogenic transformation can be associated with the abnormal activation of various signaling pathways that induce EMT; abnormal PI3‐kinase and Akt activation is frequent in various epithelial cell tumors( 32 , 33 ) and their clinical features are consistent with Akt playing roles in modification of cell morphology, tumorigenicity, cell motility, and invasiveness.( 34 ) Akt is also an upstream kinase for controlling GSK‐3β activity, so we compared Akt activity in both MCF‐7 and TAMR‐MCF‐7 cells. Akt phosphorylation was increased in TAMR‐MCF‐7 cells (Fig. 3a), but total Akt levels were lower, consistent with our previous report.( 6 ) An increase in basal Akt phosphorylation could indicate deregulation of an Akt‐targeting phosphatase. PTEN is an upstream negative regulator of Akt. PTEN levels were lower in TAMR‐MCF‐7 cells (Fig. 3b). To confirm whether defects in PTEN‐mediated Akt inactivation upregulate Snail, we measured Snail protein and the gene promoter activity after transfection of TAMR‐MCF‐7 cells with PTEN or myc‐p85 (a dominant negative mutant form of PI3‐kinase) overexpression vectors. Inhibition of PI3‐kinase by PTEN or myc‐p85 decreased both the Snail protein expression and the snail promoter activity (Fig. 3c,d). These results suggest that PTEN downregulation and subsequent activation of PI3‐kinase and Akt regulate EMT in TAMR‐MCF‐7 cells.
Figure 3.
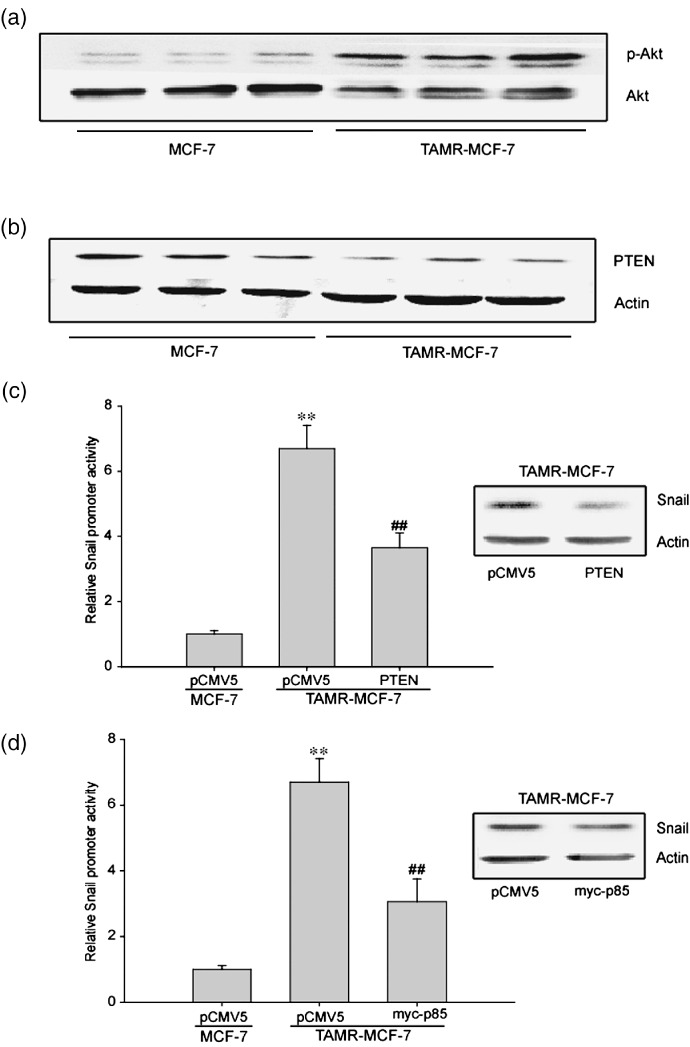
Role of phosphatase and tensin homolog depleted on chromosome ten (PTEN)–PI3‐kinase–Akt in Snail upregulation of tamoxifen‐resistant (TAMR)‐MCF‐7 cells. Immunoblot analyses of (a) phosphorylated (p)‐Akt and (b) PTEN. Equal loading of proteins was verified by actin immunoblot. (c) Inhibition of Snail transactivation by PTEN overexpression vector. Left panel: Reporter gene assay. MCF‐7 and TAMR‐MCF‐7 cells were cotransfected with –2.5 kb Snail‐Luc reporter plasmid in combination with PTEN overexpression vector (0.1 µg/well) or mock vector PCMV5 (0.1 µg/well). Data represents means ± SD with four different samples (significant vs the MCF‐7 cells, **P < 0.01; significant vs PCMV5‐transfected TAMR‐MCF‐7 cells, ## P < 0.01). Right panel: Immunoblotting of Snail in TAMR‐MCF‐7 cells transfected with PCMV5 or PTEN overexpression vector. (d) Inhibition of Snail transactivation by myc‐p85 overexpression vector. Left panel: Reporter gene assay. MCF‐7 and TAMR‐MCF‐7 cells were cotransfected with –2.5 kb Snail‐Luc reporter plasmid in combination with myc‐p85 overexpression vector (0.1 µg/well) or control mock vector PCMV5 (0.1 µg/well). Data represent means ± SD with four different samples (significant vs the MCF‐7 cells, **P < 0.01; significant vs PCMV5‐transfected TAMR‐MCF‐7 cells, ## P < 0.01). Right panel: Immunoblotting of Snail in TAMR‐MCF‐7 cells transfected with PCMV5 or myc‐p85 overexpression vector.
