

Abstract
Hepatocellular carcinoma (HCC) is one of the most common cancer‐related causes of death, and conventional treatments offer unsatisfactory response. We have previously reported that kallistatin gene therapy suppressed the growth of HCC tumors by its anti‐angiogenic activity, and meloxicam, a selective COX‐2 inhibitor, inhibited proliferation and induced apoptosis of human HCC cells in vitro. The aim of this study was to determine whether combining kallistatin gene therapy and meloxicam could offer a better therapeutic effect to combat HCC in mice. A kallistatin expression plasmid was constructed and its expression was detected after intratumoral gene transfer. Both kallistatin gene therapy and meloxicam suppressed the growth of subcutaneous human HepG2 tumors established in BALB/c nude mice, and the combinational therapy showed a stronger effect in suppressing tumor growth, tumor angiogenesis and cell proliferation, and increasing cell apoptosis, than the respective monotherapies. Gene transfer of kallistatin inhibited tumor angiogenesis, and slightly inhibited cell proliferation and increased cell apoptosis in situ, but had no effect on expression of vascular endothelial growth factor, basic fibroblast growth factor, proliferating cell nuclear antigen, Bcl‐2, Bax, or activation of caspase‐3. Meloxicam therapy inhibited cell proliferation, induced cell apoptosis, reduced expression of proliferating cell nuclear antigen, increased activation of caspase‐3, and upregulated Bax. Meloxicam also slightly inhibited tumor angiogenesis with no effect on the expression of vascular endothelial growth factor or basic fibroblast growth factor. Combining two novel anticancer agents, kallistatin targeting tumoral vascularization and meloxicam targeting cell proliferation and apoptosis, warrants investigation as a therapeutic strategy to combat HCC. (Cancer Sci 2009)
Hepatocellular carcinoma is the fifth most common cancer and the third leading cause of cancer‐related mortality worldwide with an estimated incidence of over one million new cases each year.( 1 ) Surgery offers a cure, but the rate of liver resection is <15% and recurrence rate remains as high as 50% after resection.( 2 ) The conventional treatments including chemotherapy and radiotherapy offer unsatisfactory response. Therefore, new strategies to combat HCC are urgently needed.
Solid tumors including HCC must establish an adequate vascular network to acquire necessary nutrition. Given that HCC is a hypervascular tumor, anti‐angiogenic therapy offers a promising approach to treatment. Previous studies have revealed that angiogenesis inhibitors including TNP‐470,( 3 ) angiostatin,( 4 ) endostatin,( 5 ) soluble VEGF receptor 1,( 5 ) and vasostatin( 6 ) are effective in treating HCC in experimental animal models. Kallistatin, a serine proteinase inhibitor, was first identified as a tissue kallikrein‐binding protein,( 7 ) and has recently emerged as a novel inhibitor of angiogenesis.( 8 ) Kallistatin inhibits the proliferation, migration, and adhesion of endothelial cells and angiogenesis in the rat model of hindlimb ischemia.( 9 ) It has been reported that kallikrein‐binding protein suppressed growth of gastric and hepatocellular carcinomas by its anti‐angiogenic activity.( 10 , 11 ) We have recently reported that adeno‐associated virus‐mediated expression of kallistatin inhibited angiogenesis and growth of colon and HCC tumors in mice.( 12 , 13 )
Selective COX‐2 inhibitors have been regarded as a new group of anticancer drugs based on the observation that expression of COX‐2 is elevated in a variety of human cancers( 14 , 15 , 16 , 17 , 18 ) including HCC,( 19 , 20 ) and COX‐2 has been shown to promote growth and inhibit apoptosis of cancer cells.( 21 ) A number of studies have investigated the use of meloxicam, a selective COX‐2 inhibitor, in inhibiting growth of cancers of ovarian,( 22 ) bladder,( 23 ) biliary duct,( 24 ) breast,( 25 ) lung,( 26 ) and colon.( 27 ) Meloxicam has also shown anticancer activity to treat HCC in vitro and in vivo.( 21 , 28 , 29 , 30 ) We have previously reported that meloxicam inhibited proliferation of HepG2 cells, and induced their apoptosis.( 31 )
Given that kallistatin inhibits tumor angiogenesis, and meloxicam suppresses proliferation and increases apoptosis of HCC cells, we hypothesized that these two novel anticancer agents might have synergistic activity in treating HCC. Therefore this study aims to investigate whether combining kallistatin gene therapy and meloxicam could have a better therapeutic effect to combat hepatomas in mice.
Materials and Methods
Mice, cell line, antibodies, and reagents. Male nude BALB/c mice (H‐2b), 6–8 weeks old, were obtained from the Animal Research Center, First Affiliated Hospital School of Harbin Medical University (Harbin, China). The human HCC cell line HepG2, which strongly expresses COX‐2, has been described in our previous report.( 31 ) The cells were routinely cultured at 37°C in RPMI‐1640 medium supplemented with 10% FCS. The Abs against Bcl‐2, Bax, active caspase‐3, PCNA, and bFGF were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The anti‐Ki67 and anti‐kallistatin Abs were purchased from Jingmei Biotech (Shenzhen, China). The anti‐VEGF and anti‐CD31 Abs were purchased from Lab Vision (Fremont, CA, USA) and BD Pharmingen (San Jose, CA, USA), respectively. Meloxicam injectable solution was purchased from Boehringer Ingelheim (Shanghai, China).
Kallistatin expression vector. Complementary DNA encoding kallistatin was released from a kallistatin vector,( 13 ) and subcloned into pcDNA3.1 to construct the kallistatin expression vector Kalli‐pcDNA3.1. DNA sequence analysis confirmed that the cDNA sequences were inserted in the proper reading frame, and no mutations had been incorporated.
