

Abstract
The interactions between malignant cells and the microenvironment of the local host tissue play a critical role in tumor growth, metastasis and their response to treatment modalities. We investigated the roles of smooth muscle calponin (Cnn1, also called calponin h1 or basic calponin) in the development of tumor vascul ature in vivo by analyzing mutant mice lacking the Cnn1 gene. Here we show that loss of Cnn1 in host mural cells prevents maturation of tumor vasculature. In vitro studies showed that platelet‐derived growth factor B‐induced vascular smooth muscle migration was downregulated by the Cnn1‐deficiency, and forced expression of Cnn1 restored migration. Moreover, destruction of established tumor mass by treatment with an antivascular endothelial growth factor antibody was markedly enhanced in Cnn1‐deficient mice. These data, coupled with the knowledge that structural fragility of normal blood vessels is caused by loss of the Cnn1 gene, suggest that Cnn1 plays an important role in the maturation of blood vessels, and may have implications for therapeutic strategies targeting tumor vasculature for treatment of human cancers. (Cancer Sci 2007; 98: 757–763)
Smooth muscle calponin (Cnn1) is an actin‐associated protein originally isolated from vascular smooth muscle,( 1 ) and is a major regulator of force production in smooth muscle cells (SMC).( 2 , 3 , 4 ) It is generally agreed that Cnn1 expression serves as an excellent marker for defining lineage specification, differentiation and phenotypic modulation of SMC.( 5 , 6 , 7 ) In previous studies on human cancers, we and others have reported positive and negative expression of Cnn1 in the mural SMC of tumor vasculature,( 8 , 9 , 10 ) and showed that reduced expression of Cnn1 in the tumor vasculature of patients with the early stage of hepatocellular carcinoma,( 8 ) and those with renal cell carcinoma,( 10 ) was statistically correlated with poor prognosis. Furthermore, a 17‐gene signature set was identified in the tumor–host microenvironment to predict metastatic potential and early death for a variety of human solid tumors.( 11 ) These include decreased expression of the smooth muscle genes γ2 actin (Actg2), myosin heavy chain 11 (MYH11) and Cnn1. However, as yet there is no explanation for links between tumor phenotypes and the observed downregulation of the Cnn1 gene in host cells, including vascular cells. Although a critical role of the calponin gene in the development of normal vasculature has recently been reported in zebrafish,( 12 ) the role of Cnn1 in the development and maturation of mammalian blood vessels, especially the tumor vasculature, has not been studied.
Materials and Methods
Cells, animals and antibodies. Lewis lung carcinoma cell line (LLC) was purchased from Riken Cell Bank (RCB0558; Tsukuba, Japan). B16 melanoma cells with low metastatic activity to the lung were kindly provided by Dr H. Tanaka (Osaka Medical Center, Osaka, Japan). Cnn1‐deficient mice,( 13 ) with a genetic background of C57BL6/J (Cnn1 –/– ), were generated through back‐crossing to C57BL6/J mice (Nihon SLC, Hamamatsu, Japan) for more than 15 generations. Immunoblot analysis was carried out as described previously.( 13 ) Animal procedures were approved by the Animal Care and Use Committee of Osaka Medical Center.
Antibodies and immunohistochemistry. A polyclonal antibody specific to the Cnn1 isoform was prepared as described previously.( 13 ) Anti‐smooth muscle α‐actin (α‐SMA) (clone 1A4) was purchased from Sigma Chemicals (St Louis, MO, USA). Anti‐PECAM‐1(CD31) (clone MEC13.3) was from BD PharMingen (San Diego, CA, USA). Anti‐PDGFRβ (sc‐6252, A‐3) was from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti‐mouse CD34 (clone MEC14.7) was from Hycult Biotechnology (Uden, the Netherland). Anti‐NG2 was from Chemicon International (Temacula, CA, USA).
The specimens were fixed in Bouin's solution (15%[v/v] saturated picric acid solution, 1.65%[v/v] formalin and 1%[v/v] acetic acid/phosphate‐buffered saline [PBS]) or 10% formaldehyde/PBS and embedded in paraffin. Antigen retrieval was carried out using an autoclave at 121°C for 10 min in a 10‐mM citrate buffer (pH 7.0) (Cnn1, α‐SMA, CD34 and NG2). The procedures described previously( 8 , 13 ) are available from the authors on request. For staining of CD31, excised tumor specimens were mounted in OCT compound (Miles, Elkhart, USA) and then frozen using liquid nitrogen.