Role of Pin1 overexpression in EMT of TAMR‐MCF7 cells. Pin1, a peptidyl‐prolyl isomerase, isomerizes the peptide bond pSer/Thr‐Pro to regulate cell cycle progression and other functions.( 35 , 36 ) Overexpression of Pin1 also contributes to cell transformation( 37 , 38 ) and nuclear translocation of β‐catenin, which is frequently observed in EMT.( 39 ) Therefore, we hypothesized that sustained Pin1 activation was associated with EMT of TAM‐resistant breast cancer cells. Interestingly, we observed a constitutive increase in Pin1 levels in TAMR‐MCF‐7 cells compared to MCF‐7 cells (Fig. 4a).
Figure 4.
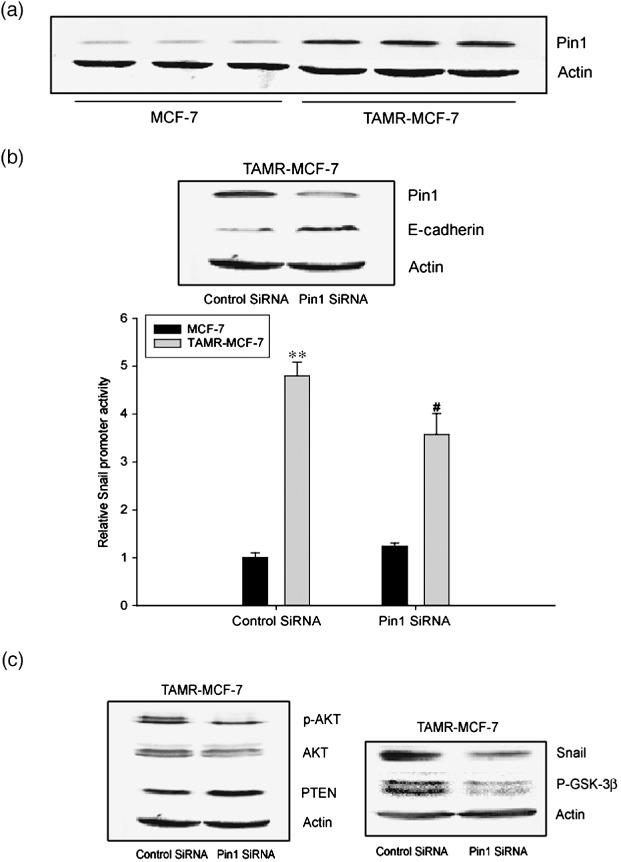
Role of Pin1 overexpression in epithelial–mesenchymal transition of tamoxifen‐resistant (TAMR)‐MCF‐7 cells. (a) Immunoblot analysis of Pin1. A representative immunoblot shows Pin1 protein in both MCF‐7 and TAMR‐MCF‐7 cells serum‐deprived for 18 h. Equal loading of proteins was verified by actin immunoblot. (b) Effects of Pin1 siRNA on the E‐cadherin expression and Snail transcription activity in TAMR‐MCF‐7 cells. Upper panel: Levels of Pin1 and E‐cadherin were determined by immunoblotting in TAMR‐MCF‐7 cells transfected with Pin1 siRNA (60 pmol) or control siRNA. Lower panel: TAMR‐MCF‐7 cells were cotransfected with –2.5 kb Snail‐Luc reporter plasmid in combination with Pin1 siRNA (20 pmol) or control siRNA. Data represent means ± SD with four different samples (significant vs the control, **P < 0.01; significant vs the control siRNA‐transfected group, # P < 0.05). (c) Decrease in the expression levels of phosphorylated (p)‐Akt, p‐glycogen synthase kinase (GSK)‐3β, Snail and increase in phosphatase and tensin homolog depleted on chromosome ten (PTEN) level by Pin1 siRNA introduction. Each protein expression was determined by immunoblotting in TAMR‐MCF‐7 cells transfected with Pin1 siRNA (60 pmol) or control siRNA.
To be certain that Pin1 overexpression in TAMR‐MCF‐7 cells was involved in EMT, we then evaluated snail promoter activity and E‐cadherin expression after exposure of TAMR‐MCF‐7 cells to control or Pin1 siRNA. Pin1 siRNA decreased Pin1 protein levels (Fig. 4b, upper panel). The increased Snail protein and the gene promoter activity were partially, but significantly, inhibited and E‐cadherin expression was also restored by Pin1 siRNA treatment (Fig. 4b,c). In addition, Pin1 siRNA reduced the phosphorylation of Akt, but increased PTEN expression in TAMR‐MCF‐7 cells (Fig. 4c). Basal Akt expression was also slightly reduced in Pin1 siRNA‐treated cells. We further determined the phosphorylation levels of GSK‐3β after introduction of Pin1 siRNA in TAMR‐MCF‐7 cells. As expected, Pin1 siRNA reduced GSK‐3β phosphorylation in TAMR‐MCF‐7 cells (Fig. 4c). However, EMT morphology was not distinctly reversed by Pin1 siRNA introduction for 24 h (data not shown). It may result from either incomplete efficiency of Pin1 siRNA or insufficient incubation time. Therefore, Pin1 affects PTEN expression and the subsequent PI3‐kinase–Akt‐dependent GSK‐3β inactivation is involved in EMT of TAMR‐MCF‐7 cells.