Animal model and treatments. All surgical procedures and care given to the animals were in accordance with institutional guidelines, and have been described previously.( 32 ) Briefly, subcutaneous HepG2 tumors were established in the mice, and volumes were estimated according to the formula: π/6 × a 2 × b, where a is the short axis, and b the long axis. When tumors reached approximately 100 mm3, the mice were assigned to four treatment groups: pcDNA3.1; Kalli‐pcDNA3.1; meloxicam; and Kalli‐pcDNA3.1 + meloxicam. To standardize the experiments, mice in each group received both intratumoral and i.p. injections. In the pcDNA3.1 and Kalli‐pcDNA3.1 groups, mice received a daily i.p. injection of 100 μL PBS, and an intratumoral injection of 200 μg pcDNA3.1 or Kalli‐pcDNA3.1 diluted in 100 μL FuGENE 6 transfection reagent (Roche, Shanghai, China), respectively. In the meloxicam group, mice received an i.p. injection of 100 μL meloxicam (diluted in PBS) at the dose of 20 mg/kg daily and an intratumoral injection of 200 μg pcDNA3.1 diluted in 100 μL FuGENE 6. In the Kalli‐pcDNA3.1 + meloxicam group, mice received an i.p. injection of 100 μL meloxicam at the dose of 20 mg/kg daily and an intratumoral injection of 200 μg Kalli‐pcDNA3.1 diluted in 100 μL FuGENE 6. FuGENE 6 was shown to be an efficient in vivo transfection reagent in our previous study.( 32 )
Immunohistochemistry. Tumor cryosections (5 μm) were fixed with acetone, rinsed with PBS, blocked with 3% BSA for 2 h, and incubated overnight with primary Abs. They were subsequently incubated for 30 min with secondary Abs using the Ultra Sensitive TMS‐P kit (Zhongshan, Beijing, China), and immunoreactivity developed with Sigma FAST 3,3′‐diaminobenzidine tetrahydrochloride and CoCl2 enhancer tablets (Sigma‐Aldrich, Shanghai, China). Sections were counterstained with hematoxylin, mounted, and examined by microscopy.
Assessment of tumor vascularity. The methodology has been described previously.( 6 ) Briefly, 5 μm tumor sections were immunostained with an anti‐CD31 Ab, followed by incubation with fluorescence‐labeled goat antirabbit IgG for 30 min, mounted and examined under fluorescence microscopy. Stained vessels were counted in 10 blindly chosen random fields at ×400 magnification, and the microvessel density was recorded.
Quantitation of Ki‐67 proliferation index. The methodology has been described previously.( 33 ) Briefly, tumor sections were immunostained with an anti‐Ki‐67 Ab as described above. The Ki‐67 positive cells were counted in 10 randomly selected ×400 high‐power fields under microscopy. The Ki‐67 proliferation index was calculated according to the following formula: number of Ki‐67 positive cells/total cell count × 100%.
In situ detection of apoptotic cells. The methodology has been described previously.( 33 ) Briefly, tumor sections were stained with the TUNEL agent (Roche) and examined by fluorescence microscopy. The total number of apoptotic cells in 10 randomly selected fields was counted. The apoptosis index was calculated as the percentage of positive staining cells according to the formula: number of apoptotic cells × 100%/total number of nucleated cells.
Western blot analysis. The tumor tissues were homogenized in protein lysate buffer. Debris was removed by centrifugation at 10 000 g for 10 min at 4°C. The lysates were resolved on 12% polyacrylamide SDS gels, and electrophoretically transferred to PVDF membranes. The membranes were blocked with 3% BSA overnight, incubated with primary Abs, and subsequently with alkaline phosphatase‐conjugated secondary Ab. They were developed with 5‐bromo‐4‐chloro‐3‐indolyl phosphate/nitroblue tetrazolium (Tiangen Biotech, Beijing, China).
Statistical analysis. The growth patterns of tumors were compared using the ANOVA test. Other results were expressed as mean values ± SD, and a Student’s t‐test was used to evaluate statistical significance. P < 0.05 was used for statistical significance.
Results
Combinational therapy with kallistatin and meloxicam shows a stronger effect to suppress hepatomas. Tumors were established by subcutaneous injection of 1 × 106 HepG2 cells into the mice. Three weeks later, when the tumors reached approximately 100 mm3, the mice were randomly assigned to four groups: pcDNA3.1 (control); Kalli‐pcDNA3.1; meloxicam; and Kalli‐pcDNA3.1 + meloxicam. As shown in Figure 1, the control tumors grew remarkably quickly, reaching 1985 ± 402 mm3 in volume six weeks after implantation, which is not significantly different from the untreated tumors (data not shown). In contrast, in the Kalli‐pcDNA3.1 group, the tumors reached only 961 ± 193 mm3 six weeks after implantation, which is significantly smaller than the control tumors (P < 0.01). Meloxicam therapy also resulted in a significant reduction in tumor volumes (1280 ± 221 mm3) compared with control tumors (P < 0.05). A combination of Kallio‐pcDNA3.1 and meloxicam further suppressed tumor growth such that tumors reached only 435 ± 149 mm3, which is highly significantly smaller than the control tumors (P < 0.001), and significantly smaller than the tumors treated with kallistatin and meloxicam monotherapies (both P < 0.05; Fig. 1).
Figure 1.
