

Abstract
Cellular levels of products from both oncogenes and tumor suppressor genes in normal cells need to be critically regulated to avoid malignant transformation. These products are often controlled by the ubiquitin proteasome pathway, the specific degradation mechanism in the cell. E3 ubiquitin ligases polyubiquitylate their specific substrates by collaborating with E1 and E2, and then the modified substrates are degraded in the proteasome. Mdm2 targets p53 and retinoblastoma protein, two major tumor suppressor gene products, for ubiquitin‐dependent degradation. SCFSkp2 targets other tumor suppressor gene products and CDK inhibitors such as p130, Tob1, p27 Kip1 , p57 Kip2 , and p21 Cip1 . Therefore, both E3 ligases act like oncogene products. In contrast, degradation of several oncogene products, such as Cyclin E, Notch, c‐Myc, c‐Jun, and c‐Myb, are mediated by SCFFbw7. Fbw7 is often deleted or mutated in human cancers and acts like a tumor suppressor. As well as growth factor receptors and signal transduction regulators, DNA repair‐related proteins are also regulated via the ubiquitin–proteasome pathway mediated by their specific E3 ligases. The stabilization of oncogene products and enhanced degradation of tumor suppressor gene products or DNA repair proteins might be associated with carcinogenesis and malignant progression, due to defects or the abnormal expression of their E3 ligases. (Cancer Sci 2009)
Ubiquitin–proteasome system in cancer
The ubiquitin–proteasome system controls the abundance of a number of cellular proteins, particularly short‐lived regulatory proteins such as p53, c‐Myc, IκBα, β‐catenin, p27 Kip1 , and cyclins.( 1 ) Polyubiquitin conjugation of lysine residues in proteins as a result of collaboration with a ubiquitin‐activating enzyme (E1), a ubiquitin‐conjugation enzyme (E2), and a ubiquitin ligase (E3) is required for selective recognition and degradation by the 26S proteasome.( 2 ) The specificity of target protein selection is defined by E3 ubiquitin ligases.( 2 )
The involvement of E1 in cancer has not been described, and only a few reports have linked E2 to cancer development. By contrast, a large amount of evidence indicates that deregulation of E3 ligase often results in cancer development. As shown in Figure 1, the stabilization of oncogene products due to inactivation of their E3 ligases and enhanced degradation of tumor suppressor gene products due to overexpression of their responsible E3 ligases seems to play a role in carcinogenesis or malignant progression. Therefore, E3 ligases that target oncogene products or tumor suppressor gene products are involved in the maintenance of a normal level of oncogene products or tumor supressor gene products, respectively.
Figure 1.
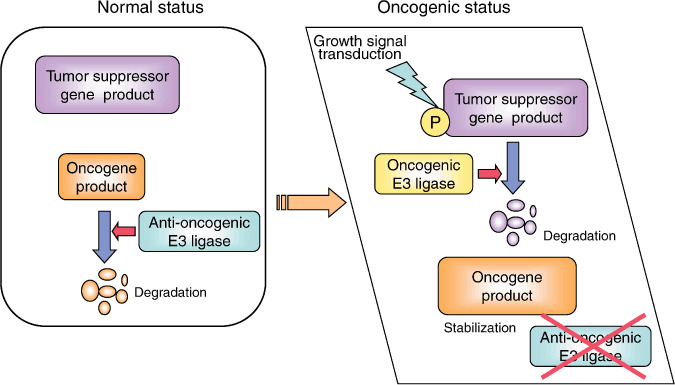
Accumulation of oncogene products and enhanced degradation of tumor suppressor gene products in cancer cells. In normal cells, tumor suppressor gene products negatively regulate transformation and cancerous growth and oncogene products are quickly degraded by their E3 ligases, which target oncogenes as anti‐oncogenic E3 ligases. Defects in the anti‐oncogenic E3 ligase and overexpression of the oncogenic E3 ligases, which target tumor suppressor gene products, often promote carcinogenesis and cancerous growth.
E3 ubiquitin ligases associated with cancer. E3 ubiquitin ligases are classified into several groups. As shown in Figure 2, E3 ligases of the HECT‐type, single RING‐type, SCF‐type, ECV‐type, and Cul4‐base selectively target oncogenic proteins or tumor suppressive proteins and are involved in carcinogenesis or tumor development.( 1 )
Figure 2.
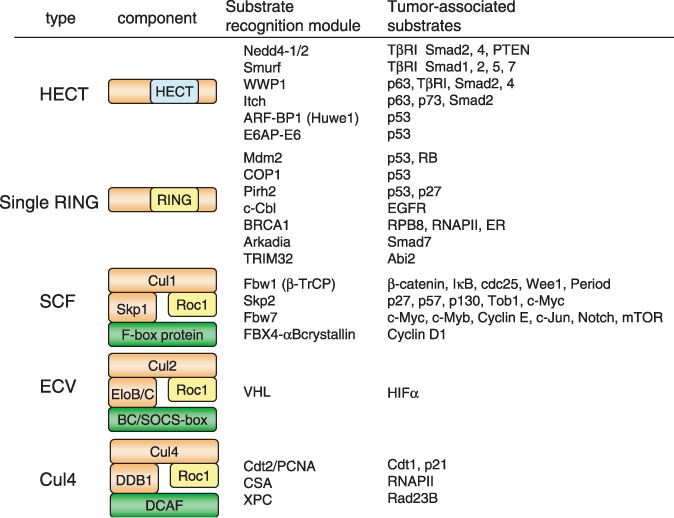
Tumor‐associated E3 ubiquitin ligases and their substrates. Type variations and their components of E3 ubiquitin ligases associated with tumors are indicated on the left side. They are classified into five types: HECT‐type, single RING‐type, SCF‐type, ECV‐type, and Cul4‐base. Specific E3 ligases and their reported substrates associated with cancer are also indicated. The green boxes are substrate recognition modules, and yellow boxes indicate the RING active domain, which receives ubiquitin from E2.