NF‐κB involvement in Pin1‐mediated EMT in TAMR‐MCF‐7 cells. NF‐κB is an essential regulator of EMT in breast cancer cells.( 40 , 41 ) NF‐κB can bind to the promoter region of the snail gene and increase its transcriptional activity.( 40 , 42 ) Because GSK‐3β activity and NF‐κB activity are related,( 43 ) we measured NF‐κB reporter activities in both cell types. The basal NF‐κB activity was 6.5‐fold enhanced in TAMR‐MCF‐7 compared to MCF‐7 cells (Fig. 5a, left panel). Moreover, the enhanced NF‐κB reporter activity in TAMR‐MCF‐7 cells was significantly reduced by GSK‐3β overexpression (Fig. 5a, right panel), which confirms that NF‐κB activity is partly dependent on GSK‐3β in TAMR‐MCF‐7 cells. Treatment of TAMR‐MCF‐7 cells for 24 h with an NF‐κB inhibitor, N‐α‐tosyl‐l‐phenylalanine chloromethyl ketone (TPCK), partially reversed the induction of Snail expression and its promoter activity (Fig. 5b). TPCK treatment also slightly increased E‐cadherin expression (Fig. 5d).
Figure 5.
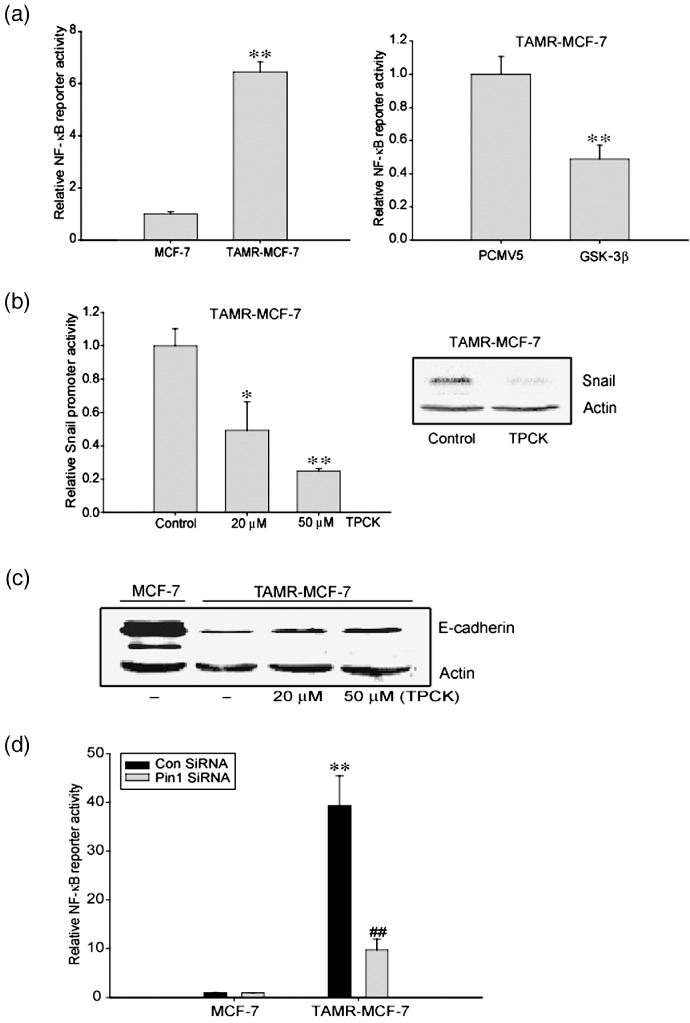
Involvement of nuclear factor (NF)‐κB in Pin1‐mediated epithelial–mesenchymal transition of tamoxifen‐resistant (TAMR)‐MCF‐7 cells. (a) Left panel: NF‐κB reporter activity in TAMR‐MCF‐7 cells. Dual luciferase reporter assays were carried out on the lysed cells co‐transfected with NF‐κB plasmid (firefly luciferase) and phRL‐SV (hRenilla luciferase) (a ratio of 100:1) 18 h after transfection. Reporter gene activation was calculated as a relative ratio of firefly luciferase to hRenilla luciferase activity. Data represent means ± SD with four different samples (significant vs the MCF‐7 cells, **P < 0.01). Right panel: Role of glycogen synthase kinase (GSK)‐3β in the activation of NF‐κB. TAMR‐MCF‐7 cells were cotransfected with NF‐κB plasmid in combination with GSK‐3β overexpression vector or PCMV5 (1 µg/well). Data represent means ± SD with four different samples (significant vs PCMV5‐transfected group, **P < 0.01). (b) Inhibition of NF‐κB transactivation by tosyl‐phenylalanine chloremethyl‐ketone (TPCK), a NF‐κB inhibitor. Left panel: Reporter gene assay. MCF‐7 and TAMR‐MCF‐7 cells were transfected with –2.5kb Snail‐Luc reporter plasmid and treated with TPCK for 24 h. Data represent means ± SD with four different samples (significant vs the control, *P < 0.05, **P < 0.01; control level = 1). Right panel: Immunoblotting of Snail in TAMR‐MCF‐7 cells treated with 20 µM TPCK. (c) Decrease in E‐cadherin level by TPCK treatment. The level of E‐cadherin was determined by immunoblotting in TAMR‐MCF‐7 cells treated with TPCK (0, 20, or 50 µM). (d) Inhibition of NF‐κB transactivation by Pin1 siRNA. TAMR‐MCF‐7 cells were cotransfected with NF‐κB reporter plasmid in combination with Pin1 siRNA (20 pmol) or control siRNA. Data represent means ± SD with four different samples (significant vs the control siRNA‐transfected MCF‐7 cells, **P < 0.01; significant vs the control siRNA‐transfected TAMR‐MCF‐7 cells, ## P < 0.01).