Quantification of the vasculature. Individual microvessel counts, as revealed by CD31 or CD34 expression in the endothelium, were carried out using ×400 fields by two independent investigators after assessing for uniformity of staining at low‐power fields (×100). Vessel density is expressed as the number of vessel profiles per mm2. In accordance with other published studies,( 14 , 15 ) the fraction of blood vessels found to be associated with α‐SMA‐positive cells in more than 50% of the vessel perimeter was defined as the pericyte coverage index. The extent of pericyte coverage on vessels was determined on 15 properly cross‐sectioned vessels in each specimen by measuring the proportion of CD31‐ or CD34‐positive vessel perimeter covered by α‐SMA‐immunoreactive cells. Thirty fields per section and at least two tissue sections were counted.
Diffusion chamber model of angiogenesis. LLC cells (4 × 107/mL cells in 200 µL of serum‐free Dulbecco's modified Eagle's medium [DMEM]) were injected into the diffusion chamber ring (PR00‐014‐00; Millipore, Bedford, MA, USA), and the chamber was inserted into the skin fold on the back of 6‐week‐old BALB/c athymic nude mice. Tissues containing angiogenic vasculature were excised 6 days after the implantation, fixed in Bouin's solution, sectioned and immunostained with the anti‐Cnn1 antibody.
Preparation of primary cultured vascular smooth muscle cells. Aortas from 4‐week‐old wild‐type and Cnn1 −/– mice were minced and incubated for 2 h at 37°C in 1.0 mg/mL collagenase (Worthington Biochemicals, Lakewood, NJ, USA), 0.375 mg/mL soybean trypsin inhibitor (Worthington Biochemicals), 0.125 mg/mL elastase III (Sigma Chemicals) and 2 mg/mL bovine serum albumin (BSA) in HEPES buffer (pH 7.5). Cells were washed, cultured in DMEM containing 10% FCS and characterized by spindle‐shaped morphology and immunostaining for α‐SMA.
Migration assay and construction of adenovirus vectors. Migration was assayed in a 48‐well chamber (Nucleopore, Bethesda, MD, USA) with a polycarbonate filter (PVP free, 8 µm pores) coated with 300 µg/mL type I collagen. SMC (1 × 104 per well) were added to the top wells of the chamber in DMEM containing 0.25% BSA. The bottom wells were filled with DMEM containing 10 ng/mL recombinant platelet‐derived growth factor (PDGF)‐B/B (Gibco BRL, Carlsbad, CA, USA). The chambers were then incubated for 4 h at 37°C in a humidified atmosphere with 5% CO2. The filters were fixed and stained with Diff‐Quik (Kokusai Shiyaku, Kobe, Japan), and migrated cells were counted. Recombinant adenovirus (Ad) vectors expressing Escherichia coli LacZ or human Cnn1 were generated using unique I‐CeuI and PI‐SceI sites in the E1 deletion region.( 16 ) Ad‐Cnn1 and Ad‐LacZ were prepared by transfection to human embryonic kidney 293 cells.
In vivo treatment of vascular endothelial growth factor (VEGF)‐neutralizing antibody. LLC cells (1 × 107/mL in 50 µL PBS) were injected subcutaneously into 6‐week‐old male wild‐type and Cnn1 −/– mice (n = 8 each), and tumor size was monitored. At 10 days after injection, the ‘VEGF ablation’ groups (n = 4) were treated with 8 µg per mouse of goat antimouse VEGF‐neutralizing antibody (AF‐493‐NA; R&D Systems, Minneapolis, MN, USA) administered intraperitoneally every 72 h. Control groups (n = 4) were treated with 8 µg per mouse of non‐immune goat IgG. Treatment was stopped after a total of four injections.
In situ apoptosis detection. Apoptosis was detected by the modified terminal deoxynucleotidyl transferase‐mediated deoxyuridine triphosphate‐biotin nick‐end labeling (TUNEL) method, using an In Situ Apoptosis Detection Kit (Takara Biomedicals, Tokyo, Japan) according to the manufacturer's methods.
Statistical analysis. Statistical differences were determined using the unpaired Student's t‐test. Differences were considered statistically significant at P < 0.05.