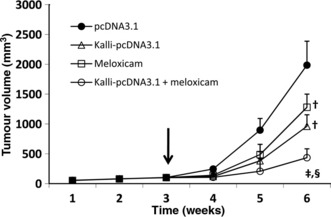
Combining kallistatin gene therapy and meloxicam shows a stronger effect in suppressing tumor growth compared to monotherapies. HepG2 hepatomas were established. When the tumors reached approximately 100 mm3 (indicated by a vertical arrow), they received pcDNA3.1 (control), Kalli‐pcDNA3.1, meloxicam, or Kalli‐pcDNA3.1 + meloxicam treatments. The sizes (mm3) of tumors were recorded. †Significant difference in tumor volumes compared to control. Significant difference in tumor volumes compared to Kalli‐pcDNA3.1 treatment (‡), and meloxicam treatment (§).
Intratumoral gene transfer results in intense in situ transgene expression. Tumor homogenates were prepared one week after treatment and subjected to Western blot analysis. As shown in Figure 2A, a weak band of kallistatin protein (49 kDa) was detected by Western blot analysis of homogenates of tumors treated with pcDNA3.1 or meloxicam. In contrast, a stronger band was detected by Western blot analysis of homogenates of tumors treated with Kalli‐pcDNA3.1 or Kalli‐pcDNA3.1 + meloxicam (Fig. 2A). The Western blot analysis results were further confirmed by immunohistochemistry. Representative photographs revealed intense expression of kallistatin throughout tumors treated with Kalli‐pcDNA3.1, whereas the sections from pcDNA3.1‐treated tumors were only slightly stained by the anti‐kallistatin Ab due to weak expression of endogenous kallistatin (Fig. 2B vs Fig. 2C).
Figure 2.
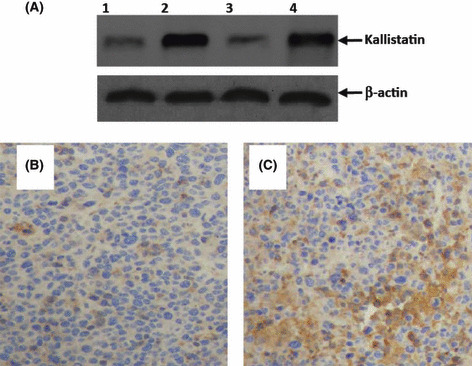
Intense expression of kallistatin in situ after intratumoral gene transfer. (A) Homogenates of tumors from mice treated with pcDNA3.1 (lane 1), Kalli‐pcDNA3.1 (lane 2), meloxicam (lane 3), or Kalli‐pcDNA3.1 + meloxicam (lane 4) one week earlier underwent Western blot analysis with either anti‐kallistatin or β‐actin Abs. Illustrated are representative tumor sections prepared one week following intratumoral gene transfer of pcDNA3.1 (B) and Kalli‐pcDNA3.1 (C) plasmids. The sections were stained with an anti‐kallistatin Ab.
Cell proliferation in situ. Tumor sections were stained with an Ab that detects the cell proliferation marker Ki‐67. Compared with the control tumors (Fig. 3A), there were fewer Ki‐67 positive cells in tumors treated with Kalli‐pcDNA3.1 (Fig. 3B) or meloxicam (Fig. 3C). The combinational therapy resulted in even fewer Ki‐67 positive cells (Fig. 3D). Ki‐67 positive cells were counted to record the proliferation index. As shown in Figure 3E, Kalli‐pcDNA3.1 therapy resulted in a reduction in the proliferation index by 16%, compared with control, but the difference did not reach significance; meloxicam therapy significantly reduced the proliferation index by 35% (P < 0.05), compared with control. The combinational therapy resulted in a highly significant reduction in the proliferation index by 51% (P < 0.001) compared to control, and a significant reduction compared to Kalli‐pcDNA3.1 and meloxicam monotherapies (both P < 0.05). We further detected tumoral expression of PCNA, which was downregulated by meloxicam and the combinational therapy, but Kalli‐pcDNA3.1 had no effect on PCNA expression (Fig. 3F).
Figure 3.
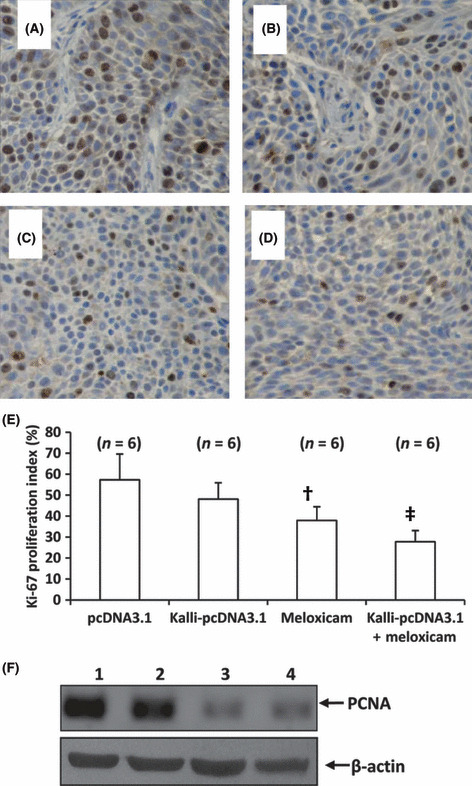
Combining the kallistatin gene and meloxicam suppresses cell proliferation in situ. Representative tumor sections prepared from mice treated with pcDNA3.1 (control) (A), Kalli‐pcDNA3.1 (B), meloxicam (C), or Kalli‐pcDNA3.1 + meloxicam (D) one week earlier. Tumor sections were stained with an anti‐Ki‐67 Ab to detect proliferating cells. (E) Ki‐67 positive cells were counted to calculate the Ki‐67 proliferation index. †Significant difference in the proliferation index from control. ‡Highly significant difference in the proliferation index from control. n, number of tumors assessed. (F) Tumors from mice treated with pcDNA3.1 (lane 1), Kalli‐pcDNA3.1 (lane 2), meloxicam (lane 3), or Kalli‐pcDNA3.1 + meloxicam (lane 4) were homogenized and subjected to Western blot analysis to detect expression of proliferating cell nuclear antigen (PCNA). β‐actin served as an internal control.
