

Abstract
Prostate cancer has its highest incidence in the USA and is becoming a major concern in Asian countries. Bufadienolides are extracts of toxic glands from toads and are used as anticancer agents, mainly on leukemia cells. In the present study, the antiproliferative and apoptotic mechanisms of bufalin and cinobufagin on prostate cancer cells were investigated. Proliferation of LNCaP, DU145, and PC3 cells was measured by 3‐(4,5‐dimethylthiazol‐2‐yle)‐2,5‐diphenyltetrazolium bromide assay and the doubling time (tD) was calculated. Bufalin and cinobufagin caused changes in the tD of three prostate cancer cell lines, which were more significant than that of human mesangial cells. In addition, bufadienolides induced prostate cancer cell apoptosis more significantly than that in breast epithelial cell lines. After treatment, the caspase‐3 activity and protein expression of caspase‐3, ‐8, and ‐9 were elevated. The expression of other apoptotic modulators, including mitochondrial Bax and cytosolic cytochrome c, were also increased. However, expression of p53 was only enhanced in LNCaP cells. Downregulation of p53 by antisense TP53 restored the cell viability suppressed by bufalienolides. Furthermore, the increased expression of Fas was more significant in DU145 and PC3 cells with mutant p53 than in LNCaP cells. Transfection of Fas small interfering RNA restored cell viability in the bufadienolide‐treated cells. These results suggest that bufalin and cinobufagin suppress cell proliferation and cause apoptosis in prostate cancer cells via a sequence of apoptotic modulators, including Bax, cytochrome c, and caspases. The upstream mediators might be p53 and Fas in androgen‐dependent LNCaP cells and Fas in androgen‐independent DU145 and PC3 cells. (Cancer Sci 2008; 99: 2467–2476)
Prostate cancer is currently showing its highest incidence for decades, and is the second‐leading cause of cancer death among men in the USA.( 1 ) It has been estimated that 186 320 new cases of prostate cancer will be diagnosed and that 28 660 deaths attributed to prostate cancer will occur in the USA in 2008.( 2 ) In addition, the mortality of prostate cancer has also increased in Asian countries during the past decade. An assay for prostate‐specific antigen has raised the early detection level of prostate cancer, which is curable by surgical and radiation therapies. Androgen ablation is the most common treatment for metastatic prostate cancer; however, 20–30% patients with prostate cancer experience recurrent disease.( 3 , 4 ) Therefore, searching for chemoprevention or chemical controls for prostate cancer has become a crucial concern.
Bufalin and cinobufagin, the major digoxin‐like components of Chan‐Su extracts from the venom of Bufo bufo gargarizan, have been reported as Na+‐K+‐ATPase inhibitors that result in elevation of the intracellular calcium concentration.( 5 , 6 ) On the basis of this mechanism, bufalin and cinobufagin increase vasoconstriction, and have long been used as a treatment for heart failure by Chinese medicine in Asian countries. Disruption of intracellular calcium homeostasis induces apoptosis in diverse cell types.( 7 ) Our previous results have illustrated that bufalin and cinobufagin are able to induce elevated calcium concentration and cell apoptosis in prostate cancer cells, but the detailed mechanism of this remains unclear.( 8 ) Bufalin is also known as a topoisomerase II inhibitor. Topoisomerase II is a nuclear enzyme that relaxes supercoiled DNA at the time of DNA replication. Its inhibitors result in protein‐linked DNA double‐strand breaks.( 9 ) Some of the topoisomerase poisons, such as etoposide and adriamycin, are efficient and widely prescribed anticancer drugs.( 10 ) According to the mechanism described above, bufalin induces leukemia cell differentiation( 11 ) and apoptosis.( 12 ) Furthermore, inhibition of solid tumor growth,( 13 ) endothelial cell proliferation, and angiogenesis by bufalin have also been reported.( 14 ) As inhibitors of topoisomerase and Na+‐K+‐ATPase have been demonstrated to induce apoptosis in some cancer cell lines, it is interesting to investigate the apoptotic mechanism induced by bufalin and cinobufagin in androgen‐dependent (LNCaP) and ‐independent (DU145 and PC3) prostate cancer cell lines.
The induction of cell apoptosis has been the mechanism behind chemotherapeutic drugs that treat a variety of cancers. Two major apoptotic mechanisms are the intrinsic pathway (mediated by p53) and the extrinsic pathway (regulated by Fas).( 15 ) The p53 protein induces apoptosis when cells experience conditions of DNA damage, cell cycle abnormality, or hypoxia.( 16 ) Binding of Fas (APO‐1/CD95) by its ligand results in the sequential recruitment of the Fas‐associated death domain protein and procaspase‐8, which form the death‐induced signaling complex. Active caspase‐8 then passes the apoptotic signal through a mitochondria‐independent or ‐dependent pathway to activate downstream caspase‐3.( 17 ) Either p53 or Fas delivers the signal to the mitochondrial pathway and mediates cell apoptosis. The Bcl‐2 protein family is divided into two functional subfamilies: pro‐apoptotic proteins (Bax and Bid) and anti‐apoptotic proteins (Bcl‐2 and Bcl‐xL). The family members translocate to the mitochondria and mediate the membrane potential to induce cytochrome c release. Cytosolic cytochrome c is further involved in caspase activation. The caspase cascade is a key pathway in apoptotic signal transduction, and can be divided into two types of subfamilies: upstream initiator caspases (caspase 8 and 9) and downstream effector caspases (caspase 3, 6, and 7), which directly induce the final events of apoptosis.( 18 )
The major objectives of the present study were to: (i) examine changes in the doubling time (tD) mediated by bufalin or cinobufagin in three prostate cancer cell lines and one normal human cell (human mesangial cell, HMC) to compare the toxic effects between normal and cancer cells; and (ii) investigate the apoptotic effects of bufadienolides by analyzing cell surface phosphatidylserine, the protein expression and activity of the caspases, and the upstream molecules, including Bax, cytochrome c, p53, and Fas. Such investigations may illustrate the antitumor mechanisms of bufalin and cinobufagin in prostate cancer.