Results and Discussion
Microvessels in the brain cortex, lung sections and retina from 6‐week‐old Cnn1 –/– mice were found to have thinner walls with fewer mural cell layers than those from wild‐type mice. Enlarged and naked endothelial tubes were scored in 16/132 cases in wild‐type brain sections (n = 3) and in 166/218 cases in Cnn1 –/– brain sections (n = 5). Representative examples from the α‐SMA‐ or hematoxylin–eosin (H&E) staining of the brain, lung and retina sections are shown in Fig. 1. Reduced numbers of α‐SMA‐positive mural cells in Cnn1 –/– mice were demonstrated in the immunohistochemistry of the brain and retina sections (Fig. 1). These findings are reminiscent of the impaired recruitment of pericytes to brain capillaries in mice lacking PDGF‐B( 17 ) or PDGF receptor (R)‐β,( 18 ) and are consistent with ultrastructural studies on the microvessels in the ocular fundus, lung and heart of Cnn1 –/– mice, demonstrating reduced mural SMC layers and increased leakiness.( 19 )
Figure 1.
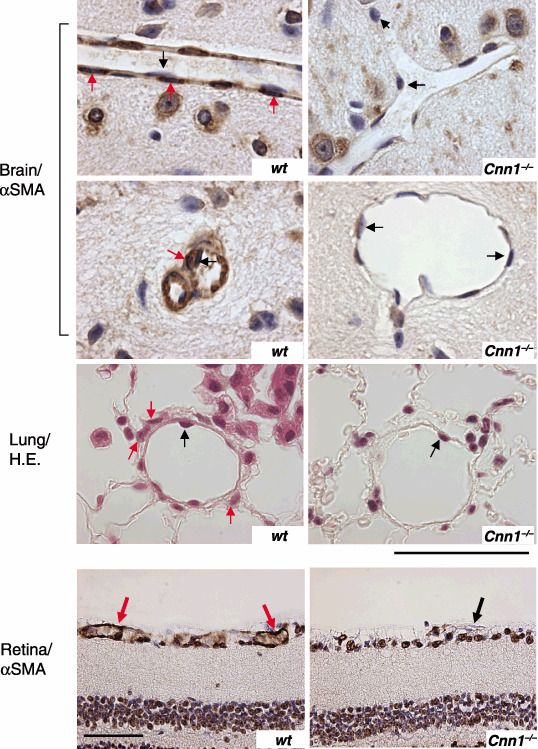
Impaired maturation of blood vessels in normal tissues in smooth muscle calponin‐deficient (Cnn1) –/– mice. Capillaries in brain (smooth muscle α‐actin [α‐SMA] immunostaining), lung (hematoxylin–eosin [H&E] staining) and retina (α‐SMA immunostaining) in Cnn1 –/– mice showed loss of pericytes/smooth muscle cells (SMC) (red arrows) attachment to the endothelium (black arrows). Note the dilation of the Cnn1 –/– capillary in brain. Scale bar = 50 µm in brain and lung, or 100 µm in retina.
LLC transplants were established in syngeneic wild‐type and Cnn1 –/– mice. Compared with wild‐type mice, the primary tumor growth was reduced in Cnn1 –/– mice (Fig. 2a). Counting the CD31‐ or CD34‐immunoreactive microvessels in vascular hot spots of LLC (microvessel density; MVD) revealed that there was a significant decrease in the MVD of tumors in Cnn1 –/– mice compared with those in wild‐type mice at 28 days of tumor transplantation (Fig. 2b). The decrease in MVD in Cnn1 –/– mice was also observed in the size‐matched tumors (approximately 600 mm3) (Fig. 2b). To quantitatively assess the maturation status of the tumor vasculature, the number of microvessels covered by α‐SMA‐immunoreactive mural cells (pericytes or SMC) was determined for LLC at 12 and 28 days after transplantation. There was a striking difference in the percentage of capillaries associated with α‐SMA‐positive mural cells (MCI; mural cell‐coverage index) between wild‐type and Cnn1 –/– tumor blood vessels at 28 days, with no difference at 12 days when the vasculatures were immature (Fig. 2c). In contrast to the wild‐type mice, most capillaries in Cnn1 –/– mice were enlarged (Fig. 2c), distorted and partially enveloped in less than 50% of the vessel surface by cells with α‐SMA immunoreactivities. Blood vessels within B16 melanoma tumors generated in Cnn1 –/– mice also showed reduced mural cell coverage and increased vascular diameter compared with those in wild‐type mice (Fig. 2d). Immunohistochemical analysis using antibodies against CD34, α‐SMA and a pericyte marker NG2 showed reduced mural cell coverage in the representative blood vessels in both the LLC and B16 melanoma xenografts in Cnn1 –/– mice (Fig. 2e). Collectively, abnormalities in the morphology of tumor vasculature as well as the phenotype of normal vessels in brain and lung tissues by Cnn1 deletion were characterized by reduced mural cell association with endothelium, indicating impaired maturation of blood vessels.( 20 )
Figure 2.