Cell apoptosis in situ. Tumor sections were stained with TUNEL for detecting apoptotic cells. A small number of apoptotic cells were detected in the control tumors (Fig. 4A), whereas a greater number of apoptotic cells were detected in tumors treated with Kalli‐pcDNA3.1 (Fig. 4B) and meloxicam (Fig. 4C). The combinational therapy resulted in even more apoptotic cells (Fig. 4D). The apoptotic cells were counted to record the apoptosis index. Kalli‐pcDNA3.1 therapy significantly increased the apoptosis index onefold (P < 0.05), compared with control, and meloxicam significantly increased the apoptosis index almost threefold (P < 0.001) (Fig. 4E). The apoptosis index in tumors treated with combinational therapy was highly significantly higher, at fivefold (P < 0.001) higher than that in control tumors, and significantly higher than Kalli‐pcDNA3.1 and meloxicam monotherapies (both P < 0.05). We further detected tumoral expression of apoptosis‐related proteins including Bcl‐2, Bax, and active caspase‐3. As shown in Figure 4F, Kalli‐pcDNA3.1 gene transfer showed no effect on any of the three molecules. Meloxicam therapy upregulated expression of Bax and active caspase‐3, but had no effect on Bcl‐2 expression. The combinational therapy showed a similar effect to meloxicam alone in regulating expression of the three proteins (Fig. 4F).
Figure 4.
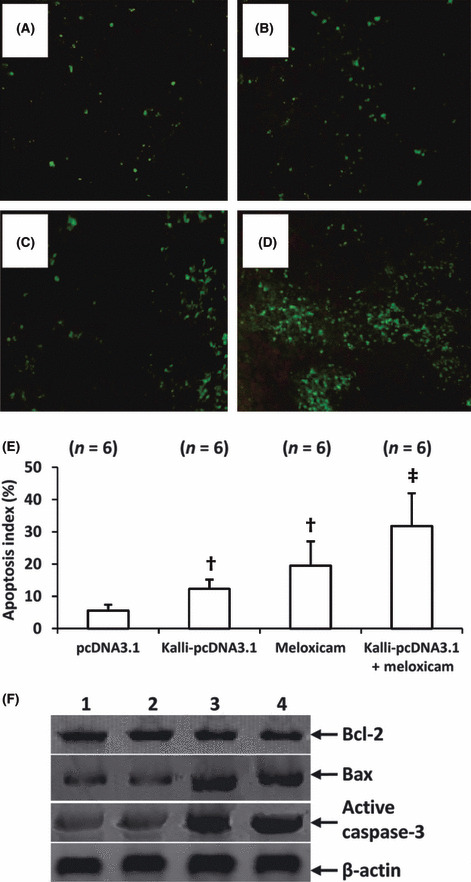
Combining kallistatin gene and meloxicam induces more apoptotic cells. Representative tumor sections prepared from mice treated with pcDNA3.1 (control) (A), Kalli‐pcDNA3.1 (B), meloxicam (C), or Kalli‐pcDNA3.1 + meloxicam (D) one week earlier. Tumor sections were stained with the TUNEL agent to view apoptotic cells. (E) TUNEL‐positive cells were counted to calculate the apoptosis index. †Significant difference in the apoptosis index from control. ‡Highly significant difference in the apoptosis index from control. n, number of tumors assessed. (F) Tumors from mice treated with pcDNA3.1 (lane 1), Kalli‐pcDNA3.1 (lane 2), meloxicam (lane 3), or Kalli‐pcDNA3.1 + meloxicam (lane 4), were homogenized and subjected to Western blot analysis to detect expression of Bcl‐2, Bax, and active caspase‐3. β‐actin served as an internal control.
Tumoral angiogenesis. Tumor sections were immunostained with an anti‐CD31 Ab for detecting tumoral vascularization. Representative tumor sections were prepared from mice treated with pcDNA3.1 (Fig. 5A), Kalli‐pcDNA3.1 (Fig. 5B), meloxicam (Fig. 5C), and Kalli‐pcDNA3.1 + meloxicam (Fig. 5D). The microvessels were counted to record the microvessel density. A significant reduction in microvessel density was observed in tumors treated with Kalli‐pcDNA3.1 (19.6 ± 5.7), compared with pcDNA3.1 (43.7 ± 9.6) (Fig. 5E). Although meloxicam therapy slightly reduced the microvessel density compared to control, the difference did not reach significance. The combinational therapy resulted in a highly significantly reduction in microvessel density by almost twofold (P < 0.001) compared to control, and a significant reduction compared to Kalli‐pcDNA3.1 and meloxicam monotherapies (both P < 0.05). Next, we investigated whether Kalli‐pcDNA3.1 or meloxicam or the combination could modulate the expression of VEGF, a key regulator of angiogenesis. However, there was little change in VEGF or bFGF expression in tumors from the four groups, detected by Western blot analysis (Fig. 5F).
Figure 5.