Materials and Methods
Cell culture and transfection. Human prostate carcinoma cell lines and normal breast epithelial cell lines were purchased from the Culture Collection and Research Center of the Food Industry Research and Development Institute (Taiwan, ROC). The primary cultured HMC were generously provided by Dr L.Y. Yang (Department of Pediatrics, Taipei Veterans General Hospital, Taipei, Taiwan, ROC). Cell lines were maintained in RPMI‐1640 (Gibco Laboratories, Buffalo, NY, USA) (LNCaP) or in Dulbecco's modified Eagle's medium (Gibco Laboratories) (DU145 and PC3) with 10% fetal calf serum (PAA Laboratories, Pasching, Austria) in an atmosphere of 5% CO2 at 37°C. HMC were cultured in RPMI‐1640 medium in the presence of 200 mmol/L glutamine, 100 mmol/L sodium‐pyruvate, 10 mmol/L nonessential amino acids, 100 IU/mL insulin, 4 mg/mL transferrin, 50 IU/mL potassium penicillin G, 50 IU/mL streptomycin sulfate, 250 µg/mL fungizone, and 10% fetal calf serum at 37°C in an atmosphere of 5% CO2. 184A1 and MCF10A were cultured in Dulbecco's modified Eagle's medium F12 with 5% horse serum, antibiotics, 10 µg/mL bovine pancrease insulin (Sigma, St Louis, MO, USA), 20 ng/mL epidermal growth factor (Sigma), 100 ng/mL cholera toxin (Calbiochem, Gibbstown, NJ, USA), 0.5 µg/mL hydrocortisone (Sigma), and 1.05 mmol/L calcium. Antisense‐TP53 and non‐specific control antisense RNA were purchased from Gibco (Carlsbad, CA, USA). Introduction of antisense TP53 into prostate cancer cells was carried out using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) with 100 nmol/L antisense RNA, 2 days before bufadienolide treatment. Fas small interfering RNA (siRNA) and non‐specific control siRNA were purchased from Ambion (Austin, TX, USA). The introduction of Fas siRNA into prostate cancer cells was carried out using the Silencer siRNA transfection II kit (Ambion) with 50 nmol/L siRNA, 2 days before bufadienolide treatment.
Cell proliferation assay. Cell proliferation was determined by the modified colorimetric 3‐(4,5‐dimethylthiazol‐2‐yle)‐2,5‐diphenyltetrazolium bromide (MTT) assay (Sigma). After cells attached they were challenged with different concentrations of bufalin (Sigma) or cinobufagin (Sigma) (day 0). The MTT assay was carried out on days 1–4 as described previously.( 8 ) The proliferation index of each day was calculated using the optical density of that day divided by the optical density of day 0. Each experimental condition was included in three preparations and repeated four times.
To calculate the tD, proliferation indices were plotted on the common log scale and fitted by linear regression using the equation:
| y = log(ODt/OD0) = (log2/tD)t, |
where ODt and OD0 represented the optical density at day t or at day 0, respectively. The doubling time was calculated using:
| tD = Log2/S (days), |
where S was the slope of the regression line.( 19 )
Annexin V apoptosis detection. To examine phosphatidylserine (PS) expression after treatment, cells were treated with bufalin and cinobufagin at concentrations of 0.1, 1, and 10 µmol/L following an incubation period for 18 h (LNCaP and DU145) or 24 h (PC3, 184A1, and MCF10A). Cell surface PS was analyzed by annexin V staining (Abcam, Cambridge, MA, USA) and flow cytometry. Cells in the fourth quadrant indicated the percentage of apoptotic cells.
Caspase activity analysis. Activation of caspase 3 was measured in the whole‐cell extracts using the peptide substrate DEVD‐pNA (R&D Systems, Minneapolis, MN, USA). Each sample was assayed following the manufacturer's procedure. Protein content in the supernatants was measured using the Bradford reagent. Results are expressed as the percentage absorbance relative to the control group (100%). Each experimental condition was repeated three times.
Immunoblotting assessment. Equal amounts of cell lysate were subjected to 10% (caspase 9) or 15% (caspase 3 and 8) sodium dodecylsulfate–polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes (Schleicher and Schuell, Keene, NH, USA). The following primary antibodies were used at 1 µg/mL: caspase 3 from Imgenex (San Diego, CA, USA); caspase 8 from BioVision (Mountain View, CA, USA), caspase 9 from Medical and Biological Laboratories (Nagoya, Japan); and β‐actin from Sigma. The blot was further incubated with horseradish peroxidase‐conjugated goat antimouse secondary antibody (1:20 000; Promega Corporation, Madison, WI, USA), and proteins were visualized using enhanced chemiluminescence detection (GE Healthcare Lifesciences, Pittsburgh, PA, USA).( 20 )
Isolation of cytosolic and mitochondrial fractions. Release of cytochrome c from mitochondria and translocation of Bax to mitochondria were measured by immunoblotting as described previously.( 21 ) The cytosolic fraction was then concentrated to 50–100 µL using a centrifugal concentration device for 10‐kDa molecular mass (Microcon YM‐10; Millipore, Bedford, MA, USA) according to the manufacturer's instructions. The resulting mitochondrial pellets were resuspended in 50 µL cell lysis buffer (20 mmol/L Tris [pH 7.5], 100 mmol/L NaCl, 1% Triton, 1 mmol/L phenylmethylsulfonyl fluride, and protease inhibitor mixture). These fractions were subjected to 15% sodium dodecylsulfate–polyacrylamide gel electrophoresis. Cytochrome c and Bax were detected using mouse monoclonal antibody at a dilution of 1:400 (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Each experimental condition was repeated three times.
Cell surface Fas analysis. The expression of Fas on the cell surface was analyzed as described previously.( 22 ) Cells were exposed to medium with or without 10 µmol/L bufalin or cinobufagin for 24 h (LNCaP and DU145) or 36 h (PC3). Following incubation with antihuman Fas monoclonal antibody (1 µg/mL; BD Transduction Laboratories, Franklin Lakes, NJ, USA) or mouse IgG as a negative control, cells were further incubated with fluorescein isothiocyanate‐conjugated rat antimouse IgG (10 µg/mL; eBioscience, San Diego, CA, USA) in the dark at 4°C for 2 h, and analyzed by flow cytometry.