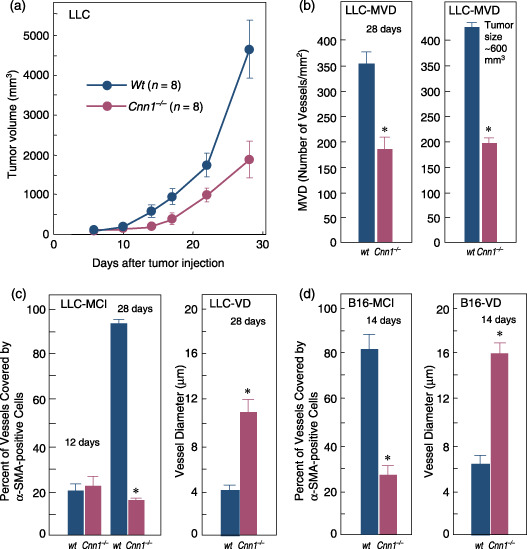
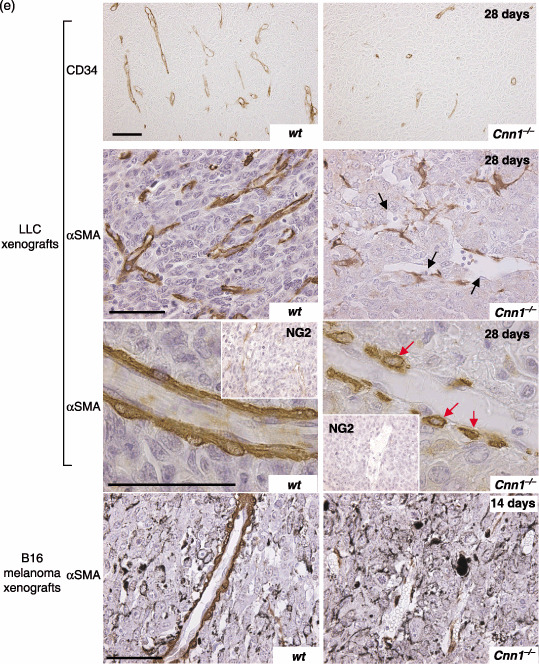
Impaired maturation of blood vessels in tumor tissues in smooth muscle calponin‐deficient (Cnn1) –/– mice. (a) Primary tumor growth of Lewis lung carcinoma (LLC) xenografts implanted into the flank (5 × 105 cells/mouse) was significantly reduced in Cnn1 –/– mice. (b–d) Immunohistochemical analysis (CD31 or CD34, smooth muscle α‐actin [α‐SMA] and NG2) showed reduced microvessel densities (MVD) (b) and fraction of vessels with mural cell‐coverage (MCI) of tumor blood vessels of LLC (n = 4) at 28 days after implantation and B16 melanoma (5 × 105 cells/mouse) (n = 4) at 14 days after implantation (c,d). The decrease in MVD in Cnn1 –/– mice was also observed in the size‐matched LLC tumors (approximately 600 mm3). Blood vessels in both tumors also showed enlarged diameter (c,d). (e) Representative tumor vessels in wild‐type and Cnn1 –/– mice showed reduced MVD and MCI. Arrows indicate endothelial cells (black) and smooth muscle cells (SMC) (red). Scale bars = 50 µm. The data are mean ± SE (30 high‐power field analyses per group). *P < 0.05.
Analysis of anti‐α‐SMA‐immunostained (Fig. 3a) and anti‐CD34‐immunostained (data not shown) vessels in LLC tumors revealed that reduction of the MCI in the vasculature of Cnn1 –/– mice was observed predominantly in the central region of tumors but not at the periphery or in the extra‐tumor connective tissue (Fig. 3a,b). During the tumor‐induced angiogenesis, Cnn1 was normally expressed by peri‐endothelial cells, most probably pericytes or SMC (Fig. 3c), as those Cnn1‐positive cells were also positive for α‐SMA (data not shown). Some of the Cnn1‐positive cells in newly formed vasculature in both mouse (not shown) and human tumors (Fig. 3c) were proliferating. Previous studies suggest that for recruitment of the mural cells to angiogenic vasculature, their migration to the endothelium via PDGF‐B and PDGFR‐β signaling is functionally important.( 17 , 18 , 20 ) Cnn1 is involved in the force generation of actin and myosin in SMC.( 2 , 3 , 4 ) We therefore investigated the effects of Cnn1 deletion on PDGF‐B‐induced migration of vascular SMC in vitro, using the Boyden chamber analysis. As illustrated in Fig. 3d, both wild‐type and Cnn1 –/– SMC cultured from aorta were found to express PDGFR‐β polypeptides, whereas only wild‐type SMC expressed Cnn1. We also prepared Cnn1 –/– SMC lines reconstituted with full‐length Cnn1 by adenovirus‐mediated gene transfer (Fig. 3d). Analysis of migration revealed that approximately 45% less Cnn1 –/– SMC than wild‐type SMC migrated through a porous membrane coated with type I collagen in response to a gradient of PDGF‐B, and also in its absence (Fig. 3e). Reconstitution of Cnn1 expression by adenovirus reversed the inhibition of PDGF‐B‐induced migration of Cnn1 –/– SMC up to levels that corresponded to wild‐type SMC, whereas the inhibition of basal migration was not reversed. Thus, Cnn1 deletion results in reduced chemotactic migration of SMC to PDGF‐B.