Pin1 directly binds to the Thr254‐Pro motif of p65( 44 ) and increases nuclear levels of p65. Basal NF‐κB promoter activity was significantly suppressed by Pin1 knockdown (Fig. 5d), implying that Pin1 overexpression in TAMR‐MCF‐7 cells is related to NF‐κB activation. To sum up, the induction of EMT in TAMR‐MCF‐7 is also partly dependent on NF‐κB activation.
No induction of EMT in Pin1‐overexpressing MCF‐7 cells. In order to test whether the sustained Pin1 overexpression alone results in EMT in breast cancer cells, we determined several EMT markers in stable Pin1‐overexpressing MCF‐7 (Pin1‐MCF‐7) cells. After multiple infections of MCF‐7 cells with Pin1 retrovirus, Pin1 expression levels in Pin‐MCF‐7 were higher than those in GFP‐MCF‐7 cells (Fig. 6a). However, the basal expression levels of E‐cadherin, N‐cadherin, and vimentin were not affected by Pin1 overexpression. Moreover, both the snail reporter activity and cell morphology of Pin1‐MCF‐7 cells were not distinct from GFP‐MCF‐7 cells (Fig. 6b,c). These results suggest that Pin1 overexpression alone is not enough to cause EMT in breast cancer cells. However, PTEN expression levels in Pin1‐MCF‐7 cells were slightly lower than those in GFP‐MCF‐7 cells (Fig. 6d), implying that PTEN expression is regulated by Pin1 activity in breast cancer cells.
Figure 6.
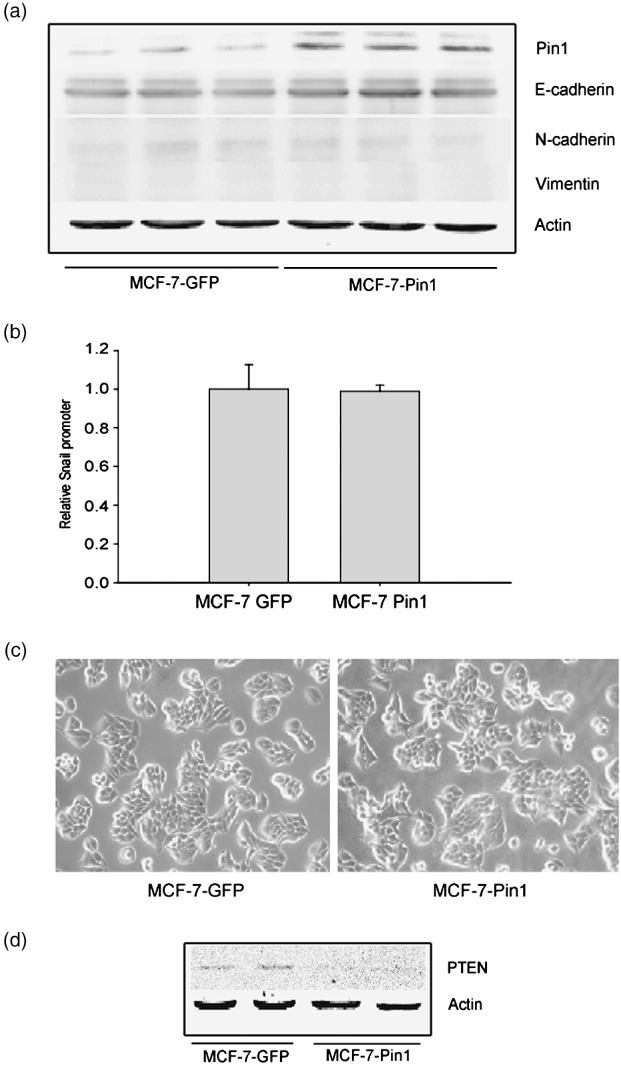
No induction of epithelial–mesenchymal transition (EMT) in Pin1‐overexpressed MCF‐7 cells. (a) Effect of Pin1 overexpression on the EMT phenotypes in MCF‐7 cells. Representative immunoblots show E‐cadherin, N‐cadherin, and vimentin protein levels in both GFP‐MCF‐7 (control) and Pin1‐MCF‐7 cells serum‐deprived for 18 h. (b) Basal reporter activity of Snail promoters (Snail‐Luc) in GFP‐MCF‐7 and Pin1‐MCF‐7 cells. Each cell type was transiently transfected with –2.5 kb Snail‐Luc plasmid. Dual luciferase reporter assays were carried out on the lysed cells cotransfected with Snail‐Luc plasmids (firefly luciferase) and phRL‐SV (hRenilla luciferase) (a ratio of 100:1) 18 h after transfection. Reporter gene activation was calculated as a relative ratio of firefly luciferase to hRenilla luciferase activity. Data represent means ± SD with six different samples. (c) Cell morphology of GFP‐MCF‐7 and Pin1‐MCF‐7 cells. (d) Phosphatase and tensin homolog depleted on chromosome ten (PTEN) expression in GFP‐MCF‐7 and Pin1‐MCF‐7 cells. A representative immunoblot shows PTEN protein in both GFP‐MCF‐7 and Pin1‐MCF‐7 cells serum‐deprived for 18 h.