Immunofluorescence detection. Cancer cells were seeded in 24‐well plates with cover glasses. After treatment with bufalin and cinobufagin, cells were fixed with 4% paraformaldehyde and permeabilized with 0.25% Triton X‐100 at room temperature for 15 min. After blocking with 10% goat serum at 37°C for 30 min, cells were incubated sequentially with Fas antibody (1:50) and fluorescein isothiocyanate‐conjugated goat antimouse IgG (1:50, Jackson Immunolab, West Grove, PA, USA) plus 4′, 6‐diamidino‐2‐phenylindole (DAPI; 0.5 µg/mL; Sigma) in the dark at 37°C for 1 h. Coverslips were then sealed onto the microscope slides using mounting solution. Fluorescence images were observed by confocal microscopy (Leica TCS SP2; Leica, Bannockburn, IL, USA). Cells treated with etoposide were used as a positive control.( 22 ) Cells incubated with mouse IgG were used as a negative control.
Examination of mRNA expression. After transfection and treatment with bufalin and cinobufagin, RNA was extracted from the cells using Trizol reagent (Invitrogen). Reverse transcription was done with 5 µg total RNA and oligo(dT) primers using SuperScript III RT (Invitrogen). Polymerase chain reaction was carried out using oligonucleotide primers and Taq DNA polymerase as follows: Fas (275 bp), 33 cycles, forward 5′‐CTGTTTCAGGATTTAAGGTTGGAGATT‐3′ and reverse 5′‐GACCCAGAATACCAA GTGCAGATGTA‐3′, annealing temperature 60°C; housekeeping gene glyceraldehyde‐3‐phosphate dehydrogenase (230 bp), 25 cycles, forward 5′‐TCAAGAAGGTGGTGAAGCAG‐3′ and reverse 5′‐CTTACTCCTTGGAGGCCATG‐3′, annealing temperature 55°C.
Statistics. All values are given as the mean ± SEM. Means were tested for homogeneity by one‐way analysis of variance (ANOVA), and the difference between specific means was tested for significance using Duncan's multiple‐range test.( 23 ) The difference between two means was considered statistically significant when P < 0.05.
Results
Change in doubling time caused by bufalin and cinobufagin in human mesangial and prostate cancer cells. Because bufalin and cinobufagin have antiproliferative effects on cancer cells,( 8 ) examination of their effects on normal human cells is necessary. The change in doubling time caused by bufalin and cinobufagin at a concentration of 0.1 µmol/L in HMC and three prostate cancer cell lines was determined by MTT assay. Proliferation indices of days 1–4 with or without treatment were calculated to form a linear regression equation (Fig. 1). The tD of the control and drug‐treated groups were calculated from the regression equations. After treatment with bufalin, the doubling time of LNCaP cells increased from 31 to 105 h, and similar effects were found in cinobufagin‐treated LNCaP cells. The increase in tD caused by bufalin and cinobufagin in LNCaP cells was approximately 3.1 times (P < 0.01). Even so, bufalin and cinobufagin were more effective in prolonging the tD in DU145 and PC3 cells, by 5.8‐ and 8.6‐fold, respectively (P < 0.01). However, the tD of HMC cells caused by bufalin and cinobufagin were lengthened only slightly. Consistent with our previous observations, the tD of bufalin‐treated prostate cancer cells increased more than that of cinobufagin‐treated cells.
Figure 1.
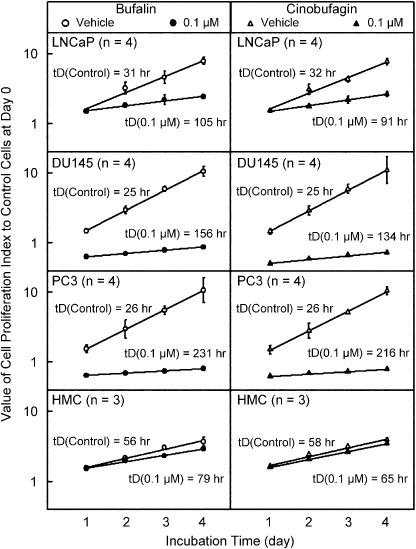
Effects of bufalin and cinobufagin on the change in doubling time (tD) in LNCaP, DU145 and PC3 cells, and human mesangial cells (HMC). Cells were treated with bufalin and cinobufagin at a concentration of 0.1 µmol/L following an incubation period of 1–4 days. The tD was calculated from the regression equation of the proliferation index. The applied equation was y = log(ODt/OD0) = (log2/tD)t, where ODt and OD0 represented the optical density at day t or day 0, respectively. The tD was calculated using tD = Log2/S (days), where S was the slope of the regression line. Each value represents mean ± SEM.
Elevation of phosphatidylserine expression by bufalin and cinobufagin in prostate cancer cells. Treatment of bufalin and cinobufagin inhibited cell growth especially in prostate cancer cells. We therefore examined the effects of cell apoptosis induced by bufadienolide in normal epithelial cells and prostate cancer cells. The expression of PS after bufalin and cinobufagin treatment of prostate cancer (LNCaP, DU145, and PC3) and normal breast epithelial cells (184A1 and MCF10A) was analyzed using annexin V staining and flow cytometry. In Figure 2a, the percentage of apoptotic cells was increased after treatment at concentrations of 0.1, 1, and 10 µmol/L for 18 h (LNCaP and DU145) or 24 h (PC3). This effect was more significant in malignant androgen‐independent prostate cancer cell lines. However, treatment with bufadienolide did not induce significant cell apoptosis in normal epithelial cells (Fig. 2b).
Figure 2.

Effects of phosphatidylserine expression after treatment with bufalin (BF) and cinobufagin (CB) in LNCaP, DU145, PC3, 184A1, and MCF10A cells. Cells were treated with BF and CB at concentrations of 0.1, 1, and 10 µmol/L following an incubation period of 18 (LNCaP and DU145) or 24 h (PC3, 184A1, and MCF10A). Cell surface PS was analyzed using annexin V staining and flow cytometry. The cells in the fourth quadrant indicate the percentage of apoptotic cells. Similar results were obtained in three other experiments.