Figure 3.
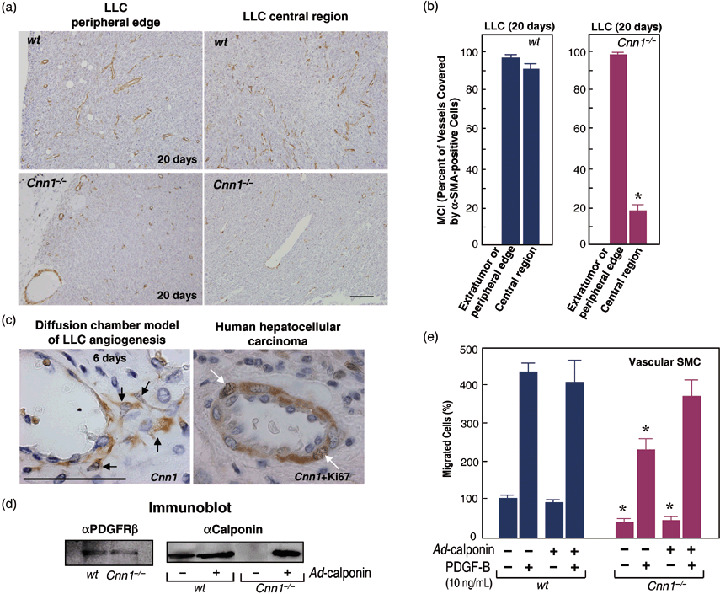
(a,b) Prominent reduction of the mural cell‐coverage (MCI) in smooth muscle calponin‐deficient (Cnn1) –/– mice in the central region of tumors but not at the periphery of tumor tissues. Lewis lung carcinoma (LLC) xenografts were analyzed at 20 days after implantation (5 × 105 cells/mouse). Scale bar = 100 µm. (c) Cnn1 was expressed in mural cells (black arrows) surrounding endothelial cells. Note the double staining of mural cells with Cnn1 (cytoplasm) and Ki‐67 (nucleus), an indicator of proliferating cells (white arrows). Scale bar = 50 µm. (d) Platelet‐derived growth factor (PDGF) receptor β was expressed in both wild‐type and Cnn1 –/– mice, whereas Cnn1 was expressed only in wild‐type mice. Add‐back of Cnn1 expression in Cnn1 –/– smooth muscle cells (SMC) was carried out by adenovirus‐mediated transfection (multiplicity of infection = 30) of Cnn1 cDNA. (e) Reduced SMC migration by Cnn1 deletion. Inhibition of PDGF‐B/B (10 ng/mL)‐induced migration was compensated by the reconstitution of Cnn1, whereas random migration was not. The experiment was repeated twice, and the data are mean ± SE (n = 4 per group). *P < 0.05.