The SCF‐type E3 ligase controls many cancer‐associated proteins including both tumor suppressor proteins and oncogene products.( 3 ) It consists of four components: the invariable subunits Skp1, Cul1, and Rbx1/Roc1 and a variable subunit F‐box protein that serves as a receptor for target proteins and thereby determines target specificity (Fig. 2).( 4 )
Generally one substrate is regulated by more than one ligase and one ligase targets several substrates, whereas the E3 ligase seems to be substrate selective. Target recognition by the E3 ligase often requires modifications of the substrates such as phosphorylation. For example, receptor subunits of SCF‐type E3 ligases such as Fbw1 (β‐TRCP), Fbw7, and Skp2 often recognize their responsible substrate in a phosphorylation‐dependent manner.( 3 , 4 )β‐TRCP recognizes the D‐pS‐G‐(X)2+n‐pS destruction motif in its target, the serines of which are phosphorylated by specific kinases.( 5 , 6 ) The interaction between Fbw7 and its substrates is also dependent on phosphorylation of the substrate within a motif called the Cdc4‐phosphodegron (CPD). The eight WD40 repeats in Fbw7 form a β‐propeller structure creating a phospho‐epitope binding pocket that can recognize phosphorylated CPD. Several reports indicate that the CPD recognized by Fbw7 contains the motif (L)‐X‐pT/pS‐P‐(P)‐X‐pS/pT/E/D (where X is any amino acid).( 3 , 7 , 8 , 9 , 10 )
Among the many F‐box proteins, Skp2 and Fbw7 have been well characterized and have been shown to control the abundance of proteins associated with human cancers.( 11 ) As shown in Figure 2, Skp2 can ubiquitylate various proteins, including p27 Kip1 ,( 12 , 13 , 14 ) p57 Kip2 ,( 15 ) p130,( 16 ) Tob1,( 17 ) and c‐Myc.( 18 ) Skp2 is mainly involved in degradation of tumor suppressor proteins such as Cdk inhibitors and p130 among others. Overexpression of Skp2 is observed in various human cancers. It has been reported that GA‐binding protein is involved in transcriptional upregulation of the Skp2 gene in tumor cells.( 19 ) Moreover, gene amplifications of the Skp2 gene were found in human gastric cancers.( 20 ) Therefore, Skp2 is suggested to be an oncogene. Fbw7 mainly targets the degradation of oncogenic proteins such as Cyclin E,( 7 ) c‐Myc,( 8 , 9 ) c‐Jun,( 10 ) c‐Myb,( 21 , 22 ) Notch,( 23 ) and mTOR.( 24 ) Moreover, deletion and mutation of Fbw7 is found in human cancers.( 25 ) Conditional inactivation of Fbw7 manifests thymic hyperplasia because of c‐Myc accumulation, resulting in the development of thymic lymphoma.( 26 ) Therefore, Fbw7 is believed to be a tumor suppressor gene that contributes to the negative regulation of the cellular content of oncogene products.
Breast cancer susceptibility gene (BRCA) 1, a RING finger E3 ligase, plays a crucial role in the DNA damage response.( 27 ) The BRCA1–BRCA1‐associated RING domain (BARD) 1 heterodimeric RING finger complex has ubiquitin ligase activity, whereas individually, BRCA1 and BARD1 have very low ubiquitin ligase activities. It has been reported that RNA polymerase II (RNAPII) and RPB8 are polyubiquitylated by BRCA1–BRCA1 in response to DNA damage without a decrease in protein amount.( 27 ) Somatic mutation of BRCA1 or BARD1 is often identified in breast cancer and ovarian cancer. Ubiquitin ligase activity of the complex is important for the prevention of breast and ovarian cancer development.( 28 )
As shown in Figure 2, HECT‐type E3 ligases control the TGFβ‐Smad pathway,( 29 , 30 ) whereas some HECT‐type E3 ligases target degradation of the p53 family of proteins. Because these HECT‐type E3 ligases are often overexpressed in human cancers, they may be associated with carcinogenesis or tumor cell growth. E3 ligases with an intrinsic single RING‐finger domain such as Mdm2, Pirh2 and COP1 also target p53 (Fig. 2).( 31 ) These single RING‐type E3 ligases are often overexpressed in human cancers and thereby carcinogenesis or tumor cell growth should be promoted. In the next portion of this review, we focus on the degradation mechanism of tumor suppressive proteins and oncogenic proteins.
Degradation of tumor suppressive proteins
p53 and retinoblastoma family proteins.
-
1
The tumor suppressor gene product p53 plays an important role in apoptosis, DNA repair, and the cell cycle. Its cellular expression level is differentially regulated by transcriptional control and the ubiquitin–proteasome system.( 31 ) In the presence of human papilloma virus E6 protein, a cellular HECT domain‐containing protein, E6AP, ubiquitylates p53 in collaboration with E6 protein.( 32 ) Human papilloma virus promotes the degradation of p53 so that it can promote its own amplification in the host cell. On the other hand, the HECT‐type E3 ligase ARF‐BP1 (Huwe1) is also involved in the ubiquitin‐dependent degradation of p53 (Fig. 3).( 33 ) Mdm2, COP1, and Pirh2, all RING‐finger‐type ubiquitin ligases, ubiquitylate p53 without accompanied virus infection.( 34 , 35 , 36 ) Following DNA damage, ATM/ATR kinase is activated and phosphorylates p53, Mdm2, and COP1. In addition to the phosphorylation of p53, phosphorylation of Mdm2 and COP1 reduces their interaction with p53. p53 autonomously stimulates transcription of its E3 ligase genes, Mdm2, Pirh2, and COP1, for feedback attenuation. Overexpression of Pirh2 in human lung cancer has been reported and thus is thought to contribute to the downregulation of p53, which promotes carcinogenesis.( 37 ) This evidence is contradictory because Pirh2 can promote degradation of not only wild‐type p53 but also mutant p53, which functions as an oncoprotein in a dominant‐negative manner against wild‐type p53, and enhancement of the degradation of mutant p53 may contribute to tumor suppression. Thus, further study is required to clarify the involvement of Pirh2 in human cancers.
Figure 3.
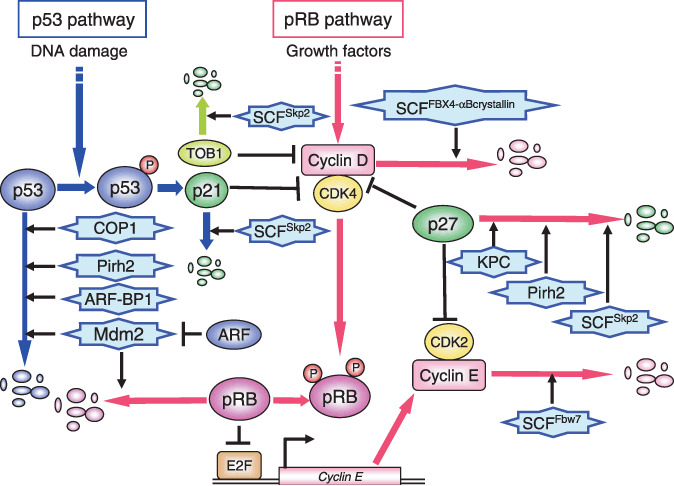
E3 ubiquitin ligases in the regulation of the p53 and retinoblastoma protein (pRB) pathway. The tumor suppressor gene product p53 is an unstable protein that is degraded by several E3 ligases such as Mdm2, COP1, Pirh2, and ARF‐BP1 in the normal cell cycle. After DNA damage, p53 is stabilized by phosphorylation via the ATM/ATR pathway. In addition, ARF, one of the alternative products of the INK4/ARF locus, binds to Mdm2 and thereby prevents it from antagonizing p53. Mdm2 promotes ubiquitylation and degradation of not only p53 but also pRB. At late G1 phase, the CDK inhibitor p27 Kip1 is degraded and Cyclin D–Cdk4/6 kinases are activated to phosphorylate and inactivate pRB. Then the target transcription factors are released from pRB and activate the transcription of growth‐promoting genes such as Cyclin E, thus promoting cell cycle progression from G1 to S phase. SCFSkp2 KPC complex and Pirh2 target p27 for ubiquitin‐dependent degradation. SCFFBX4‐αB crystallin promotes cyclin D1 degradation in S phase. SCFFbw7 targets cyclin E for degradation.