Discussion
Phosphorylation of proteins on serine/threonine residues preceding proline (Ser/Thr‐Pro) is a key signaling mechanism in breast cancer cell lines.( 45 , 46 , 47 ) Pin1 is essential for diverse cellular processes, such as proliferation, differentiation, and apoptosis.( 20 , 21 , 37 ) Moreover, Pin1 overexpression influences oncogenic pathways and/or contributes to the transformation of breast epithelial cells. Our findings suggest a new pathological role of Pin1 in EMT induction of TAM‐resistant breast cancer. The overexpressed Pin1 in TAMR‐MCF‐7 cells seems to be related with EMT phenomena, as evidenced by reversal of E‐cadherin expression by Pin1 siRNA. Although increased migration occurs in TAMR‐MCF‐7 cells,( 18 ) the role of Pin1 in EMT in these cells is unclear, but Pin1 can partially transform mammary epithelial cells.( 48 ) It has been shown that the acquisition of TAM resistance accelerates cell invasion and migration.( 7 ) Because EMT is a critical event for efficient invasion and motility of cancer cells,( 9 ) our findings provide evidence for the involvement of Pin1 in the migratory phenotype of TAMR‐MCF‐7 cells.
The Pin1‐mediated EMT is associated with increases in the transcription and protein stability of Snail, as Snail blocks E‐cadherin expression at multiple levels. GSK‐3β constitutively suppresses transactivation of the snail gene as well as Snail protein stability by phosphorylating Snail to promote its degradation and cytoplasmic localization.( 26 , 49 ) Here, we found that Ser9‐phosphorylated GSK‐3β, an inactive form of the kinase, was enhanced in TAMR‐MCF‐7 cells. An important implication of our findings is that endogenous suppressors of GSK‐3β, such as Akt, which are frequently activated in carcinoma cells,( 29 ) may also inhibit E‐cadherin transcription and promote an EMT. Also, Akt activation in epithelial cells leads to substantial morphological modification, and induction of cell migration and invasiveness.( 50 ) Akt can phosphorylate and inactive GSK‐3β,( 51 ) and we found that the basal Akt phosphorylation was increased in TAMR‐MCF‐7 cells, suggesting that its regulation is disrupted. PTEN is an upstream antagonistic phosphatase of PI3‐kinase. It removes the 3′ phosphate of phosphatidylinositol phosphate (PIP)3 and inhibits downstream signaling of activated PI3‐kinase.( 52 ) We confirmed that PTEN expression in TAMR‐MCFR‐7 cells is downregulated. Through these data, we may infer that PTEN loss induced the consistent activation of Akt. Furthermore, PTEN loss and subsequent Akt activation may significantly reduce GSK‐3β activity and transcriptional activation of the snail gene. Activation of oncogenes, including Pin1, controls PI3‐kinase activity during EMT.( 53 ) Indeed, blocking Pin with siRNA treatment disrupted PI3‐kinase activity and Pin1‐mediated EMT. Moreover, Pin1‐mediated EMT seems to be derived from the consistent PTEN downregulation, as both Akt phosphorylation and expression of its upstream negative regulator PTEN were partially reversed by Pin1 siRNA (Fig. 4c). Pin1 may change PTEN expression, which subsequently promotes the activation of PI3‐kinase–Akt–GSK‐3β‐dependent EMT in TAMR‐MCF‐7 cells.
NF‐κB is essential for EMT and metastasis in a breast cancer cell model.( 40 ) NF‐κB affects cell proliferation and various cancers, including breast cancer cells.( 54 , 55 ) High levels of NF‐κB are found in the majority of primary human and rodent breast tumor tissue samples, breast cancer cell lines, and carcinogen‐transformed mammary epithelial cells.( 54 , 56 ) There are reports that GSK‐3β regulates Snail via NF‐κB,( 43 ) and NF‐κB directly binds to the snail promoter to increase its activity.( 42 ) NF‐κB is a central mediator of EMT,( 40 ) and NF‐κB function is regulated by Pin1‐mediated prolyl isomerization and ubiquitin‐mediated proteolysis of the p65/RelA subunit.( 44 ) Upon cytokine treatment, Pin1 binds to the pThr254‐Pro motif in p65 and inhibits p65 binding to ΙκΒα, resulting in increased nuclear accumulation and protein stability of p65 and enhanced NF‐κB activity.( 44 ) Our study also showed that NF‐κB is partly associated with Pin1‐mediated EMT.
Although Pin1 is involved in the EMT phenotype of TAMR‐MCF‐7 cells, Pin1 overexpression alone is not sufficient to induce EMT or snail transcription in MCF‐7 cells. In fact, Pin1 overexpression alone does not seem to affect cell growth or cell morphology under normal growth conditions.( 48 ) Pin1 may become oncogenic only after activation of other oncogenic pathways leading to phosphorylation of its substrates. Indeed, Pin1 greatly enhances and facilitates transformation induced by oncogenic Her‐2/Neu and Ras in mammary epithelial cells.( 48 ) Thus, Pin1 can cooperate with oncogenic pathways in ‘post‐phosphorylation’ regulation where the prompt cis–trans isomerization of prolyl residues adjacent to phosphorylated serine or threonine residues promotes cell proliferation and transformation. EMT is therefore under the control of several signaling pathways. It has also been shown that overall development of organs in Pin1‐null mice is normal. However, Pin1‐null mice show a developmental defect during pregnancy in preparation for lactation.( 57 ) Hence, Pin1 seems not to be essential for the development of organs, but it could be required for the rapid expansion of mammary epithelial cells under specific conditions. Our data demonstrating that Pin1 overexpression alone is not enough for the induction of EMT in MCF‐7 cells thus imply that Pin1 may be partly involved in the specific phenotype change during the acquisition of TAM resistance.