Given the reduced mural cell association, implicating immaturity of blood vessels, it is likely that Cnn1 –/– tumor vessels display VEGF‐dependent remodeling of capillary structures, and endothelial and cancer cell survival.( 21 , 22 ) We therefore tested whether the tumor vasculature in Cnn1 –/– mice is sensitized to anti‐angiogenesis treatment targeting VEGF. Implanted LLC cells in wild‐type and Cnn1 –/– mice were allowed to grow for 10 days, forming flank tumors (n = 8 each). Animals in the wild‐type and Cnn1 –/– groups were divided randomly and injections of mouse VEGF‐specific neutralizing antibody or normal goat IgG were then given intraperitoneally twice a week for 2 weeks. Strikingly, at 18 days after initial anti‐VEGF antibody injection, the treated tumors in Cnn1 –/– mice were found to have prominent degeneration when compared with those in wild‐type mice. Representative examples from macroscopic examination of the H&E‐stained sections of the treated and untreated tumors are shown in Fig. 4a. It depicts extensive necrosis, vulnerability of immature vessels with hemorrhage, and more prominent obliteration of tumor vasculature in Cnn1 –/– mice than in wild‐type mice (Fig. 4b). An in situ apoptosis analysis (TUNEL) revealed that TUNEL‐positive endothelial cells and cancer cells were rarely detected in untreated tumors in wild‐type mice. In contrast, cancer cells transplanted in Cnn1 –/– mice, even in untreated tumors, displayed significant propensity of apoptosis (Fig. 4c). In addition, the anti‐VEGF antibody treatment increased TUNEL‐positive endothelial cells more potently in Cnn1 –/– mice than in wild‐type mice (data not shown). The anti‐VEGF antibody can decrease MVD in both wild‐type and Cnn1 –/– mice (Fig. 4d). The MCI of the tumor vessels in Cnn1 –/– mice increased significantly after treatment (Fig. 4e), indicating selective destruction of the fraction of vessels negative for α‐SMA and normalization of tumor vasculature( 23 ) (Fig. 4f). The results suggest that, under the experimental conditions used, reduced MCI in LLC tumor vasculature in Cnn1 –/– mice may be caused by VEGF. The relationship between Cnn1 deficiency and VEGF production in mural cells should be clarified in the future study. Moreover, consistent with the notion that angiogenesis inhibitors control tumor growth by increasing apoptosis of tumor cells, the anti‐VEGF antibody was found to be more efficacious in killing pre‐existing cancer cells in Cnn1 –/– mice than in wild‐type mice.
Figure 4.
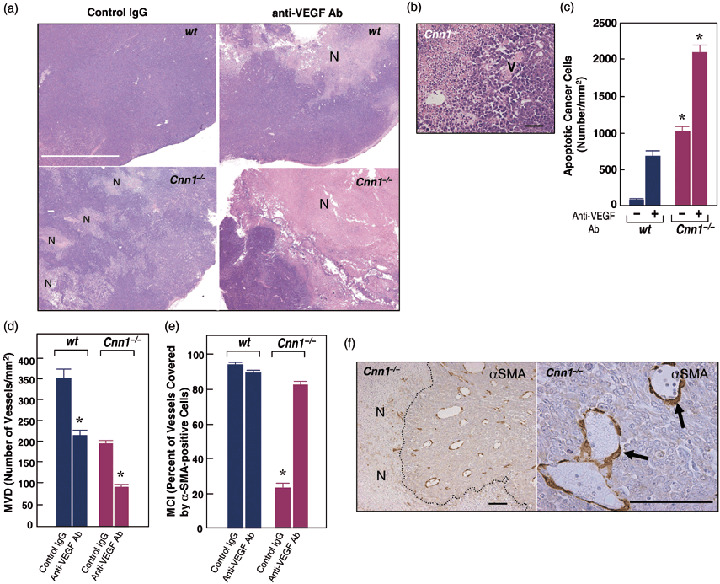
Deletion of smooth muscle calponin (Cnn1) –/– leads to sensitization of cancer cells to anti‐vascular endothelial growth factor (VEGF) antibody treatment. (a) Macroscopic view of hematoxylin–eosin (H&E)‐stained sections of Lewis lung carcinoma (LLC) tumors 18 days after initial anti‐angiogenesis therapy showed the most extensive necrosis of cancer cells (N) in anti‐VEGF antibody‐treated Cnn1 –/– mice. Note that in Cnn1 –/– mice, even non‐treated tumor showed focal necrosis of cancer cells (N). White bar = 5 mm. (b) Higher magnification pictures show prominent destruction of tumor vasculature (V) in anti‐VEGF antibody‐treated LLC tumors in Cnn1 –/– mice. Black bar = 50 µm. (c) Quantitation of TUNEL assay of tissue sections from the tumors in part (a), showing a striking increase in the number of apoptotic cancer cells in anti‐VEGF antibody‐treated LLC tumors in Cnn1 –/– mice. Mean ± SE (30 high‐power field analyses from four mice). *P < 0.05. (d–f) Treatment with anti‐VEGF antibody significantly reduced the microvessel densities (MVD) in both wild‐type and Cnn1 –/– mice (d) and normalized the pericyte coverage index (PCI) in Cnn1 –/– mice (e). Blood vessels covered by smooth muscle α‐actin (α‐SMA)‐positive mural cells in Cnn1 –/– mice were refractory to the anti‐VEGF antibody therapy (f). Scale bar = 100 µm.