Mdm2 also binds to p73, which is a p53 family protein, but does not promote its degradation. p73 and p63 but not p53 are regulated by Itch, a Hect‐type E3 ligase.( 38 , 39 ) p73 is induced to accumulate in response to DNA damage, which in turn downregulates Itch expression, whereas accumulated p73 promotes G1/S cell cycle arrest and/or apoptosis. p63 is not mutated during cancer development and is often amplified in cancer. Using alternative promoters, the p63 gene expresses two proteins, δNp63 and TAp63. TAp63 induces cell apoptosis, whereas δNp63 has the opposite function. Both the p63 gene products are also targets of WWP1, which is expressed under DNA damage conditions in a p53‐dependent manner. WWP1 may govern cell survival by targeting different p63 proteins.( 40 )
Another tumor suppressor gene product, retinoblastoma protein (pRB), plays critical roles in the regulation of the G1 phase of the cell cycle and tumor suppression.( 41 ) During late G1 phase, Cyclin D–Cdk4/6 kinases phosphorylate and inactivate pRB, which plays a crucial role in the pRB pathway (Fig. 3).( 41 , 42 ) Then target transcription factors such as E2F are released and activate the transcription of growth‐promoting genes such as Cyclin E and DHFR, thus promoting cell cycle progression from G1 to S phase. Mdm2 regulates the function of not only p53 but also pRB via the ubiquitin‐dependent degradation of pRB (Fig. 3).( 43 ) pRB (but not other RB family proteins such as p107 and p130) was efficiently ubiquitylated by Mdm2 and thereby degraded via the proteasome.( 43 ) p130 is targeted by SCFSkp2 E3 ligase,( 16 ) whereas the E3 ligase for p107 is still unknown.
The Mdm2 gene is an oncogene that is amplified in 30–40% of human sarcomas without mutations in the p53 gene.( 44 ) Downregulation of pRB expression has been shown to be inversely correlated with the expression level of Mdm2 in human lung cancer.( 44 , 45 ) Because the binding region for p53 in Mdm2 is different from that for pRB, pRB may bind to Mdm2 without competition by p53. Therefore, it is expected that degradation of both p53 and pRB are accelerated by overexpression of Mdm2 in the tumor. In addition, ARF, one of the alternative products of the INK4/ARF locus, binds to Mdm2 and prevents its E3 activity (Fig. 3).( 43 , 46 , 47 ) In many human cancers, mutation or deletion frequently occurs in the INK4/ARF gene.( 48 ) Altogether, accelerated degradation of both p53 and pRB via these mechanisms may efficiently promote carcinogenesis.
MdmX, a structural homolog of Mdm2, is known to negatively regulate p53 function by enhancing the Mdm2‐mediated ubiquitylation and degradation of p53.( 49 ) In contrast, MdmX inhibited the Mdm2‐mediated ubiquitylation and destabilization of pRB.( 50 ) Therefore, MdmX may have different roles in the regulation of Mdm2 activity for ubiquitylation of pRB and p53.
CDK inhibitors. p27 Kip1 , a Cip/Kip type CDK inhibitor that inhibits both cyclin D–CDK4/6 and cyclin E/A–CDK2, is a critical negative regulator of G1/S phase progression.( 51 ) Degradation of p27 Kip1 promotes activation of these cyclin–CDK complexes and thereby the cell cycle progresses from the G1 to S phase. KPC has been reported as a ubiquitin ligase that targets cytoplasmic p27 Kip1 , which is exported from the nucleus in G0 and G1 phases.( 52 ) Pirh2, a RING‐type ubiquitin ligase, was also identified as a p27 Kip,1 ‐interacting protein, promoting the ubiquitylation and destabilization of p27 Kip1 .( 53 ) Pirh2 expression is increased from late G1 to S phase, and its distribution is in both the nucleus and cytoplasm. SCFSkp2 was the first‐identified E3 ligase for p27 Kip1 .( 12 ) It localizes to the nucleus and mediates p27 Kip1 degradation from early S phase. Therefore, from G1 to G2 phase, the degradation of p27 Kip1 is sequentially regulated by three systems that are coupled with cellular location and cell phase (Fig. 4). Reduction in Pirh2 results in an impairment of p27 Kip1 degradation and inhibition of cell cycle progression at the G1/S transition in a p53‐independent manner.( 53 ) Skp2 recognizes Thr187‐phosphorylated p27 Kip1 in collaboration with cdc kinase subunit 1 (Cks1) to promote polyubiquitylation of p27 Kip1 .( 14 ) p27 Kip1 has been found to accumulate in Skp2‐knockout mouse tissues and embryonic fibroblast cells.( 13 ) Many studies have shown that a low expression level of p27 Kip1 is associated with poor prognosis in a variety of human cancers, such as colon, breast, lung, stomach, head, and neck cancer.( 54 ) Some studies have shown high expression levels of Skp2 and Cks1 proteins in some human cancers such as non‐small cell lung carcinomas.( 54 , 55 ) Therefore, Skp2 and Cks1 are thought to be involved in the degradation of p27 Kip1 in human cancers. Overexpression of Pirh2 is also frequently observed and inversely correlated with p27 Kip1 levels in human head and neck cancers; moreover, high expression levels of Pirh2 have been significantly associated with the poor prognosis of these patients.( 56 ) Pirh2 may be involved in the enhanced degradation of p27 Kip1 in malignant cancers. The correlation of KPC with human cancers has not yet been reported.
Figure 4.
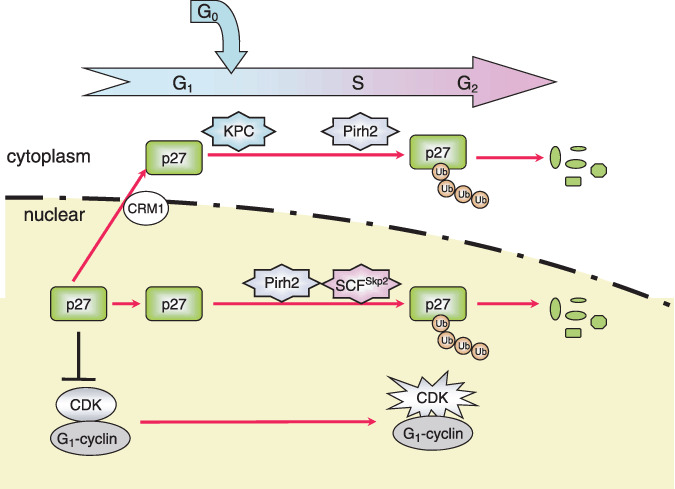
E3 ligases involved in p27 Kip1 degradation. The CDK inhibitor p27 Kip1 is degraded in late G1 phase by the ubiquitin–proteasome pathway, allowing cells to enter into S phase. The RING‐type E3 KPC complex targets cytoplasmic p27 Kip1 exported from the nucleus by the nuclear exporter, CRM1 in G0/G1 transition phases. Pirh2 promotes degradation of p27 Kip1 during G1/S transition. SCFSkp2 is mainly implicated in degradation of p27 during the S and G2 phases in the nucleus. Ub, Ubiquitin.