Changes in enzymatic and proteolytic processing of caspases and induction of Bax translocation and cytochrome c release by bufalin and cinobufagin. An apoptotic mechanism was involved in the death of bufadienolide‐treated cancer cells.( 8 ) To examine whether bufalin and cinobufagin activated the caspase cascade, we measured the activity of caspase 3 at different time points in three prostate cancer cell lines (Fig. 3a). A colorimetric assay was carried out after 8, 12, 18, and 24 h of treatment in three cell lines. In LNCaP cells, bufalin at 10 µmol/L increased caspase 3 activity from 100% to 230% (P < 0.01) after 12 h of treatment. The peak response appeared at approximately 18 h after treatment. Bufalin at 1 µmol/L also caused elevated caspase activity after 18 h of treatment. Cinobufagin had similar effects to bufalin on LNCaP cells, but the increased ranges were less than those caused by bufalin. Bufalin and cinobufagin induced similar results in DU145 cells. In PC3 cells, after treatment for 18 h, 1 and 10 µmol/L bufalin elevated caspase 3 activity to ~150–180% (P < 0.01). The peak response occurred after 24 h of treatment. Cinobufagin had similar effects to bufalin in PC3 cells.
Figure 3.
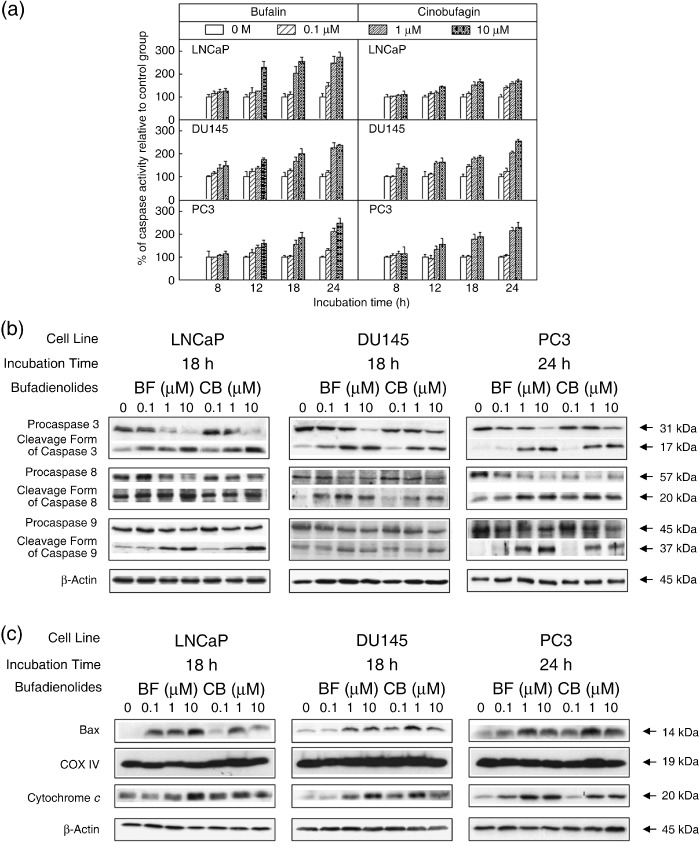
Caspase 3 activity and the protein expression of apoptosis‐related molecules in LNCaP, DU145, and PC3 cell lines after administration of bufalin (BF) or cinobufagin (CB). (a) Activity of caspase 3 was assayed using a colorimetric kit, and was determined after treatment at concentrations of 0, 0.1, 1, and 10 µmol/L for 8, 12, 18, and 24 h in three prostate cancer cell lines. Each value represents mean ± SEM. Control value = 100%. (b) After treatment with BF or CB for 18 (LNCaP and DU145) or 24 h (PC3), cell lysates were sized by 10 (caspase 9) or 15% (caspase 3 and 8) sodium dodecylsulfate–polyacrylamide gel electrophoresis (SDS‐PAGE). (c) Cell lysates were separated into mitochondrial and cytosolic fractions and subjected to 15% SDS‐PAGE and analyzed by western blotting. Each lane was loaded with 40 µg protein sample. Similar results were obtained in three other experiments.
Based on the above results, immunoblotting analysis of whole‐cell lysates obtained from three prostate cancer cells were carried out using antibodies for caspase 3, 8, and 9 at the time point of the peak response of caspase 3 activity (Fig. 3b). Activation of a specific caspase results in a reduction in its proenzyme form.( 24 ) We found that protein levels of procaspase 3, 8, and 9 were downregulated when the concentration of bufalin or cinobufagin was increased. Meanwhile, the proteolytic forms of these caspases were detectable in cell lysates obtained after treatment for 18, 18, and 24 h in LNCaP, DU145, and PC3 cells, respectively. The active forms of these caspases were elevated when the treatment concentration was increased.
Changes in mitochondrial membrane potential leading to the release of cytochrome c are thought to mediate caspase activation in many models of cell death programs. The role of mitochondria is crucial in cell apoptosis as Bcl‐2 family members are targeted to the mitochondrial outer membrane.( 15 ) Western blotting was used to analyze the protein levels of mitochondrial Bax and cytosolic cytochrome c. As in the mechanism described above, treatment of bufalin resulted in an elevation of Bax in the mitochondrial fraction of three prostate cancer cell lines (Fig. 3c). The release of cytochrome c into the cytosolic fraction was also found in three prostate cancer cell lines after bufalin or cinobufagin treatment. Significant results were found at time points of 18 h for LNCaP and DU145 cells and 24 h for PC3 cells. The increase in cytochrome c release and Bax translocation paralleled the dose effects.
Involvement of p53 in bufalin‐ and cinobufagin‐induced apoptosis in LNCaP cells. p53 mediates cell apoptosis stimulated by many chemotherapeutic drugs. To examine whether p53 mediates cell apoptosis after treatment with bufalin and cinobufagin, the protein expression of p53 in whole‐cell lysates was analyzed by western blotting in three prostate cancer cell lines. Expression of p53 was elevated by 12 h treatment with bufalin or cinobufagin in LNCaP cells, which express wild‐type p53 (Fig. 4a). This effect was more significant after 18 h of treatment. There was no increase in p53 expression in DU145 or PC3 cells, which express mutant or null p53 (data not shown).
Figure 4.