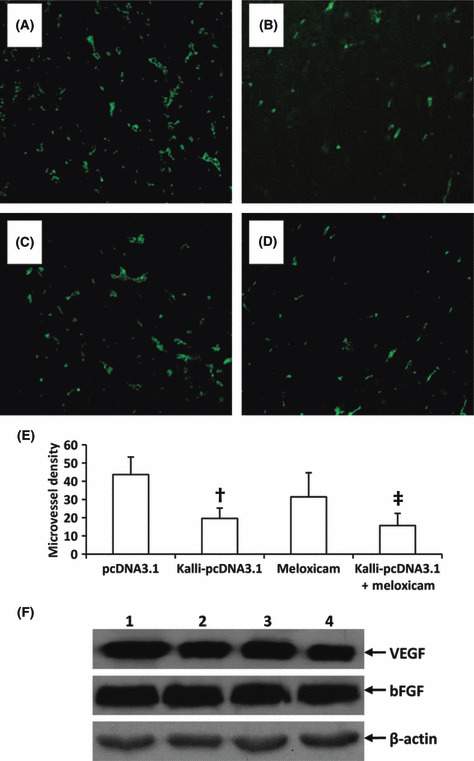
Kallistatin gene and the combinational therapies inhibit tumor angiogenesis. Representative tumor sections prepared from mice two weeks after treatment with pcDNA3.1 (control) (A), Kalli‐pcDNA3.1 (B), meloxicam (C), or Kalli‐pcDNA3.1 + meloxicam (D). Tumor microvessels in sections were fluorescently stained with anti‐CD31 Ab and counted in blindly chosen random fields to record microvessel density (E). †Significant difference in microvessel density from control. ‡Highly significant difference in microvessel density from control. Five tumors accessed per group. (F) Homogenates of tumors from mice treated with pcDNA3.1 (lane 1), Kalli‐pcDNA3.1 (lane 2), meloxicam (lane 3), or meloxicam + Kalli‐pcDNA3.1 (lane 4) were subjected to Western blot analysis with Abs against vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), and β‐actin.
Discussion
Kallistatin has been shown to inhibit VEGF or bFGF through induced proliferation, migration, and adhesion of endothelial cells and to attenuate bFGF through induced capillary density in mice.( 8 , 34 ) We have previously reported that gene transfer of recombinant adeno‐associated virus–kallistatin inhibited proliferation of vascular endothelial cells in vitro.( 13 ) Accordingly the present study has shown that kallistatin gene transfer inhibited tumor angiogenesis. Our results are also supported by two reports where kallistatin significantly reduced spontaneous angiogenesis in a rat model of hind limb ischemia,( 9 ) and a single intratumoral injection of kallistatin gene inhibited growth and angiogenesis of human breast tumor xenografts in athymic mice.( 8 ) The present study has also shown that kallistatin displayed little effect on the expression of VEGF or bFGF. Its anti‐angiogenic activity may be explained by its molecular feature as a heparin binding factor, which is similar to VEGF and bFGF.( 35 ) Kallistatin can compete with VEGF and bFGF binding to heparan‐sulfate proteoglycans, a low affinity‐binding site, thus inhibiting the binding activity of VEGF and bFGF and the angiogenesis signaling cascades induced by VEGF and bFGF.( 8 ) In addition, kallistatin has shown multiple biologic functions including blood pressure regulation and vasculature relaxation.( 8 , 9 ) Kallistatin shares a considerable sequence homology with serine proteinase inhibitors.( 35 ) Many serine proteinase inhibitors have been shown to have anti‐angiogenic activity.( 36 , 37 , 38 ) All of these features have made kallistatin a promising angiogenesis inhibitor, although further exploration for the mechanism is required.
In the present study, kallistatin gene transfer inhibited the proliferation and apoptosis of HCC cells in vivo, in accordance with our previous in vitro assays.( 13 ) But the mechanisms are not clear as kallistatin showed no effect on expression of PCNA, nor the apoptosis‐related proteins including Bal‐2, Bax, and caspase‐3. As caspase‐3 is the key ‘effector’ protease in the apoptotic cascade, the results might suggest that kallistatin‐induced cell apoptosis may be caspase‐independent. There is increasing evidence that VEGF has paracrine functions in tumor biology, which is able to directly stimulate growth of tumor cells.( 39 , 40 ) Kallistatin may act by competing with VEGF and binding to heparan‐sulfate proteoglycans, and inhibit VEGF binding activity and cell proliferation. Kallistatin is originally identified as a tissue kallikrein‐binding protein.( 41 ) Tissue kallikreins play important roles in tumor growth, angiogenesis, and invasion.( 42 ) By binding to tissue kallikreins, kallistatin might inhibit cell viability. In addition, the inhibition of vascularization by kallistatin might also restrict the supply of tumor cells with survival factors provided by endothelial cells and/or the circulation, as endothelial cells produce at least 20 paracrine factors, such as platelet‐derived growth factor, interleukin 6, and heparin‐binding epithelial growth factor.( 13 ) Kallistatin inhibits the proliferation of endothelial cells,( 14 ) thus being able to reduce production of paracrine factors.
Several selective COX‐2 inhibitors, including meloxicam, have been shown to inhibit proliferation and/or induce apoptosis of cells from a variety of tumors including HCC in vitro and in vivo.( 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 ) The present study has shown that meloxicam inhibited cell proliferation in situ, supported by our previous report where meloxicam inhibited proliferation of HepG2 cells in vitro.( 31 ) Both studies have shown expression of PCNA, a key cell proliferation marker, was reduced by meloxicam. The mechanisms by which meloxicam induces apoptosis are not entirely clear, although it has been reported that inhibition of COX‐2 resulted in activation of caspase‐9 and downstream execution‐caspases in liver tumor cells.( 28 ) Cellular apoptosis is triggered by the mitochondrial intrinsic and/or death‐receptor extrinsic pathway.( 43 ) A number of anticancer drugs induce apoptosis through regulation of the Bcl‐2 family, the most prominent regulators of apoptosis in cancer cells.( 44 ) Bcl‐2, located on the membrane of mitochondria, is an anti‐apoptotic protein, whereas Bax directly binds to Bcl‐2 and inhibits its function.( 44 ) In the present study, meloxicam upregulated Bax expression and increased activation of caspase‐3, indicating the intrinsic pathway was involved in meloxicam‐induced apoptosis, although it had little effect on Bcl‐2 expression. However, our previous report( 31 ) showed that Fas, a molecule in the death‐receptor extrinsic pathway, was upregulated by meloxicam, suggesting the extrinsic pathway might also participate in the apoptosis induced by meloxicam. In addition, the caspase‐independent pathways might be also involved in COX‐2‐inhibition‐induced apoptosis, as Yamanaka et al. ( 45 ) reported that selective COX‐2 inhibitors NS398 and CAY10404 induced apoptosis of human HCC SK‐Hep1 cells by upregulating TRAIL receptors and downregulating survivin.