Assembly of mural cells to endothelial cells is well known to play a critical role in vessel maturation,( 17 , 18 , 20 , 21 , 22 ) and it has been demonstrated that treatments targeted at killing both endothelial and mural cells are more effective in cancer treatment in preclinical animals.( 22 ) Thus, an understanding of the molecular mechanism of mural cell recruitment to tumor endothelial cells is important for cancer treatment. However, genes as well as molecular pathways in mural cells controlling their assembly to the tumor vasculature are not fully understood. The results presented here, for the first time, demonstrate an important role for the Cnn1 gene in vascular maturation at the tumor–host interface. Recent studies have demonstrated that knockdown of calponin in zebrafish blocks the proper migration of endothelial cells during formation of intersegmental vessels.( 12 ) We now report that loss of Cnn1 expression in SMC results in reduced chemotactic migration of SMC to PDGF‐B, suggesting that it may cause defective SMC coverage of the tumor microvessels in Cnn1 –/– mice. Our results also demonstrated that the properties of tissue vasculature affected by Cnn1 expression in mural cells play an important role in determining tumor phenotypes such as apoptosis and susceptibility to the treatment modalities.
Based on a current model of vessel assembly and remodeling,( 20 , 21 ) our results imply that when tumor cells continue to express high levels of VEGF, reduced expression of Cnn1 in local mural cells may lead to a rapid growth of leaky tumor vessels. Previous studies have demonstrated that when fluorescein is injected intravenously into mice, Cnn1 –/– mice exhibit a greater and more rapid leakage of fluorecein from the blood vessels of the ocular fundus compared with wild‐type mice.( 19 ) These blood vessel phenotypes in Cnn1 –/– mice may help to promote metastasis of the tumor cells as implicated by identification of Cnn1 downregulation as a metastasis signature of solid tumors.( 11 ) The Cnn1 –/– phenotype of the host tissues, in fact, promotes lung metastasis of solid tumors.( 19 ) We suggest that the status of vessel maturation evaluated by Cnn1 expression in the mural cells of a given tumor may predict the efficacy of anti‐angiogenesis treatments aimed at VEGF ablation, currently being used in the clinical stage.( 21 , 22 )
The finding that the vessel maturity changes in Cnn1 –/– mice affect the vasculature in the central region of the tumor more than at the periphery and in the extra‐tumor connective tissue raises the intriguing possibility that the effects may instead be due to a response to hypoxia of Cnn1 –/– SMC at the tumor–host interface.
The results of the present study indicate that there is the potential for development of a new stromal therapy as a strategy for anticancer treatment. For example, viral replication controlled by the Cnn1 promoter can destroy activated mural cells with Cnn1 expression while sparing normal quiescent SMC.( 24 ) We envision that, at least for certain human cancers with strong angiogenic capacity, combinatorial use of the viral agent targeting mural cells may potentiate the efficacy of anti‐VEGF antibody treatment. Prevention of vessel maturation in expanding tumors by silencing Cnn1 expression may also be exploited for enforcing vessel regression via VEGF‐withdrawal therapy, and may provide clues for solving the prevention of rarefaction of mature vessels that are refractory to VEGF in retinopathy of prematurity.( 25 )
Acknowledgments
This work was supported in part by Grants‐in‐Aids for Scientific Research from the Ministry of Education, Science, Sports and Culture, Japan, the Ministry of Health and Welfare, Japan, the Princes Takamatsu Cancer Research Fund (Tokyo).
The authors declare that they have no competing financial interests.