In the present study, we have revealed that Pin1 overexpression partly contributes to the EMT process via PTEN–PI3‐kinase–Akt–GSK‐3β‐dependent and/or NF‐κB‐dependent Snail activation and suggest potential Pin1 involvement in EMT during acquisition of resistance to anti‐hormones in breast cancers (Fig. 7).
Figure 7.
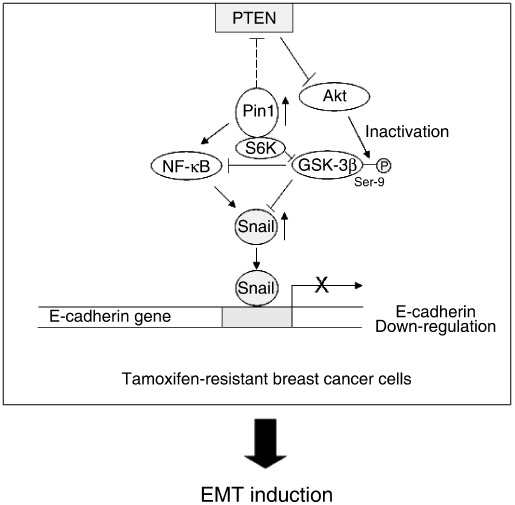
Possible mechanism of epithelial–mesenchymal transition (EMT) induction in tamoxifen‐resistant (TAMR) breast cancer. Pin1 is overexpressed in TAMR‐MCF‐7 cells. The increased Pin1 activity persistently activates Akt presumably through phosphatase and tensin homolog depleted on chromosome ten (PTEN) downregulation and subsequently inactivates glycogen synthase kinase (GSK)‐3β. GSK‐3β inactivation enhances cellular Snail level via transcriptional activation or inhibition of proteasomal degradation. Direct (p65 accumulation in nucleus) or indirect (GSK‐3β‐mediated) nuclear factor (NF)‐κB activation is also involved in Snail upregulation in TAMR‐MCF‐7 cells. The increased Snail suppresses transcription of the E‐cadherin gene and finally causes EMT in TAMR‐MCF‐7 cells.
Acknowledgments
This study was financially supported by a Korean Science and Engineering Foundation grant funded by the Korea government (MEST) through the Research Center for Resistant Cells (R13‐2003‐009).
References
- 1. Mueller SO, Clark JA, Mvers PH, Korach KS. Mammary gland development in adult mice requires epithelial and stromal estrogen receptor alpha. Endocrinol 2002; 143: 2357–65. [DOI] [PubMed] [Google Scholar]
- 2. Petrangeli E, Lubrano C, Ortolani F et al . Estrogen receptors: new perspectives in breast cancer management. J Steroid Biochem Mol Biol 1994; 49: 327–31. [DOI] [PubMed] [Google Scholar]
- 3. Rose C, Thorpe SM, Andersen KW et al . Beneficial effect of adjuvant tamoxifen therapy in primary breast cancer patients with high oestrogen receptor values. Lancet 1985; 1: 16–9. [DOI] [PubMed] [Google Scholar]
- 4. Ali S, Coombes RC. Endocrine‐responsive breast cancer cell and strategies for combating resistance. Nat Rev Cancer 2002; 2: 101–12. [DOI] [PubMed] [Google Scholar]
- 5. Osborne CK, Fuqua SA. Mechanism of tamoxifen resistance. Breast Cancer Res Treat 1994; 32: 49–55. [DOI] [PubMed] [Google Scholar]
- 6. Choi HK, Yang JW, Roh SH, Han CY, Kang KW. Induction of multidrug resistance associated protein 2 in tamoxifen‐resistant breast cancer cells. Endocr Relat Cancer 2007; 14: 293–303. [DOI] [PubMed] [Google Scholar]
- 7. Knowlden JM, Hutcheson IR, Jones HE et al . Elevated levels of epidermal growth factor receptor/c‐erbB2 heterodimers mediate an autocrine growth regulatory pathway in tamoxifen‐resistant MCF‐7 cells. Endocrinology 2003; 144: 1032–44. [DOI] [PubMed] [Google Scholar]
- 8. Thiery JP. Epithelial–mesenchymal transitions in tumour progression. Nat Rev Cancer 2002; 2: 442–54. [DOI] [PubMed] [Google Scholar]
- 9. Thompson EW, Newgreen DF, Tarin D. Carcinoma invasion and metastasis: a role for epithelial–mesenchymal transition? Cancer Res 2005; 65: 5991–5. [DOI] [PubMed] [Google Scholar]
- 10. Friedl P, Wolf K. Tumour‐cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer 2003; 3: 362–74. [DOI] [PubMed] [Google Scholar]
- 11. Birchmeier W, Behrens J. Cadherin expression in carcinomas: role in the formation of cell junctions and the prevention of invasiveness. Biochim Biophys Acta 1994; 1198: 11–26. [DOI] [PubMed] [Google Scholar]
- 12. Christofori G, Semb H. The role of the cell‐adhesion molecule E‐cadherin as a tumour‐suppressor gene. Trends Biochem Sci 1999; 24: 73–6. [DOI] [PubMed] [Google Scholar]
- 13. Peinado H, Portillo F, Cano A. Transcriptional regulation of cadherins during development and carcinogenesis. Int J Dev Biol 2004; 48: 365–75. [DOI] [PubMed] [Google Scholar]
- 14. Batlle E, Sancho E, Franci C et al . The transcription factor snail is a repressor of E‐cadherin gene expression in epithelial tumour cells. Nat Cell Biol 2000; 2: 84–9. [DOI] [PubMed] [Google Scholar]
- 15. Cano A, Perez‐Moreno MA, Rodrigo I et al . The transcription factor snail controls epithelial–mesenchymal transitions by repressing E‐cadherin expression. Nat Cell Biol 2000; 2: 76–83. [DOI] [PubMed] [Google Scholar]