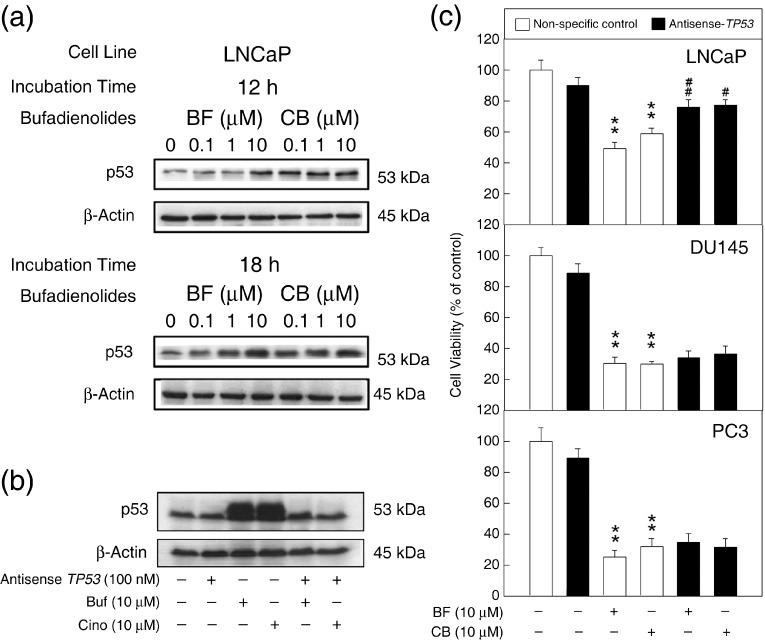
The role of p53 in the apoptotic pathway induced by bufalin (BF) and cinobufagin (CB) in LNCaP cells. (a) LNCaP cells were treated with BF or CB at concentrations of 0, 0.1, 1, and 10 µmol/L for 12 and 18 h. Whole‐cell lysates were sized by 10% sodium dodecylsulfate–polyacrylamide gel electrophoresis. (b) Cells were transfected with 100 nmol/L antisense TP53 for 48 h and then treated with BF or CB at a concentration of 10 µmol/L for 18 h. After administration, cancer cells were harvested and western blotting was carried out for the expression of p53. Each lane was loaded with 100 µg protein. Similar results were obtained in three other experiments. (c) Cell viability was measured by MTT assay. BF and CB were added to the medium after a 48‐h transfection. Open columns represent the non‐specific antisense RNA‐transfected group; closed columns represent the antisense TP53‐treated group. Control value = 100%; *P < 0.05, **P < 0.01 versus non‐specific antisense RNA‐control group; # P < 0.05, ## P < 0.01 versus non‐specific antisense RNA plus BF or CB groups.
To determine whether p53 plays an important role in bufalin‐ and cinobufagin‐induced cell apoptosis, LNCaP cells were transfected with 100 mmol/L antisense TP53 to inhibit the transcription of p53 mRNA. The control groups were transfected with a non‐specific antisense sequence. After 48 h transfection, cells were treated with bufalin and cinobufagin at 10 µmol/L, and then p53 expression was examined by western blotting (Fig. 4b). Treatment with bufalin and cinobufagin caused the increase and phosphorylation of p53. After the transfection of antisense TP53, the p53 expression induced by the treatment was inhibited (lanes 5 and 6).
We next examined whether cell viability was affected by the downregulation of p53. The cell viability of three prostate cancer cell lines was analyzed by MTT assay after transfection and bufadienolide treatment (Fig. 4c). After treatment with bufalin and cinobufagin at 10 µmol/L, cell viability was inhibited to approximately 53, 30, and 28% in LNCaP, DU145, and PC3 cells, respectively. With the combination of antisense TP53 transfection and bufadienolide treatment, the viability of LNCaP cells was significantly reversed to approximately 76% (P < 0.01). However, inhibition of p53 expression in DU145 and PC3 cells did not show significantly increased cell viability.
Fas expression in bufalin‐ and cinobufagin‐treated prostate cancer cells. Fas, a death receptor on the cell surface, is activated by death stimuli to induce cell apoptosis.( 17 ) Previous results show that p53 may be involved in the signal transduction of cell apoptosis induced by bufalin or cinobufagin in LNCaP cells, which have wild‐type p53. To investigate whether Fas mediates cell apoptosis induced by bufalin or cinobufagin in prostate cancer cells with mutant or null p53, flow cytometry and immunocytochemistry were used to examine the expression of Fas on the cell surface. In Figure 5a, the percentage of Fas expression on the cell surface was elevated after treatment for 18 (LNCaP and DU145) or 24 h (PC3). The increased amount of Fas was more significant in DU145 (from 19% in the control group to approximately 42 and 37% in the treated groups) and PC3 cells (from 7 to approximately 25 and 16%) than in LNCaP cells (from 22 to approximately 31 and 26%). Mouse IgG was used as a negative control instead of antiFas antibody. Treatment with etoposide at 40 µmol/L did induce Fas expression on the cell surface (positive control). Under the detection of confocal microscopy, the expression of Fas on the cell surface was increased after 18 and 24 h treatment at a concentration of 10 µmol/L bufalin and cinobufagin. The elevated expression of Fas was more significant in DU145 and PC3 cells than in LNCaP cells (Fig. 5b).
Figure 5.
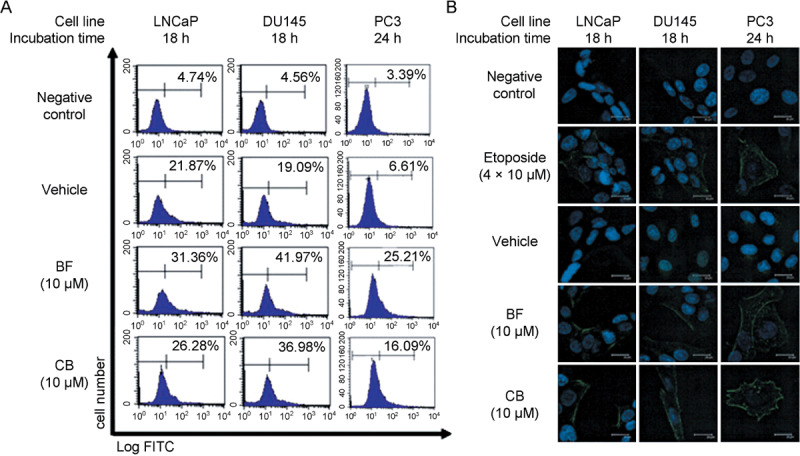
Fas expression on the surface of bufalin (BF)‐ and cinobufagin (CB)‐treated prostate cancer cells. (a) Prostate cancer cells were incubated with BF or CB at a concentration of 10 µmol/L for 18 (LNCaP and DU145) or 24 h (PC3). Cells were washed and incubated with mouse antihuman Fas monoclonal antibody or antimouse IgG as a negative control at 4°C for 2 h. After washing twice, cells were further incubated with fluorescein isothiocyanate (FITC)‐conjugated rat antimouse IgG and analyzed by flow cytometry. Similar results were obtained in three other experiments. (b) After administration, cancer cells on the coverslips were fixed and permeabilized. Coverslips were then incubated sequentially with antiFas antibody and FITC‐conjugated goat antimouse IgG. Fluorescence images were observed by confocal microscopy. Cells treated with etoposide were used as the positive control group. Scale bar = 20 µm.