Our results have shown that meloxicam had weak anti‐angiogenic activity and little effect on expression of VEGF and bFGF. COX‐2 inhibitors block the activation of MAPK induced by VEGF in endothelial cells.( 46 ) Inhibition of COX‐2 suppressed integrin αVβ3‐dependent migration of endothelial cells and angiogenesis induced by fibroblast growth factor‐2.( 47 ) It has been reported that the COX‐2 inhibitor etodolac downregulated expression of MMP‐9, and reduced the degradation of extracellular matrix, thus inhibiting tumor angiogenesis and hepatic metastases of colon cancer.( 48 )
The present study has for the first time shown that concomitant treatment of mice bearing subcutaneous human HCC tumors with kallistatin gene therapy and meloxicam results in increased inhibition of tumor growth. The major role of kallistatin was its anti‐angiogenic activity, whereas meloxicam inhibited cell proliferation by downregulating expression of PCNA and induced cell apoptosis by increasing Bax expression and caspase‐3 activation. Whether the interaction between kallistatin and meloxicam is synergistic or additive is debatable, but at least each agent did not impair the other’s anticancer activity. Irrespective of the mechanism, the enhanced therapeutic efficacy and benefit obtained by combining kallistatin with meloxicam is without question. By targeting vascular endothelial cells, kallistatin increased the antiproliferative and pro‐apoptotic activities mediated by meloxicam. Some authors have speculated that anti‐angiogenic therapy targeting the tumor vasculature could hinder blood‐borne therapeutic agents from reaching tumor cells.( 49 , 50 ) However, a combination of the anti‐angiogenic agent, tumor necrosis factor‐α, and the cytotoxic agent, melphalan, was shown to generate a higher intratumoral concentration of melphalan than melphalan alone.( 51 ) Anti‐angiogenic therapy might ‘normalize’ the tumor vasculature, thereby helping other anticancer agents to reach tumor cells.( 52 ) However, anti‐angiogenic therapy itself is probably best carried out in combination with other forms of therapy, considering the wide range of angiogenic factors produced by tumor cells and the biological heterogeneity of tumor‐induced blood vessels.( 53 ) In the present study, meloxicam augmented the anticancer activity of kallistatin by inhibiting the proliferation and inducing apoptosis of tumor cells.
Abbreviations
- bFGF
basic fibroblast growth factor
- HCC
hepatocellular carcinoma
- PCNA
proliferating cell nuclear antigen
- VEGF
vascular endothelial growth factor
Acknowledgment
This work was supported in part by grants from the National Natural Scientific Foundation of China (30571808, 30872987).
References
- 1. Befeler AS, Di Bisceglie AM. Hepatocellular carcinoma: diagnosis and treatment. Gastroenterology 2002; 122: 1609–19. [DOI] [PubMed] [Google Scholar]
- 2. Schafer DF, Sorrell MF. Hepatocellular carcinoma. Lancet 1999; 353: 1253–7. [DOI] [PubMed] [Google Scholar]
- 3. Kinoshita S, Hirai R, Yamano T et al. Angiogenesis inhibitor TNP‐470 can suppress hepatocellular carcinoma growth without retarding liver regeneration after partial hepatectomy. Surg Today 2004; 34: 40–6. [DOI] [PubMed] [Google Scholar]
- 4. Ishikawa H, Nakao K, Matsumoto K et al. Antiangiogenic gene therapy for hepatocellular carcinoma using angiostatin gene. Hepatology 2003; 37: 696–704. [DOI] [PubMed] [Google Scholar]
- 5. Graepler F, Verbeek B, Graeter T et al. Combined endostatin/sFlt‐1 antiangiogenic gene therapy is highly effective in a rat model of HCC. Hepatology 2004; 41: 879–86. [DOI] [PubMed] [Google Scholar]
- 6. Ma L, Luo L, Qiao H et al. Vasostatin synergizes with B7H3‐mediated immunotherapy to eradicate hepatocellular carcinomas. J Hepatol 2006; 46: 98–106. [DOI] [PubMed] [Google Scholar]
- 7. Chao J, Chai KX, Chen LM et al. Tissue kallikrein‐binding protein is a serpin, I: purification,characterization, and distribution in normotensive and spontaneously hypertensive rats. J Biol Chem 1990; 265: 16394–401. [PubMed] [Google Scholar]
- 8. Miao RQ, Agata J, Chao L et al. Kallistatin is a new inhibitor of angiogenesis and tumor growth. Blood 2002; 100: 3245–52. [DOI] [PubMed] [Google Scholar]
- 9. Chao J, Miao RQ, Chen V, Chen LM, Chao L. Novel roles of kallistatin, a specific tissue kallikrein inhibitor, in vascular remodeling. Biol Chem 2001; 382: 15–21. [DOI] [PubMed] [Google Scholar]
- 10. Zhu B, Lu L, Cai W et al. Kallikrein‐binding protein inhibits growth of gastric carcinoma by reducing vascular endothelial growth factor production and angiogenesis. Mol Cancer Ther 2007; 6: 3297–306. [DOI] [PubMed] [Google Scholar]