Another Cip/Kip CDK inhibitor, p57 Kip2 , is targeted for ubiquitin‐dependent degradation by SCFSkp2.( 15 ) It is reported that p21 Cip1 is targeted by not only SCFSkp2 but also adenoma polyposis coli (APC)/Ccdc20 and the CUL4 CRL ubiquitin ligase (CRL4) complex containing Cdt2 in cooperation with PCNA.( 57 , 58 , 59 ) The INK4 family CDK inhibitors p16INK4a and p19INK4d were thought to be relatively stable proteins because they have no lysine residues. However, it has been reported that the N‐terminal amino residues of not only p16INK4a and p19INK4d but also p19ARF are ubiquitylated, and thereby degraded by the proteasome.( 60 ) Further elucidation and identification of their E3 ligases is required.
Other tumor suppressor proteins. The tumor suppressor phosphatase and tensin homolog (PTEN), a critical regulator for multiple cellular processes and an antagonist of phosphatidylinositol 3‐kinase, is frequently mutated or deleted in various human cancers. The PTEN level is regulated by the HECT‐type E3 ligase NEDD4‐1 via ubiquitin‐mediated proteasomal degradation.( 61 ) Elimination of NEDD4‐1 expression inhibited the growth of a xenotransplanted tumor in a PTEN‐dependent manner.
Tob1, a member of the Tob/BTG family, is involved in the control of G1/S progression by suppressing cyclin D1 expression and acts as a tumor suppressor gene. SCFSkp2 E3 ligase is involved in the ubiquitin‐dependent degradation of Tob1 (Fig. 3).( 17 ) Tob1 is stabilized and cyclin D1 expression is suppressed in the Skp2‐depleted cells. Overexpression of Skp2 should destroy Tob1 and induce hyperexpression of cyclin D1, which may be involved in carcinogenesis or tumor cell growth.
The TGFβ–Smad pathway plays a crucial role in tumor progression and metastasis as well as normal development. The TGFβ family binds to heteromeric serine/threonine kinase receptors (TβRI/II) and transduces the signals to intracellular Smad proteins. TβRI and Smad proteins are regulated by the ubiquitin–proteasome pathway. Various E3 ligases including Smurf, WWP1, Itch, Nedd4, and Arkadia are involved in their regulation.( 30 )
Abl‐interactor (Abi) 2, a tumor suppressor and a cell migration inhibitor, specifically binds to Abl, which has been shown to be a non‐receptor tyrosine kinase involved in the regulation of intercellular signals. Kano et al. reported that Abi2 is targeted for ubiquitin‐dependent degradation by tripartite motif (TRIM) 32, which is a TRIM protein containing a RING finger.( 62 ) TRIM32 is highly expressed in human head and neck squamous cell carcinomas and may be a novel oncogene that promotes tumor growth, metastasis, and resistance to anticancer drugs.
Degradation of oncogenic proteins
Cyclins. Cyclins are often overexpressed in cancer cells, and thereby accelerate cancer cell growth. They are typically unstable, and their cellular amounts are controlled by both transcriptional induction and ubiquitin–proteasomal degradation. In G1 phase, the cyclin D–CDK4/6 complex is activated resulting in the phosphorylation of pRB, thereby promoting the transition from the G1 to S phase of the cell cycle.( 41 , 42 ) During S phase, cyclin D1 is phosphorylated by glycogen synthase kinase (GSK) 3β, which promotes nuclear export and ubiquitylation of cyclin D1 by the SCFFBX4‐αBcrystalline complex, leading to proteasome‐dependent degradation of cyclin D1 (Fig. 3).( 63 ) Cyclin E, which is involved in G1/S progression, is periodically induced in the late G1 portion of the cell cycle and degraded in mid‐S phase by SCFFbw7 (Fig. 3).( 7 ) Cyclins A and B act as the mitotic cyclins. Both are ubiquitylated by the APC/C complex, which targets many M/G1‐regulating factors, and are degraded in the proteasome to complete mitosis.( 1 )
Myc and Myb.
-
1
c‐Myc is controlled by two SCF‐type E3 ligases, SCFSkp2 and SCFFbw7. Skp2 interacts with c‐Myc and promotes its ubiquitylation and degradation. The interaction between Skp2 and c‐Myc occurs during the G1 to S phase transition of the cell cycle in normal lymphocytes.( 18 ) Skp2 mediates c‐Myc ubiquitylation by binding to the Myc box 2 domain. On the other hand, Fbw7 interacts with and thereby destabilizes c‐Myc in a manner dependent on phosphorylation of Thr‐58 and Ser‐62 in Myc box 1.( 8 ) Phosphorylation of c‐Myc on Thr‐58, the most frequent site of c‐myc mutations in lymphoma cells, is mediated by GSK3.( 9 ) Fbw7 and Skp2 differentially regulate c‐Myc stability by targeting Myc box 1 and Myc box 2 respectively. Recently it has been reported that c‐Myc is directly bound to Skp2, which recruits Elongin C, Elongin B, and Cullin 2, thereby forming a novel ubiquitin E3 ligase.( 64 ) In addition, a novel c‐Myc‐binding protein, MM‐1, bound directly to Rpt3, a subunit of the 26S proteasome.( 64 ) MM‐1 may connect c‐Myc to the ubiquitin E3 ligase and the proteasome.