Effects of Fas small interfering RNA on Fas expression and cell viability in bufalin‐ and cinobufagin‐treated prostate cancer cells. To examine whether the expression of Fas was the essential factor that inhibited cell proliferation in bufalin‐ and cinobufagin‐treated cells, Fas siRNA was transfected into three prostate cancer cell lines. Cells transfected with non‐specific siRNA were used as the control group. In Figure 6a, 10 µmol/L bufalin and cinobufagin caused elevated Fas expression in three cancer cell lines. After transfection of Fas siRNA, Fas expression was downregulated significantly. The Fas mRNA expression of the three prostate cancer cell lines is shown in Figure 6b. Fas siRNA inhibited the expression of Fas mRNA. Treatment with bufalin and cinobufagin significantly elevated the expression of Fas mRNA, but transfection with Fas siRNA inhibited this increase in Fas mRNA induced by bufalin and cinobufagin.
Figure 6.
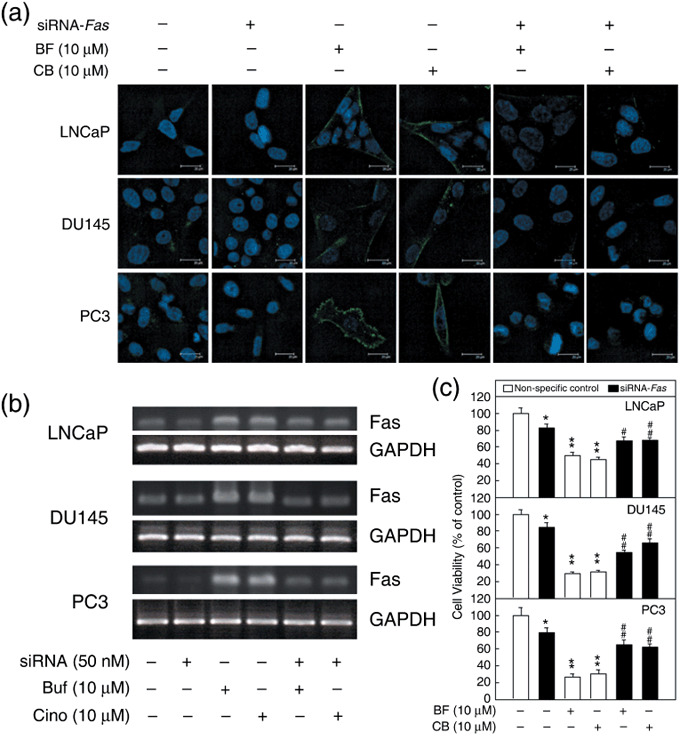
Fas expression and cell viability in Fas small interfering RNA (siRNA)‐transfected prostate cancer cells after treatment with bufalin (BF) and cinobufagin (CB). (a) Cells were transfected with 50 nmol/L siRNA for 48 h then treated with BF or CB at a concentration of 10 µmol/L for 18 (LNCaP and DU145) or 24 h (PC3). The protein expression of Fas induced by BF and CB was inhibited after Fas siRNA transfection. Scale bar = 20 µm. (b) The mRNA of transfected cells was extracted and analyzed by reverse transcription–polymerase chain reaction. The mRNA expression of Fas elevated by BF and CB was downregulated. (c) Cell viability was measured by MTT assay. BF and CB were added to the medium after a 48‐h‐transfection. Open columns represent the non‐specific siRNA‐transfected group; closed columns represent the Fas siRNA‐treated group. Control value = 100%; *P < 0.05, **P < 0.01 versus non‐specific siRNA control group; # P < 0.05, ## P < 0.01 versus non‐specific siRNA plus BF or CB groups. GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase.
The viability of Fas siRNA‐transfected cells was detected by MTT assay after 24 h of treatment with bufalin and cinobufagin (Fig. 6c). The control groups were transfected with non‐specific siRNA. There was no significant variation in cell viability between the non‐specific control group and the non‐transfected groups (data not shown). After treatment with 10 µmol/L bufalin and cinobufagin, the cell viabilities were decreased significantly from 100% to approximately 47, 30, and 28% in LNCaP, DU145, and PC3 cells, respectively (P < 0.01). In comparing bufalin‐ and cinobufagin‐treated cells transfected with either non‐specific siRNA or Fas siRNA, we found that transfection with Fas siRNA caused an increase in cell viability from approximately 47 to 67% in LNCaP cells, from 30 to 60% in DU145 cells, and from 28 to 63% in PC3 cells (Fig. 6c; compare bars 2 and 3 with bars 5 and 6, P < 0.05). Although cell viability was affected by transfection with Fas siRNA (Fig. 6c; compare bars 4 and 1, P < 0.05), the data indicate that reduction of Fas protein expression reduces the cell death caused by bufalin and cinobufagin treatment.
Discussion
Through the present study we have demonstrated a number of findings:
-
1
Bufalin and cinobufagin prolong the tD of cell proliferation selectively in prostate cancer cells but not in normal HMC.
-
2
Bufadienolides induce cell apoptosis in prostate cancer cells but not in normal breast epithelial cells.
-
3
Bufalin and cinobufagin induce cell apoptosis through the mitochondrial pathway and the activation of caspases.
-
4
Bufadienolides elevate the expression of p53 in LNCaP cells, which express wild‐type p53, and downregulation of p53 reverses the cell viability inhibited by bufalin and cinobufagin in LNCaP cells.