- 11. Lu L, Yang Z, Zhu B et al. Kallikrein‐binding protein suppresses growth of hepatocellular carcinoma by anti‐angiogenic activity. Cancer Lett 2007; 257: 97–106. [DOI] [PubMed] [Google Scholar]
- 12. Diao Y, Ma J, Xiao WD et al. Inhibition of angiogenesis and HCT‐116 xenograft tumor growth in mice by kallistatin. World J Gastroenterol 2007; 13: 4615–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tse LY, Sun X, Jiang H et al. Adeno‐associated virus‐mediated expression of kallistatin suppresses local and remote hepatocellular carcinomas. J Gene Med 2008; 10: 508–17. [DOI] [PubMed] [Google Scholar]
- 14. Ristimäki A, Honkanen N, Jänkälä H, Sipponen P, Härkönen M. Expression of cyclooxygenase‐2 in human gastric carcinoma. Cancer Res 1997; 57: 1276–80. [PubMed] [Google Scholar]
- 15. Wolff H, Saukkonen K, Anttila S, Karjalainen A, Vainio H, Ristimäki A. Expression of cyclooxygenase‐2 in human lung carcinoma. Cancer Res 1998; 58: 4997–5001. [PubMed] [Google Scholar]
- 16. Hwang D, Scollard D, Byrne J, Levine E. Expression of cyclooxygenase‐1 and cyclooxygenase‐2 in human breast cancer. J Natl Cancer Inst 1998; 90: 455–60. [DOI] [PubMed] [Google Scholar]
- 17. Zimmermann KC, Sarbia M, Weber AA, Borchard F, Gabbert HE, Schrör K. Cyclooxygenase‐2 expression in human esophageal carcinoma. Cancer Res 1999; 59: 198–204. [PubMed] [Google Scholar]
- 18. Tucker ON, Dannenberg AJ, Yang EK et al. Cyclooxygenase‐2 expression is up‐regulated in human pancreatic cancer. Cancer Res 1999; 59: 987–90. [PubMed] [Google Scholar]
- 19. Shiota G, Okubo M, Noumi T et al. Cyclooxygenase‐2 expression in hepatocellular carcinoma. Hepatogastroenterology 1999; 46: 407–12. [PubMed] [Google Scholar]
- 20. Bae SH, Jung ES, Park YM et al. Expression of cyclooxygenase‐2 (COX‐2) in hepatocellular carcinoma and growth inhibition of hepatoma cell lines by a COX‐2 inhibitor, NS‐398. Clin Cancer Res 2001; 7: 1410–8. [PubMed] [Google Scholar]
- 21. Kern MA, Schöneweiss MM, Sahi D et al. Cyclooxygenase‐2 inhibitors suppress the growth of human hepatocellular carcinoma implants in nude mice. Carcinogenesis 2004; 25: 1193–9. [DOI] [PubMed] [Google Scholar]
- 22. Xin B, Yokoyama Y, Shigeto T, Futagami M, Mizunuma H. Inhibitory effect of meloxicam, a selective cyclooxygenase‐2 inhibitor, and ciglitazone, a peroxisome proliferator‐activated receptor gamma ligand, on the growth of human ovarian cancers. Cancer 2007; 110: 791–800. [DOI] [PubMed] [Google Scholar]
- 23. Hattori K, Iida K, Joraku A, Tsukamoto S, Akaza H, Oyasu R. Chemopreventive effects of cyclooxygenase‐2 inhibitor and epidermal growth factor‐receptor kinase inhibitor on rat urinary bladder carcinogenesis. BJU Int 2006; 97: 640–3. [DOI] [PubMed] [Google Scholar]
- 24. Tsuchida A, Itoi T, Kasuya K et al. Inhibitory effect of meloxicam, a cyclooxygenase‐2 inhibitor, on N‐nitrosobis (2‐oxopropyl) amine induced biliary carcinogenesis in Syrian hamsters. Carcinogenesis 2005; 26: 1922–8. [DOI] [PubMed] [Google Scholar]
- 25. Teh SH, Hill AK, Foley DA, McDermott EW, O’Higgins NJ, Young LS. COX inhibitors modulate bFGF‐induced cell survival in MCF‐7 breast cancer cells. J Cell Biochem 2004; 91: 796–807. [DOI] [PubMed] [Google Scholar]
- 26. Tsubouchi Y, Mukai S, Kawahito Y et al. Meloxicam inhibits the growth of non‐small cell lung cancer. Anticancer Res 2000; 20: 2867–72. [PubMed] [Google Scholar]
- 27. Goldman AP, Williams CS, Sheng H et al. Meloxicam inhibits the growth of colorectal cancer cells. Carcinogenesis 1998; 19: 2195–9. [DOI] [PubMed] [Google Scholar]
- 28. Kern MA, Schubert D, Sahi D et al. Proapoptotic and antiproliferative potential of selective cyclooxygenase‐2 inhibitors in human liver tumor cells. Hepatology 2002; 36: 885–94. [DOI] [PubMed] [Google Scholar]
- 29. Kern MA, Schirmacher P, Breinig M. Significance of cyclooxygenase‐2 as a chemotherapeutic target in hepatocellular carcinoma [Article in German]. Verh Dtsch Ges Pathol 2007; 91: 257–68. [PubMed] [Google Scholar]
- 30. Dohmen K, Okabe H, Ishibashi H. Regression of hepatocellular carcinoma due to cyclooxygenase (COX)‐2 inhibitor. Am J Gastroenterol 2006; 101: 2437–8. [DOI] [PubMed] [Google Scholar]
- 31. Li J, Chen X, Dong X, Xu Z, Jiang H, Sun X. Specific COX‐2 inhibitor, meloxicam, suppresses proliferation and induces apoptosis in human HepG2 hepatocellular carcinoma cells. J Gastroenterol Hepatol 2006; 21: 1814–20. [DOI] [PubMed] [Google Scholar]