c‐Myb is a protooncogene product that is involved in the regulation of proliferation, differentiation, and survival of various hematopoietic lineages and also acts as an inhibitor of terminal differentiation of both erythroblastic and myeloblastic leukemia cell lines. It is extensively phosphorylated and then degraded by the ubiquitin–proteasome pathway. Recently, it was reported that Fbw7 targets c‐Myb for degradation in a Wnt‐1‐ and NLK‐dependent manner, or GSK‐dependent manner.( 21 , 22 ) There have been reports that mutation and deletion of Fbw7 in T‐cell acute lymphoblastic leukemia might increase the stability and level of c‐Myb, thereby affecting transformation and cell growth.( 25 )
β‐Catenin. The cellular amount of β‐catenin is critically regulated. In the absence of Wnt signal stimulation, GSK3β phosphorylates β‐catenin in collaboration with APC and Axin, and then β‐catenin is degraded by SCFβ‐TrCP/Fbw1 E3 ligase.( 6 ) The stimulation of Wnt signal induces the inhibition of GSK3β via Frizzled. Deletion or mutation of the APC or Axin gene has been found in familial adenoma in colon and gastric cancers, and β‐catenin mutation in the phosphorylation sites has also been reported.( 65 , 66 ) In these cells, the GSK3β‐mediated phosphorylation of β‐catenin is decreased and thereby degradation of β‐catenin inhibits the enhanced transcription of Cyclin D1 and c‐Myc, which promotes cell growth from adenomas. SCFβ–TrCP/Fbw1 also ubiquitylates IκB, Cdc25, and Wee1 in a phosphorylation‐dependent manner (Fig. 2).( 3 )
Hypoxia‐inducible factor. Accumulation of hypoxia‐inducible factor (HIF)‐1 is observed in most types of solid tumor and is frequently associated with poor prognosis and cancer progression, in particular via angiogenesis through VEGF in cancer.( 67 ) The protein stability of the catalytic subunit HIFα is regulated by the von Hippel‐Lindau (VHL) E3 ligase for oxygen‐dependent ubiquitin‐mediated destruction (Fig. 2).( 68 ) VHL, a tumor suppressor E3 ligase, is a component of ECV, consisting of elongin B, elongin C, Rbx1/ROC1, and Cullin 2 (Fig. 2). Structurally and functionally, ECV is analogous to the SCF complex. Tumor hypoxia or an inactivating mutation in VHL results in the stabilization of HIFα and in the consequential activation of HIF, triggering expression of its various targets, such as glucose uptake and metabolism, angiogenesis, extracellular matrix formation and turnover, chemotaxis, and cell proliferation‐ or survival‐regulating factors.( 67 )
Other oncogenic proteins. It has been reported that the stability of c‐Jun is regulated by SCFFbw7, which ubiquitylates phosphorylated c‐Jun and facilitates c‐Jun degradation.( 10 ) Fbw7 depletion resulted in accumulation of phosphorylated c‐Jun by JNK, stimulation of AP1 activity, and apoptosis induction. Another AP1 component, c‐Fos, is ubiquitylated by the E3 ligase UBR1 in the cytoplasm during interleukin‐6 and its receptor subunit, gp130 stimulation under MEK inhibition.( 69 ) UBR1 contains RING‐type and UBR‐type zinc fingers, and it binds to substrates bearing specific amino‐terminal residues that are destabilizing according to the N‐end rule, leading to their ubiquitylation and subsequent degradation. Phosphorylation of Thr232 in c‐Fos by ERK5 inhibits the nuclear export of c‐Fos and disrupts the interaction of c‐Fos with UBR1.( 69 )
EGFR and other protein tyrosine kinases are also involved in carcinogenesis and tumor cell growth. c‐Cbl, a RING finger type E3 ubiquitin ligase, promotes downregulation of EGFR and some protein tyrosine kinases (Fig. 2).( 70 )
Ubiquitin system in DNA repair
DNA repair is an important process aimed at the correction of DNA damage before it is fixed as a mutation, which would lead to tumorigenesis or cell death. Recent studies have shown that the DNA repair process is regulated by various ubiquitin ligases, such as BRCA1 and CUL4–DDB1.( 71 , 72 ) BRCA1 interacts predominantly with hyperphosphorylated RNAPII in undamaged cells, then it dissociates from RNAPII and localizes to sites of DNA damage. CUL4–DDB1 complexes have been well‐characterized in terms of the regulation of nucleotide excision repair (NER), in which damaged DNA bases are removed and subsequently DNA polymerase and ligase activities fill in the gaps. There are two distinct pathways of NER: transcription‐coupled repair (TCR), which removes damage from the actively transcribed strand of DNA, and global genomic repair (GGR), which repairs non‐transcribed DNA (Fig. 5). CUL4–DDB1 complexes carry out their respective functions via both TCR and GGR by interacting with different substrate‐specific receptors, which are called DDB1‐binding WD40 (DWD) proteins (also called DCAF, for Ddb1‐ and CUl4‐associated factors, or CDW, for CUL4‐ and DDB1‐associated WD40 repeat proteins).( 73 , 74 , 75 , 76 ) At the initiation of TCR, Cockayne Syndrome B (CSB) is recruited to the lesion‐stalled RNAPII. Cockayne Syndrome A (CSA), a DWD protein, assembles with CUL4–DDB1 to RNAPII stalled at sites of DNA damage as part of the TCR process.( 77 ) Interaction of COP9 signalosome (CSN) with CUL4–DDB1–CSA negates their ubiquitylation ability. CSB subsequently recruits NER proteins, histone acetyltransferase p300, and CSN‐bound CUL4–DDB1–CSA to RNAPII. After the DNA repair, CSN dissociates from CUL4–DDB1–CSA, leading to the activation of ligase and polyubiquitylation of CSB. The ubiquitylated CSB is degraded in a proteasome‐dependent manner, which is crucial for recovery of the post‐TCR process.
Figure 5.
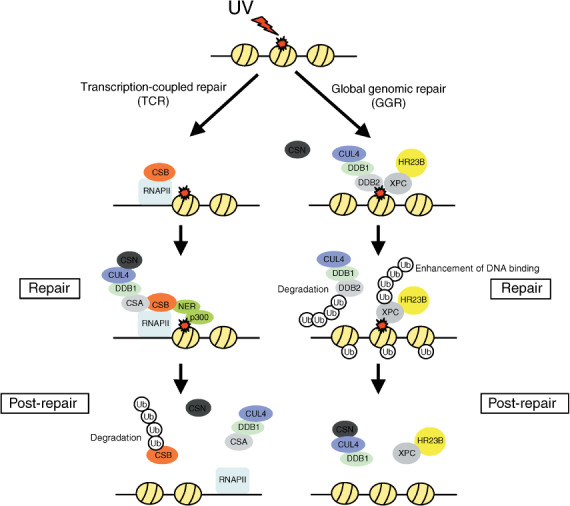
The regulation of nucleotide excision repair (NER) by the CUL4–DDB1 complex. Cockayne Syndrome B (CSB) and CUL4–DDB1–Cockayne Syndrome A (CSA) are recruited to the lesion‐stalled RNA polymerase II (RNAPII) as part of the transcription‐coupled repair (TCR) process. After DNA repair, COP9 signalosome (CSN) dissociates from CUL4–DDB1–CSA, leading to the degradation of CSB in a proteasome‐dependent manner. CUL4–DDB1–DDB2 and the repair complex, Xeroderma pigmentosum group C protein (XPC) and HR23B, recognize and bind to the site of DNA damage as part of the global genomic repair (GGR) process. During the GGR process, CUL4–DDB1 ubiquitylates and degrades DDB2. CUL4–DDB1 also ubiquitylates histones and XPC, but this does not promote degradation. These modulations lead to the displacement of DDB2 by XPC. Ub, Ubiquitin.
DDB2, the other DWD protein, binds and recruits CUL4–DDB1 to UV‐damaged DNA as part of the GGR process.( 78 , 79 ) Once bound to chromatin, DDB2 also recruits the repair complex composed of the Xeroderma pigmentosum group C protein (XPC) and HR23B to the site of DNA damage. CSN subsequently dissociates from CUL4–DDB1–DDB2, leading to the activation of ligase and polyubiquitylation of the substrates XPC and DDB2. The ubiquitylated DDB2 is degraded in a proteasome‐dependent manner, whereas the ubiquitylated XPC enhances its affinity for DNA, resulting in XPC displacing DDB2 at sites of DNA damage. After the completion of DNA repair, CSN reassociates with CUL4–DDB1 and inactivates the ligase. XPC is de‐ubiquitylated and subsequently released from the DNA.