-
5
The expression of Fas on the cell surface is increased more significantly in DU145 and PC3 cells than in LNCaP cells after treatment with bufalin or cinobufagin.
-
6
Transfection of Fas siRNA elevates cell viability in three bufadienolide‐treated prostate cancer cell lines.
Bufalin and cinobufagin, which are digitalis‐like molecules from an animal source, have been shown to induce apoptosis in leukemic cells by the activation of activating protein (AP)‐1,( 25 ) c‐Jun N‐terminal protein kinase, Rac1, and by the expression of Tiam1.( 26 ) However, the apoptotic signaling of bufadienolides in prostate cancer remains unclear. It is known that prostate cancer cells express Na+‐K+‐ATPase, which acts as a target for digitalis‐like drugs.( 27 ) As Na+‐K+‐ATPase inhibitors, bufalin and cinobufagin cause an increase in intracellular calcium in prostate cancer cells. Toxin‐induced cell apoptosis is usually provoked by sustained upregulation of cytosolic Ca2+.( 28 , 29 ) Therefore, it is reasonable to treat cancer cells with cardioactive steroids. Our previous data have shown that digitalis‐like molecule‐induced apoptosis in prostate cancer is related to the elevation of intracellular Ca2+.( 8 , 30 , 31 ) In addition, bufalin also functions as an inhibitor of topoisomerase II. Topoisomerase II is the target of several antitumor drugs, such as etoposide, adriamycin, genistein, and 4‐(2‐[3.5‐Dioxo‐1‐piperazinyl]‐1‐methylpropyl) piperazine‐2,6‐drone (ICRF)‐193.( 32 ) By blocking the enzyme function of topoisomerase II, DNA double‐strand breaks remain and DNA damage occurs. Cells are able to recognize such DNA damage and to eliminate the injured cells by apoptosis.
Digitalis has a narrow therapeutic range of 0.8–2.0 ng/mL in plasma.( 33 ) Bufalin has similar toxicity to digitalis, and can be eliminated with digitalis‐specific antibody fragment.( 34 ) Because bufalin and cinobufagin might be toxic to cells, their cytotoxic effects on normal human cells and prostate cancer cells were examined. In Figure 1, the increased tD of prostate cancer cells indicated that bufadienolides at a concentration of 0.1 µmol/L had significant antiproliferative effects on prostate cancer cells but not non‐cancerous HMC. Previous results demonstrated that digitalis had no significant effects on normal glomerular epithelial cells.( 31 ) In addition, Jing et al. reported that bufalin does not induce apoptosis in normal mononuclear and polymorphonuclear cells, indicating that the antiproliferative effects of bufalin may be cell type specific.( 35 ) Moreover, the antiproliferative effects were more significant in androgen‐independent prostate cancer cells than in androgen‐dependent cells. With a relatively short growth cycle, the occurrence of DNA damage in DU145 and PC3 cells would be more than that in LNCaP cells. Although human mesangial and glomerular epithelial cells may not represent every kind of normal human cell, it is apparent that bufalin has antiproliferative effects on rapidly proliferating cancer cells rather than on normal cells with longer growth periods.
Apoptosis‐inducing molecules, such as cisplatin, paclitaxel, camptothecin, etoposide, and all‐trans‐retinoic acid, which are specific for cancer cells, are able to be used as antitumor drugs because programmed cell death does not cause an inflammatory response.( 36 ) In Figure 2, bufalin and cinobufagin significantly induced cell apoptosis in prostate cancer cells, especially in malignant cell lines. The apoptotic signal was not significant in breast epithelial cell lines. However, the rapidly growing 184A1 cells were more sensitive to bufadienolide treatment. These data suggest that the apoptotic effects induced by bufalin and cinobufagin were more specific to rapidly growing malignant cells. Caspase activation is involved in the apoptotic signal transduction induced by digitalis and bufalin.( 8 , 30 , 31 ) Figure 3a illustrates that the time point of the peak response of caspase activation differed in three kinds of prostate cancer cells. Our results suggest that LNCaP and DU145 cells were more sensitive to treatment with higher concentrations over a shorter time period than PC3 cells. However, the change in tD indicates that a lower concentration of bufalin is more effective in PC3 cells after prolonging the time of treatment. These differences might result from the differing nature of these three prostate cancer cell lines. LNCaP cells have wild‐type p53 and release prostate‐specific antigen. They seem more sensitive to chemotherapeutic drugs than androgen‐independent prostate cancer cells with mutant p53.( 37 ) Also, topoisomerase II is expressed at highest levels in rapidly proliferating prostate cancer cells.( 38 ) The loss of enzyme activity caused by bufalin treatment resulted in dramatic DNA damage and cell apoptosis in rapidly proliferating androgen‐independent prostate cancer cells. These two mechanisms might explain why LNCaP cells were more sensitive to higher concentrations and shorter time treatments, whereas proliferation of PC3 cells was decreased after lower concentrations and longer time treatments with bufadienolides.
DNA damage might induce Bax translocation to the mitochondria, in turn resulting in release of cytochrome c.( 39 , 40 ) Consistent with previous reports, our results showed that bufalin and cinobufagin stimulate the translocation of Bax to the mitochondria. In addition, cytosolic cytochrome c was also elevated after treatment (Fig. 3c). These results indicate that the mitochondrial pathway is involved in the signal transduction of bufalin‐ and cinobufagin‐induced apoptosis. Although treatment with 0.1 µmol/L for 18 and 24 h did not induce significant effects on cell apoptosis (Fig. 3), the results shown in the supplementary figure suggest that the concentration of 0.1 µmol/L used in Figure 1 could induce cancer cell apoptosis after lengthening the treatment time. DNA damage caused by topoisomerase II inhibitor might induce the phosphorylation of p53. The active form of p53 prompted translocation of Bax to the mitochondria and release of cytochrome c into the cytosol.( 39 ) Figure 4a shows that the expression of p53 was increased in LNCaP cells after treatment with bufalin and cinobufagin. The mechanism described above might explain the signal transduction caused by bufalin and cinobufagin in LNCaP cells, which express wild‐type p53. In DU145 and PC3 cells, which express mutant and null p53, c‐Jun N‐terminal protein kinase‐dependent Fas‐activated apoptosis may act for the p53‐dependent apoptotic pathway.( 22 ) In Figure 4b, the protein expression of p53 was not suppressed after the transfection of antisense TP53. This might result from the longer half‐life of endogenous p53.( 41 ) The expression of p53 in DU145 and PC3 is mutated and null, respectively. These malignant prostate cancer cells escape the p53‐regulated apoptotic pathway.( 41 ) Therefore, the downregulation of p53 did not reverse the decrease in cell viability caused by bufalin and cinobufagin in DU145 and PC3 cells (Fig. 4c).