References
- 1. Takahashi K, Hiwada K, Kokubu T. Vascular smooth muscle calponin: a novel troponin T‐like protein. Hypertension 1988; 11: 620–6. [DOI] [PubMed] [Google Scholar]
- 2. Takahashi K, Yamamura H. Studies and perspectives of calponin in smooth muscle regulation and cancer gene therapy. Adv Biophys 2003; 37: 91–111. [DOI] [PubMed] [Google Scholar]
- 3. Matthew JD, Khromov AS, McDuffie MJ et al. Contractile properties and proteins of a calponin knockout mouse. J Physiol (London) 2000; 529: 811–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Babu GJ, Celia G, Rhee AY et al. Effects of h1‐calponin ablation on the contractile properties of bladder vs vascular smooth muscle in SM‐B null mice. J Physiol (London) 2006; 577: 1033–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Takahashi K, Nadal‐Ginard B. Molecular cloning and sequence analysis of smooth muscle calponin. J Biol Chem 1991; 266: 13 284–8. [PubMed] [Google Scholar]
- 6. Miano JM, Olsen EN. Expression of the smooth muscle cell calponin gene marks the early cardiac and smooth muscle cell lineages during mouse embryogenesis. J Biol Chem 1996; 271: 7095–103. [DOI] [PubMed] [Google Scholar]
- 7. Yamashita J, Itoh H, Hirashima M et al. Flk1‐positive cells derived from embryonic stem cells serve as vascular progenitors. Nature 2000; 408: 92–6. [DOI] [PubMed] [Google Scholar]
- 8. Sasaki Y, Yamamura H, Kawakami Y et al. Expression of smooth muscle calponin in tumor vessels of human hepatocellular carcinoma and its possible association with prognosis. Cancer 2002; 94: 1777–86. [DOI] [PubMed] [Google Scholar]
- 9. Koganeshita Y, Takeoka M, Ehara T et al. Reduced expression of actin‐binding proteins, h‐caldesmon and calponin h1, in the vascular smooth muscle inside melanoma lesions: an adverse prognostic factor for malignant melanoma. Br J Dermatol 2003; 148: 971–80. [DOI] [PubMed] [Google Scholar]
- 10. Islam AH, Ehara T, Kato H et al. Calponin h1 expression in renal tumor vessels: correlation with multiple pathological factors of renal cell carcinoma. J Urol 2004; 171: 1319–23. [DOI] [PubMed] [Google Scholar]
- 11. Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nature Genet 2003; 33: 49–54. [DOI] [PubMed] [Google Scholar]
- 12. Tang J, Hu G, Hanai J et al. A critical role for calponin 2 in vascular development. J Biol Chem 2006; 281: 6664–72. [DOI] [PubMed] [Google Scholar]
- 13. Yoshikawa H, Taniguchi S, Yamamura H et al. Mice lacking smooth muscle calponin display increased bone formation that is associated with enhancement of bone morphogenetic protein responses. Genes Cells 1998; 3: 685–95. [DOI] [PubMed] [Google Scholar]
- 14. Eberhard A, Kahlert S, Goede V, Hemmerlein B, Plate KH, Augustin HG. Heterogeneity of angiogenesis and blood vessel maturation in human tumors: implication for antiangiogenic tumor therapies. Cancer Res 2000; 60: 1388–93. [PubMed] [Google Scholar]
- 15. Morikawa S, Baluk P, Kaidoh T, Haskell A, Jain RK, McDonald DM. Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am J Pathol 2002; 160: 985–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Morioka T, Koyama H, Yamamura H et al. Role of h1‐calponin in pancreatic AR42J cell differentiation into insulin‐producing cells. Diabetes 2003; 52: 760–6. [DOI] [PubMed] [Google Scholar]
- 17. Lindahl P, Johansson BR, Leveen P, Betsholtz C. Pericyte loss and microaneurysm formation in PDGF‐B‐deficient mice. Science 1997; 277: 242–5. [DOI] [PubMed] [Google Scholar]
- 18. Hellstrom M, Kalen M, Lindahl P, Abramsson A, Betsholtz C. Role of PDGF‐B and PDGFR‐β in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development 1999; 126: 3047–55. [DOI] [PubMed] [Google Scholar]
- 19. Taniguchi S, Takeoka M, Ehara T et al. Structural fragility of blood vessels and peritoneum in calponin h1‐deficient mice, resulting in an increase in hematogenous metastasis and peritoneal dissemination of malignant tumor cells. Cancer Res 2001; 61: 7627–34. [PubMed] [Google Scholar]
- 20. Jain RK, Booth MF. What brings pericytes to tumor vessels? J Clin Invest 2003; 112: 1134–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Benjamin LE, Golijanin D, Itin A, Pode D, Keshet E. Selective ablation of immature blood vessels in established human tumors follows vascular endothelial growth factor withdrawal. J Clin Invest 1999; 103: 159–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bergers G, Song S, Meyer‐Morse N, Bergsland E, Hanahan D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest 2003; 111: 1287–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Willet CG, Boucher Y, Di Tomaso E et al. Direct evidence that the VEGF‐specific antibody bevacizumab has antivascular effects in human rectal cancer. Nature Med 2004; 10: 145–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yamamura H, Hashio M, Noguchi M et al. Identification of the transcriptional regulatory sequences of human calponin promoter and their use in targeting a conditionally replicating herpes vector to malignant human soft tissue and bone tumors. Cancer Res 2001; 61: 3969–77. [PubMed] [Google Scholar]
- 25. Keshet E. Preventing pathological regression of blood vessels. J Clin Invest 2003; 112: 27–9. [DOI] [PMC free article] [PubMed] [Google Scholar]