In addition to the role of CUL4–DDB1–DDB2 in recruiting XPC to damaged DNA, it has been shown that CUL4–DDB1–DDB2 mono‐ubiquitylates histone H2A,( 80 ) H3, and H4( 81 ) in response to DNA damage. Its modulation as well as methylation and acetylation of the histone may contribute to the ‘histone code’ for the regulation of gene expression through DNA repair. Recently, CUL4–DDB1 has been shown to interact with the methyltransferase MLL1 and it is also involved in p16 induction by oncogenic signaling via enhancement of H3K4 methylation on the p16 locus.( 82 )
Conclusion and perspectives
Tumor suppressor gene products suppress transformation and cancerous growth in normal cells. Moreover, oncogene products are quickly degraded by their responsible E3 ligases, which target oncogenes as antioncogenic E3 ligases. In cancer cells, defects of the antioncogenic E3 ligases or overexpression of the oncogenic E3 ligases, which target tumor suppressor gene products, are often observed, and thereby carcinogenesis and cancerous growth are likely promoted. Inhibitors which target oncogenic E3 ligases to prevent degradation of tumor suppressor gene products may be useful for cancer therapy. Although it is reported that Bortezomib exibits anticancer effect via inhibition of nuclear factor κB function, it should inhibits 26S proteasome and thereby many other proteins should be accumulated in normal cells. It is desirable to find more specific molecular targets for the evasion of toxicity. In addition to the identification of a novel substrate to the known E3 ligase, recently, the novel E3 complex has also gradually been found. Moreover, ubiquitin modification plays a diverse role, not only in the quantitative regulation of target proteins. Therefore, further clinicopathological study of E3 ligases is important for the development of prognostic strategies and cancer therapy.
Abbreviations
| BTG | B‐cell translocation gene |
| BRCA1/BRCA1 | BRCA1‐BARD1 |
| CDK | cyclin‐dependent kinase |
| Cdk | cyclin‐dependent kinase |
| COP | constitutive photomorphogenic |
| CRL | Cullin‐RING ubiquitin ligase |
| DDB | Damage specific DNA binding protein |
| DHFR | dihydrofolate reductase |
| ECV | Elongins/Cul2/VHL |
| EGFR | epidermal growth factor receptor |
| HECT | homologous to E6‐AP C‐terminus |
| Huwe | HECT, UBA and WWE domain containing |
| KPC | Kip1 ubiquitylation Promoting Complex |
| Mdm | mouse double minute |
| MLL | mixed‐lineage leukemia |
| MM | Myc modulator |
| mTOR | mammalian target of rapamycin |
| NLK | Nemo like kinase |
| PCNA | Proliferating Cell Nuclear Antigen |
| Pirh2 | p53‐induced RING‐H2 |
| RPB | common subunit to three RNA polymerases |
| Rpt | regulatory particle ATPase |
| SCF | Skp1‐Cul1‐F‐box‐protein |
| TGF | transforming growth factor |
| Tob | transducer of ERBB2 |
| VEGF | vascular endothelial growth factor |
| WWP | WW domain containing E3 ubiquitin protein ligase |
References
- 1. Hershko A. The ubiquitin system for protein degradation and some of its roles in the control of the cell division cycle. Cell Death Differ 2005; 12: 1191–7. [DOI] [PubMed] [Google Scholar]
- 2. Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem 1998; 67: 425–79. [DOI] [PubMed] [Google Scholar]
- 3. Nakayama K, Nakayama KI. Ubiquitin ligases: cell‐cycle control and cancer. Nat Rev Cancer 2006; 6: 369–81. [DOI] [PubMed] [Google Scholar]
- 4. Cardozo T, Pagano M. The SCF ubiquitin ligase: insights into a molecular machine. Nat Rev Mol Cell Biol 2004; 5: 739–51. [DOI] [PubMed] [Google Scholar]
- 5. Hatakeyama S, Kitagawa M, Nakayama K et al . Ubiquitin‐dependent degradation of IκBα is mediated by a ubiquitin ligase Skp1/Cul 1/F‐box protein FWD1. Proc Natl Acad Sci USA 1999; 96: 3859–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kitagawa M, Hatakeyama S, Shirane M et al . An F‐box protein, FWD1, mediates ubiquitin‐dependent proteolysis of β‐catenin. EMBO J 1999; 18: 2401–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Koepp DM, Schaefer LK, Ye X et al . Phosphorylation‐dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science 2001; 294: 173–7. [DOI] [PubMed] [Google Scholar]
- 8. Yada M, Hatakeyama S, Kamura T et al . Phosphorylation‐dependent degradation of c‐Myc is mediated by the F‐box protein Fbw7. EMBO J 2004; 23: 2116–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Welcker M, Orian A, Jin J et al . The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation‐dependent c‐Myc protein degradation. Proc Natl Acad Sci USA 2004; 101: 9085–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wei W, Jin J, Schlisio S, Harper JW, Kaelin WG Jr. The v‐Jun point mutation allows c‐Jun to escape GSK3‐dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer Cell 2005; 8: 25–33. [DOI] [PubMed] [Google Scholar]
- 11. Kipreos ET, Pagano M. The F‐box protein family. Genome Biol 2000; 1: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin‐mediated degradation of the CDK inhibitor p27. Nat Cell Biol 1999; 1: 193–9. [DOI] [PubMed] [Google Scholar]
- 13. Nakayama K, Nagahama H, Minamishima YA et al . Targeted disruption of Skp2 results in accumulation of cyclin E and p27 Kip1 , polyploidy and centrosome overduplication. EMBO J 2000; 19: 2069–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ganoth D, Bornstein G, Ko TK et al . The cell‐cycle regulatory protein Cks1 is required for SCFSkp2‐mediated ubiquitinylation of p27. Nat Cell Biol 2001; 3: 321–4. [DOI] [PubMed] [Google Scholar]
- 15. Kamura T, Hara T, Kotoshiba S et al . Degradation of p57 Kip2 mediated by SCFSkp2‐dependent ubiquitylation. Proc Natl Acad Sci USA 2003; 100: 10 231–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tedesco D, Lukas J, Reed SI. The pRb‐related protein p130 is regulated by phosphorylation‐dependent proteolysis via the protein‐ubiquitin ligase SCFSkp2 . Genes Dev 2002; 16: 2946–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hiramatsu Y, Kitagawa K, Suzuki T et al . Degradation of Tob1 mediated by SCFSkp2‐dependent ubiquitination. Cancer Res 2006; 66: 8477–83. [DOI] [PubMed] [Google Scholar]