- 32. Liu F, Wang P, Jiang X et al. Antisense hypoxia‐inducible factor 1 α gene therapy enhances the therapeutic efficacy of doxorubicin to combat hepatocellular carcinomas. Cancer Sci 2008; 99: 2055–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen H, Sun B, Pan S, Jiang H, Sun X. Dihydroartemisinin inhibits growth of pancreatic cancer cells in vitro and in vivo. Anticancer Drugs 2009; 20: 131–40. [DOI] [PubMed] [Google Scholar]
- 34. Miao RQ, Chen V, Chao L et al. Structural elements of kallistatin required for inhibition of angiogenesis. Am J Physiol Cell Physiol 2003; 284: C1604–13. [DOI] [PubMed] [Google Scholar]
- 35. Zhou GX, Chao L, Chao J. Kallistatin: a novel human tissue kallikrein inhibitor: purification, characterization, and reactive center sequence. J Biol Chem 1992; 267: 25873–80. [PubMed] [Google Scholar]
- 36. Dawson DW, Volpert OV, Gillis P et al. Pigment epithelium‐derived factor: a potent inhibitor of angiogenesis. Science 1999; 285: 245–8. [DOI] [PubMed] [Google Scholar]
- 37. O’Reilly MS, Piri e‐Shepherd S, Lane WS, Folkman J. Antiangiogenic activity of the cleaved conformation of the serpin antithrombin. Science 1999; 285: 1926–8. [DOI] [PubMed] [Google Scholar]
- 38. Zhang M, Volpert O, Shi YH, Bouck N. Maspin is an angiogenesis inhibitor. Nat Med 2000; 6: 196–9. [DOI] [PubMed] [Google Scholar]
- 39. Schoeffner DJ, Matheny SL, Akahane T et al. VEGF contributes to mammary tumor growth in transgenic mice through paracrine and autocrine mechanisms. Lab Invest 2005; 85: 608–23. [DOI] [PubMed] [Google Scholar]
- 40. Barr MP, Byrne AM, Duffy AM et al. A peptide corresponding to the neuropilin‐1‐binding site on VEGF (165) induces apoptosis of neuropilin‐1‐expressing breast tumour cells. Br J Cancer 2005; 92: 328–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chao J, Tillman DM, Wang MY et al. Identification of a new tissue‐kallikrein‐binding protein. Biochem J 1986; 239: 325–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Borgono CA, Diamandis EP. The emerging roles of human tissue kallikreins in cancer. Nat Rev Cancer 2004; 4: 876–90. [DOI] [PubMed] [Google Scholar]
- 43. Hengartner MO. The biochemistry of apoptosis. Nature 2000; 407: 770–6. [DOI] [PubMed] [Google Scholar]
- 44. Thomadaki H, Scorilas A. BCL2 family of apoptosis‐related genes: functions and clinical implications in cancer. Crit Rev Clin Lab Sci 2006; 43: 1–67. [DOI] [PubMed] [Google Scholar]
- 45. Yamanaka Y, Shiraki K, Inoue T et al. COX‐2 inhibitors sensitize human hepatocellular carcinoma cells to TRAIL‐induced apoptosis. Int J Mol Med 2006; 18: 41–7. [PubMed] [Google Scholar]
- 46. Jones MK, Wang H, Peskar BM et al. Inhibition of angiogenesis by nonsteroidal anti‐inflammatory drugs: insight into mechanisms and implications for cancer growth and ulcer healing. Nat Med 1999; 5: 1418–23. [DOI] [PubMed] [Google Scholar]
- 47. Dormond O, Foletti A, Paroz C, Ruegg C. NSAIDs inhibit alphaVbeta3 integrin‐mediated and Cdc42/Rac‐dependent endothelial cell spreading, migration and angiogenesis. Nat Med 2001; 7: 1041–7. [DOI] [PubMed] [Google Scholar]
- 48. Ishizaki T, Katsumata K, Tsuchida A et al. Etodolac, a selective cyclooxygenase‐2 inhibitor, inhibits liver metastasis of colorectal cancer cells via the suppression of MMP‐9 activity. Int J Mol Med 2006; 17: 357–62. [PubMed] [Google Scholar]
- 49. Yuan F, Chen Y, Dellian M, Safabakhsh N, Ferrara N, Jain RK. Time‐dependent vascular regression and permeability changes in established human tumor xenografts induced by an anti‐vascular endothelial growth factor/vascular permeability factor antibody. Proc Natl Acad Sci USA 1996; 93: 14765–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Griscelli F, Li H, Bennaceur‐Griscelli A et al. Angiostatingene transfer: inhibition of tumor growth in vivo by blockage of endothelial cell proliferation associated with a mitosis arrest. Proc Natl Acad Sci USA 1998; 95: 6367–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nooijen PT, Manusama ER, Eggermont AM et al. Synergistic effects of TNF‐alpha and melphalan in an isolated limb perfusion model of rat sarcoma: a histopathological, immunohistochemical and electron microscopical study. Br J Cancer 1996; 74: 1908–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jain RK. Normalizing tumor vasculature with anti‐angiogenic therapy: a new paradigm for combination therapy. Nat Med 2001; 7: 987–9. [DOI] [PubMed] [Google Scholar]
- 53. Abdollahi A, Hlatky L, Huber PE. Endostatin: the logic of antiangiogenic therapy. Drug Resist Updat 2005; 8: 59–74. [DOI] [PubMed] [Google Scholar]