- 16. Peinado H, Marin F, Cubillo E et al . Snail and E47 repressors of E‐cadherin induce distinct invasive and angiogenic properties in vivo . J Cell Sci 2000; 117: 2827–39. [DOI] [PubMed] [Google Scholar]
- 17. Nieto MA. The snail superfamily of zinc‐finger transcription factors. Nat Rev Mol Cell Biol 2002; 3: 155–66. [DOI] [PubMed] [Google Scholar]
- 18. Hiscox S, Morgan L, Barrow D, Dutkowskil C, Wakeling A, Nicholson RI. Tamoxifen resistance in breast cancer cells accompanied by an enhanced motile and invasive phenotype: inhibition by gefitinib (‘Iressa’ ZD1839). Clin Exp Metastasis 2004; 21: 201–12. [DOI] [PubMed] [Google Scholar]
- 19. Hiscox S, Jiang WG, Obermeier K et al . Tamoxifen resistance in MCF7 cells promotes EMT‐like behaviour and involves modulation of beta‐catenin phosphorylation. Int J Cancer 2006; 118: 290–301. [DOI] [PubMed] [Google Scholar]
- 20. Ayala G, Wang D, Wulf G et al . The prolyl isomerase Pin1 is a novel prognostic marker in human prostate cancer. Cancer Res 2003; 63: 6244–51. [PubMed] [Google Scholar]
- 21. Wulf GM, Ryo A, Wulf GG et al . Pin1 is overexpressed in breast cancer and cooperates with Ras signaling in increasing the transcriptional activity of c‐Jun towards cyclin D1. EMBO J 2001; 20: 3459–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fujita N, Jave DL, Kajita M, Geigeman C, Moreno CS, Wade PA. MTA3, a Mi‐2/NuRD complex subunit, regulates an invasive growth pathway in breast cancer. Cell 2003; 113: 207–19. [DOI] [PubMed] [Google Scholar]
- 23. Moody SE, Perez D, Pan TC et al . The transcriptional repressor Snail promotes mammary tumor recurrence. Cancer Cell 2005; 8: 197–209. [DOI] [PubMed] [Google Scholar]
- 24. Olmeda D, Jorda M, Peinado H, Fabra A, Cano A. Snail silencing effectively suppresses tumour growth and invasiveness. Oncogene 2007; 6: 1862–74. [DOI] [PubMed] [Google Scholar]
- 25. Blanco MJ, Moreno‐Bueno G, Sarrio D et al . Correlation of Snail expression with histological grade and lymph node status in breast carcinomas. Oncogene 2002; 21: 3241–6. [DOI] [PubMed] [Google Scholar]
- 26. Zhou BP, Deng J, Xia W et al . Dual regulation of Snail by GSK‐3beta‐mediated phosphorylation in control of epithelial–mesenchymal transition. Nat Cell Biol 2004; 6: 931–40. [DOI] [PubMed] [Google Scholar]
- 27. Doble BW, Woodgett JR. Role of glycogen synthase kinase‐3 in cell fate and epithelial–mesenchymal transitions. Cells Tissues Organs 2007; 185: 73–84. [DOI] [PubMed] [Google Scholar]
- 28. Chen H, Liu J, Zhao CQ, Diwan BA, Merrick BA, Waalkes MP. Association of c‐myc overexpression and hyperproliferation with arsenite‐induced malignant transformation. Toxicol Appl Pharmacol 2001; 175: 260–8. [DOI] [PubMed] [Google Scholar]
- 29. Doble BW, Woodgett JR. GSK‐3: tricks of the trade for a multi‐tasking kinase. J Cell Sci 2003; 116: 1175–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mallikarjuna G, Dhanalakshmi S, Singh RP, Agarwal C, Agarwal R. Silibinin protects against photocarcinogenesis via modulation of cell cycle regulators, mitogen‐activated protein kinases and Akt signaling. Cancer Res 2004; 64: 6349–56. [DOI] [PubMed] [Google Scholar]
- 31. Mitsuuchi Y, Johnson SW, Selvakumaran M, Williams SJ, Hamilton TC, Testa JR. The phophatidylinositol 3‐kinase/AKT signal transduction pathway plays a critical role in the expression of p21WAF1/CIP1/SDI1 induced by cisplatin and paclitaxel. Cancer Res 2000; 60: 5390–4. [PubMed] [Google Scholar]
- 32. Testa JR, Bellacosa A. AKT plays a central role in tumorigenesis. Proc Natl Acad Sci USA 2001; 98: 10 983–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bellacosa A, Testa JR, Moore R, Larue L. A portrait of AKT kinase: human cancer and animal models depict a family with strong individualities. Cancer Biol Ther 2004; 3: 268–75. [DOI] [PubMed] [Google Scholar]
- 34. Grille SJ, Bellacosa A, Upson J et al . The protein kinase Akt induces epithelial mesenchymal transition and promotes enhanced motility and invasiveness of squamous cell carcinoma lines. Cancer Res 2003; 63: 2172–8. [PubMed] [Google Scholar]
- 35. Bayer E, Goettsch S, Mueller JW et al . Structural analysis of the mitotic regulator hPin1 in solution: insights into domain architecture and substrate binding. J Biol Chem 2003; 278: 26 183–93. [DOI] [PubMed] [Google Scholar]