- 18. Von Der Lehr N, Johansson S, Wu S et al . The F‐box protein Skp2 participates in c‐Myc proteosomal degradation and acts as a cofactor for c‐Myc‐regulated transcription. Mol Cell 2003; 11: 1189–200. [DOI] [PubMed] [Google Scholar]
- 19. Imaki H, Nakayama K, Delehouzee S et al . Cell cycle‐dependent regulation of the Skp2 promoter by GA‐binding protein. Cancer Res 2003; 63: 4607–13. [PubMed] [Google Scholar]
- 20. Yokoi S, Yasui K, Saito‐Ohara F et al . A novel target gene, SKP2, within the 5p13 amplicon that is frequently detected in small cell lung cancers. Am J Pathol 2002; 161: 207–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kanei‐Ishii C, Nomura T, Takagi T, Watanabe N, Nakayama KI, Ishii S. Fbxw7 acts as an E3 ubiquitin ligase that targets c‐Myb for nemo‐like kinase (NLK)‐induced degradation. J Biol Chem 2008; 283: 30 540–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kitagawa K, Hiramatsu Y, Uchida C et al . Fbw7 promotes ubiquitin‐dependent degradation of c‐Myb: involvement of GSK3‐mediated phosphorylation of Thr‐572 in mouse c‐Myb. Oncogene in press. [DOI] [PubMed]
- 23. Wu G, Lyapina S, Das I et al . SEL‐10 is an inhibitor of notch signaling that targets notch for ubiquitin‐mediated protein degradation. Mol Cell Biol 2001; 21: 7403–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mao JH, Kim IJ, Wu D et al . FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science 2008; 321: 1499–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thompson BJ, Buonamici S, Sulis ML et al . The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. J Exp Med 2007; 204: 1825–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Onoyama I, Tsunematsu R, Matsumoto A et al . Conditional inactivation of Fbxw7 impairs cell‐cycle exit during T cell differentiation and results in lymphomatogenesis. J Exp Med 2007; 204: 2875–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Starita LM, Parvin JD. Substrates of the BRCA1‐dependent ubiquitin ligase. Cancer Biol Ther 2006; 5: 137–41. [DOI] [PubMed] [Google Scholar]
- 28. Hashizume R, Fukuda M, Maeda I et al . The RING heterodimer BRCA1‐BARD1 is a ubiquitin ligase inactivated by a breast cancer‐derived mutation. J Biol Chem 2001; 276: 14 537–40. [DOI] [PubMed] [Google Scholar]
- 29. Bernassola F, Karin M, Ciechanover A, Melino G. The HECT family of E3 ubiquitin ligases: multiple players in cancer development. Cancer Cell 2008; 14: 10–21. [DOI] [PubMed] [Google Scholar]
- 30. Inoue Y, Imamura T. Regulation of TGF‐β family signaling by E3 ubiquitin ligases. Cancer Sci 2008; 99: 2107–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brooks CL, Gu W. p53 ubiquitination: Mdm2 and beyond. Mol Cell 2006; 21: 307–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Scheffner M, Huibregtse JM, Vierstra RD, Howly PM. The HPV‐16 E6 and E6‐AP complex functions as a ubiquitin‐protein ligase in the ubiquitination of p53. Cell 1993; 75: 495–505. [DOI] [PubMed] [Google Scholar]
- 33. Chen D, Kon N, Li M, Zhang W, Qin J, Gu W. ARF‐BP1/Mule is a critical mediator of the ARF tumor suppressor. Cell 2005; 121: 1071–83. [DOI] [PubMed] [Google Scholar]
- 34. Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett 1997; 420: 25–7. [DOI] [PubMed] [Google Scholar]
- 35. Leng RP, Lin Y, Ma W et al . Pirh2, a p53‐induced ubiquitin‐protein ligase, promotes p53 degradation. Cell 2003; 112: 779–91. [DOI] [PubMed] [Google Scholar]
- 36. Dornan D, Wertz I, Shimizu H et al . The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature 2004; 429: 86–92. [DOI] [PubMed] [Google Scholar]
- 37. Duan W, Gao L, Druhan LJ et al . Expression of Pirh2, a newly identified ubiquitin protein ligase, in lung cancer. J Natl Cancer Inst 2004; 96: 1718–21. [DOI] [PubMed] [Google Scholar]
- 38. Rossi M, De Laurenzi V, Munarriz E et al . The ubiquitin‐protein ligase Itch regulates p73 stability. EMBO J 2005; 24: 836–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Melino G, Knight RA, Cesareni G. Degradation of p63 by Itch. Cell Cycle 2006; 5: 1735–9. [DOI] [PubMed] [Google Scholar]
- 40. Li Y, Zhou Z, Chen C. WW domain‐containing E3 ubiquitin protein ligase 1 targets p63 transcription factor for ubiquitin‐mediated proteasomal degradation and regulates apoptosis. Cell Death Differ 2008; 15: 1941–51. [DOI] [PubMed] [Google Scholar]
- 41. Weinberg RA. The retinoblastoma protein and cell cycle control. Cell 1995; 81: 323–30. [DOI] [PubMed] [Google Scholar]
- 42. Kitagawa M, Higashi H, Jung HK et al . The consensus motif for phosphorylation by cyclin D1‐Cdk4 is different from that for phosphorylation by cyclin A/E‐Cdk2. EMBO J 1996; 15: 7060–9. [PMC free article] [PubMed] [Google Scholar]
- 43. Uchida C, Miwa S, Kitagawa K et al . Enhanced Mdm2 activity inhibits pRB function via ubiquitin‐dependent degradation. EMBO J 2005; 24: 160–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Oliner JD, Kinzler KW, Meltzer PS et al . Amplification of a gene encoding a p53‐associated protein in human sarcomas. Nature 1992; 358: 80–3. [DOI] [PubMed] [Google Scholar]
- 45. Miwa S, Uchida C, Kitagawa K et al . Mdm2‐mediated pRB downregulation is involved in carcinogenesis in a p53‐independent manner. Biochem Biophys Res Commun 2006; 340: 54–61. [DOI] [PubMed] [Google Scholar]
- 46. Pomerantz J, Schreiber‐Agus N, Liégeois NJ et al . The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2's inhibition of p53. Cell 1998; 92: 713–23. [DOI] [PubMed] [Google Scholar]
- 47. Honda R, Yasuda H. Association of p19Arf with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. EMBO J 1999; 18: 22–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell 1995; 83: 993–1000. [DOI] [PubMed] [Google Scholar]
- 49. Toledo F, Wahl GM. MDM2 and MDM4: p53 regulators as targets in anticancer therapy. Int J Biochem Cell Biol 2007; 39: 1476–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Uchida C, Miwa S, Isobe T et al . Effects of MdmX on Mdm2‐mediated downregulation of pRB. FEBS Lett 2006; 580: 1753–8. [DOI] [PubMed] [Google Scholar]
- 51. Nakayama KI, Nakayama K. Cip/Kip cyclin‐dependent kinase inhibitors: brakes of the cell cycle engine during development. Bioessays 1998; 20: 1020–9. [DOI] [PubMed] [Google Scholar]