- 36. Lu KP. Pinning down cell signaling, cancer and Alzheimer's disease. Trends Biochem Sci 2003; 29: 200–9. [DOI] [PubMed] [Google Scholar]
- 37. Bao L, Kimzey A, Sauter G, Sowadski JM, Lu KP, Wang DG. Prevalent overexpression of prolyl isomerase Pin1 in human cancers. Am J Pathol 2004; 164: 1727–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lu KP. Prolyl isomerase Pin1 as a molecular target for cancer diagnostics and therapeutics. Cancer Cell 2003; 4: 175–80. [DOI] [PubMed] [Google Scholar]
- 39. Ryo A, Nakamura M, Wulf G, Liou YC, Lu KP. Pin1 regulates turnover and subcellular localization of beta‐catenin by inhibiting its interaction with APC. Nat Cell Biol 2001; 3: 793–801. [DOI] [PubMed] [Google Scholar]
- 40. Huber MA, Kraut N, Beug H. Molecular requirements for epithelial–mesenchymal transition during tumor progression. Curr Opin Cell Biol 2005; 17: 548–58. [DOI] [PubMed] [Google Scholar]
- 41. Chua HL, Bhat‐Nakshatri P, Clare SE, Morimiva A, Badve S, Nakshatri H. NF‐kappaB represses E‐cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: potential involvement of ZEB‐1 and ZEB‐2. Oncogene 2007; 26: 711–24. [DOI] [PubMed] [Google Scholar]
- 42. Barbera MJ, Puig I, Dominguez D et al . Regulation of Snail transcription during epithelial to mesenchymal transition of tumor cells. Oncogene 2004; 23: 7345–54. [DOI] [PubMed] [Google Scholar]
- 43. Bachelder RE, Yoon SO, Franci C, De Herreros AG, Mercurio AM. Glycogen synthase kinase‐3 is an endogeneous inhibitor of Snail transcription: implication for the epithelial–mesenchymal transition. J Cell Biol 2005; 168: 29–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ryo A, Suizu F, Yoshida Y et al . Regulation of NF‐kappaB signaling by Pin1‐dependent prolyl isomerization and ubiquitin‐mediated proteolysis of p65/RelA. Mol Cell 2003; 12: 1413–26. [DOI] [PubMed] [Google Scholar]
- 45. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100: 57–70. [DOI] [PubMed] [Google Scholar]
- 46. Blume‐Jensen P, Hunter T. Oncogenic kinase signaling. Nature 2001; 411: 355–65. [DOI] [PubMed] [Google Scholar]
- 47. Lu KP, Liou YC, Zhou XZ. Pinning down the proline‐directed phosphorylation signaling. Trends Cell Biol 2002; 12: 164–72. [DOI] [PubMed] [Google Scholar]
- 48. Ryo A, Liou YC, Wulf G, Nakamura N, Lee SW, Lu KP. Pin1 is an E2F target gene essential for the Neu/Ras‐induced transformation of mammary epithelial cells. Mol Cell Biol 2002; 22: 5281–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhou BP, Deng J, Xia W et al . Dual regulaton of Snail by GSK‐3‐mediataed phosphorylation in control of epithelial–mesenchymal transition. Nat Cell Biol 2004; 6: 931–40. [DOI] [PubMed] [Google Scholar]
- 50. Julien S, Puig I, Caretti E et al . Activation of NF‐kappaB by Akt upregulates Snail expression and induces epithelium mesenchyme transition. Oncogene 2007; 26: 7445–56. [DOI] [PubMed] [Google Scholar]
- 51. Vivanco I, Sawyers CL. The phosphatidylinositol 3‐kinase AKT pathway in human cancer. Nat Rev Cancer 2002; 2: 489–501. [DOI] [PubMed] [Google Scholar]
- 52. Byun DS, Cho K, Ryu BK et al . Frequent monoallelic deletion of PTEN and its reciprocal association with PIK3CA amplification in gastric carcinoma. Int J Cancer 2003; 104: 318–27. [DOI] [PubMed] [Google Scholar]
- 53. Xu GP, Li QQ, Cao XX et al . The Effect of TGF‐beta1 and SMAD7 gene transfer on the phenotypic changes of rat alveolar epithelial cells. Cell Mol Biol Lett 2007; 87: 1918–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cogswell PC, Guttridge DC, Funkhouser WK, Baldwin Jr AS. Selective activation of NF‐kappa B subunit in human breast cancer: potential roles for NF‐kappa B2/p52 and for Bcl‐3. Oncogene 2000; 19: 1123–31. [DOI] [PubMed] [Google Scholar]
- 55. Orlowski RZ, Baldwin Jr AS. NF‐kappaB as a therapeutic target in cancer. Trends Mol Med 2002; 8: 385–9. [DOI] [PubMed] [Google Scholar]
- 56. Kim DW, Sovak MA, Zanieski G et al . Activation of NF‐kappaB/Rel occurs early during neoplastic transformation of mammary cells. Carcinogenesis 2000; 21: 871–9. [DOI] [PubMed] [Google Scholar]
- 57. Liou YC, Ryo R, Huang HK et al . Loss of Pin1 function in the mouse resembles the cyclin D1‐null phenotypes. Proc Natl Acad Sci USA 2002; 99: 1335–40. [DOI] [PMC free article] [PubMed] [Google Scholar]