- 52. Kamura T, Hara T, Matsumoto M et al . Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27 Kip1 at G1 phase. Nat Cell Biol 2004; 6: 1229–35. [DOI] [PubMed] [Google Scholar]
- 53. Hattori T, Isobe T, Abe K et al . Pirh2 promotes ubiquitin‐dependent degradation of the cyclin‐dependent kinase inhibitor p27 kip1 . Cancer Res 2007; 67: 10 789–95. [DOI] [PubMed] [Google Scholar]
- 54. Frescas D, Pagano M. Deregulated proteolysis by the F‐box proteins SKP2 and β‐TrCP: tipping the scales of cancer. Nat Rev Cancer 2008; 8: 438–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Inui N, Kitagawa K, Miwa S et al . High expression of Cks1 in human non‐small cell lung carcinomas. Biochem Biophys Res Commun 2003; 303: 978–84. [DOI] [PubMed] [Google Scholar]
- 56. Shimada M, Kitagawa K, Dobashi Y et al . High expression of Pirh2, an E3 ligase for p27, is associated with low expression of p27 and poor prognosis in head and neck cancers. Cancer Sci 2009; 100: 866–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bornstein G, Bloom J, Sitry‐Shevah D, Nakayama K, Pagano M, Hershko A. Role of the SCFSkp2 ubiquitin ligase in the degradation of p21Cip1 in S phase. J Biol Chem 2003; 278: 25 752–7. [DOI] [PubMed] [Google Scholar]
- 58. Amador V, Ge S, Santamaría PG, Guardavaccaro D, Pagano M. APC/CCdc20 controls the ubiquitin‐mediated degradation of p21 in prometaphase. Mol Cell 2007; 27: 462–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Abbas T, Sivaprasad U, Terai K, Amador V, Pagano M, Dutta A. PCNA‐dependent regulation of p21 ubiquitylation and degradation via the CRL4Cdt2 ubiquitin ligase complex. Genes Dev 2008; 22: 2496–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kuo ML, Den Besten W, Bertwistle D, Roussel MF, Sherr CJ. N‐terminal polyubiquitination and degradation of the Arf tumor suppressor. Genes Dev 2004; 18: 1862–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wang X, Trotman LC, Koppie T et al . NEDD4‐1 is a proto‐oncogenic ubiquitin ligase for PTEN. Cell 2007; 128: 129–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kano S, Miyajima N, Fukuda S, Hatakeyama S. Tripartite motif protein 32 facilitates cell growth and migration via degradation of Abl‐interactor 2. Cancer Res 2008; 68: 5572–80. [DOI] [PubMed] [Google Scholar]
- 63. Lin DI, Barbash O, Kumar KG et al . Phosphorylation‐dependent ubiquitination of cyclin D1 by the SCFFBX4‐αBcrystalline complex. Mol Cell 2006; 24: 355–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kimura Y, Nagao A, Fujioka Y et al . MM‐1 facilitates degradation of c‐Myc by recruiting proteasome and a novel ubiquitin E3 ligase. Int J Oncol 2007; 31: 829–36. [PubMed] [Google Scholar]
- 65. Segditsas S, Tomlinson I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene 2006; 25: 7531–7. [DOI] [PubMed] [Google Scholar]
- 66. Kolligs FT, Bommer G, Göke B. Wnt/beta‐catenin/tcf signaling: a critical pathway in gastrointestinal tumorigenesis. Digestion 2002; 66: 131–44. [DOI] [PubMed] [Google Scholar]
- 67. Ohh M. Ubiquitin pathway in VHL cancer syndrome. Neoplasia 2006; 8: 623–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Maxwell PH, Wiesener MS, Chang GW et al . The tumour suppressor protein VHL targets hypoxia‐inducible factors for oxygen‐dependent proteolysis. Nature 1999; 399: 271–5. [DOI] [PubMed] [Google Scholar]
- 69. Sasaki T, Kojima H, Kishimoto R, Ikeda A, Kunimoto H, Nakajima K. Spatiotemporal regulation of c‐Fos by ERK5 and the E3 ubiquitin ligase UBR1, and its biological role. Mol Cell 2006; 24: 63–75. [DOI] [PubMed] [Google Scholar]
- 70. Thien CB, Walker F, Langdon WY. RING finger mutations that abolish c‐Cbl‐directed polyubiquitination and downregulation of the EGF receptor are insufficient for cell transformation. Mol Cell 2001; 7: 355–65. [DOI] [PubMed] [Google Scholar]
- 71. Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 2002; 108: 171–82. [DOI] [PubMed] [Google Scholar]
- 72. O’Connell BC, Harper JW. Ubiquitin proteasome system (UPS): what can chromatin do for you? Curr Opin Cell Biol 2007; 19: 206–14. [DOI] [PubMed] [Google Scholar]
- 73. He YJ, McCall CM, Hu J, Zeng Y, Xiong Y. DDB1 functions as a linker to recruit receptor WD40 proteins to CUL4‐ROC1 ubiquitin ligases. Genes Dev 2006; 20: 2949–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Angers S, Li T, Yi X, MacCoss MJ, Moon RT, Zheng N. Molecular architecture and assembly of the DDB1‐CUL4A ubiquitin ligase machinery. Nature 2006; 443: 590–3. [DOI] [PubMed] [Google Scholar]
- 75. Higa LA, Wu M, Ye T, Kobayashi R, Sun H, Zhang H. CUL4‐DDB1 ubiquitin ligase interacts with multiple WD40‐repeat proteins and regulates histone methylation. Nat Cell Biol 2006; 8: 1277–83. [DOI] [PubMed] [Google Scholar]
- 76. Jin J, Arias EE, Chen J, Harper JW, Walter JC. A family of diverse Cul4‐Ddb1‐interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol Cell 2006; 23: 709–21. [DOI] [PubMed] [Google Scholar]
- 77. Fousteri M, Vermeulen W, Van Zeeland AA, Mullenders LH. Cockayne syndrome A and B proteins differentially regulate recruitment of chromatin remodeling and repair factors to stalled RNA polymerase II in vivo . Mol Cell 2006; 23: 471–82. [DOI] [PubMed] [Google Scholar]
- 78. Groisman R, Polanowska J, Kuraoka I et al . The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell 2003; 113: 357–67. [DOI] [PubMed] [Google Scholar]
- 79. Sugasawa K, Okuda Y, Saijo M et al . UV‐induced ubiquitylation of XPC protein mediated by UV‐DDB‐ubiquitin ligase complex. Cell 2005; 121: 387–400. [DOI] [PubMed] [Google Scholar]
- 80. Kapetanaki MG, Guerrero‐Santoro J, Bisi DC, Hsieh CL, Rapiæ‐Otrin V, Levine AS. The DDB1‐CUL4ADDB2 ubiquitin ligase is deficient in xeroderma pigmentosum group E and targets histone H2A at UV‐damaged DNA sites. Proc Natl Acad Sci USA 2006; 103: 2588–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wang H, Zhai L, Xu J et al . Histone H3 and H4 ubiquitylation by the CUL4‐DDB‐ROC1 ubiquitin ligase facilitates cellular response to DNA damage. Mol Cell 2006; 22: 383–94. [DOI] [PubMed] [Google Scholar]
- 82. Kotake Y, Zeng Y, Xiong Y. DDB1‐CUL4 and MLL1 mediate oncogene‐induced p16 INK4a activation. Cancer Res 2009; 69: 1809–14. [DOI] [PMC free article] [PubMed] [Google Scholar]