In the Fas‐mediated apoptotic pathway, activated caspase 8 activates downstream caspase 3 directly, and also participates in crosstalk with the mitochondrial pathway through cleavage of Bid, a Bcl‐2 family protein. Truncated Bid translocates to the mitochondria, inducing cytochrome c release, which sequentially activates caspase 9 and 3.( 42 ) Our results indicate that Fas might mediate the bufalin‐ and cinobufagin‐induced apoptosis seen in prostate cancer cells (5, 6). The elevated Fas levels were found to be more significant in malignant androgen‐independent prostate cancer cells, expressing mutant p53, than in LNCaP cells. Bufalin and cinobufagin are topoisomerase II inhibitors. Many chemotherapeutic drugs, such as etoposide, also have this characteristic. According to other studies on etoposide, topoisomerase II inhibitors may stimulate Fas‐mediated cell apoptosis independently of Fas ligand.( 43 ) Treatment with etoposide induces c‐Jun N‐terminal protein kinase activation, and then mediates Fas aggregation and subsequent death‐induced signaling complex formation in p53‐mutated prostate cancer cells.( 44 ) Therefore, bufalin‐ and cinobufagin‐induced Fas‐mediated death signaling might not require the participation of Fas ligand. If Fas ligand is involved in the cell apoptosis induced by bufalin and cinobufagin, the ligand might be from neighboring cells. Treatment with bufalin and cinobufagin might induce the expression of Fas ligand and the aggregation of Fas. Then, neighboring cells could cooperate to trigger Fas‐mediated cell apoptosis.( 45 ) Androgen‐dependent LNCaP cells have wild‐type p53, thus the death stimuli from bufadienolides may go through either p53‐mediated or Fas‐mediated apoptotic pathways. This might explain why there is less Fas expressed on the surface of LNCaP cells. Transfection of Fas siRNA significantly increased the viability of bufadienolide‐treated prostate cancer cells. This phenomenon was more obvious in DU145 and PC3 cells with mutant p53 than in LNCaP cells with wild‐type p53 (Fig. 6c), which implies that Fas might be a major factor in bufadienolide‐induced apoptosis in androgen‐independent prostate cancer cells. These results suggest that bufalin and cinobufagin induce cell apoptosis through p53 and Fas in androgen‐dependent prostate cancer cells, which have wild‐type p53, or via Fas in androgen‐independent prostate cancer cells, which have mutant p53.
In summary, bufalin and cinobufagin caused antiproliferative effects and cell apoptosis in androgen‐dependent and ‐independent prostate cancer cells. Caspase activation, Bax translocation, and cytochrome c release were involved in p53‐ and Fas‐mediated apoptotic pathways after treatment with bufalin or cinobufagin.
Supporting information
Fig. S1. The percentage of the prostate cancer cell population in sub‐G0–G1 after treatment with bufalin and cinobufagin. Cancer cells were treated with bufalin and cinobufagin at a concentration of 0.1 µmol/L for 2–4 days. The cell cycle distribution was examined by flow cytometry. The percentage of the population in sub‐G0–G1 is indicated at the upper‐right corner of each figure. The percentage increased after 2–4 days of treatment in three cancer cell lines. Although treatment with 0.1 µmol/L for 18 and 24 h didn’t induce significant apoptotic effects (Fig. 3), the results of S1 suggest that the concentration of 0.1 µmol/L used in Figure 1 could induce cancer cell apoptosis after a longer treatment time.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Acknowledgments
This work was supported by a grants from the Committee on Chinese Medicine and Pharmacy, Department of Health, Executive Yuan, Taiwan (CCMP97‐RD‐010) and from the National Yang‐Ming University, Tapei, Taiwan (96A‐D‐T189, 97A‐C‐T189).
References
- 1. Gronberg H. Prostate cancer epidemiology. Lancet 2003; 361: 859–64. [DOI] [PubMed] [Google Scholar]
- 2. Jemal A, Siegel R, Ward E et al . Cancer statistics, 2008. CA Cancer J Clin 2008; 58: 71–96. [DOI] [PubMed] [Google Scholar]
- 3. Feldman BJ, Feldman D. The development of androgen‐independent prostate cancer. Nat Rev Cancer 2001; 1: 34–45. [DOI] [PubMed] [Google Scholar]
- 4. Han M, Partin AW, Piantadosi S et al . Era specific biochemical recurrence free survival following radical prostatectomy for clinically localized prostate cancer. J Urol 2001; 166: 416–19. [PubMed] [Google Scholar]
- 5. Bagrov AY, Roukoyatkina NI, Fedorova OV, Pinaev AG, Ukhanova MV. Digitalis‐like and vasoconstrictor effects of endogenous digoxin‐like factor(s) from the venom of Bufo matinus toad. Eur J Pharmacol 1993; 234: 165–72. [DOI] [PubMed] [Google Scholar]
- 6. Krenn L, Kopp B. Bufadienolides from animal and plant sources. Phytochemistry 1998; 48: 1–29. [DOI] [PubMed] [Google Scholar]
- 7. McConkey DJ, Orrenius S. The role of calcium in the regulation of apoptosis. Biochem Biophys Res Commun 1997; 239: 357–66. [DOI] [PubMed] [Google Scholar]
- 8. Yeh JY, Huang WJ, Kan SF, Wang PS. Effects of bufalin and cinobufagin on the proliferation of androgen dependent and independent prostate cancer cells. Prostate 2003; 54: 112–24. [DOI] [PubMed] [Google Scholar]
- 9. Hsieh T. Mechanistic aspects of type‐II DNA topoisomerases. In: Cozzarell: NR , Wang JC, eds. DNA Topology and its Biological Effects. New York: Cold Spring Harbor Laboratory Press, 1990; 243–63. [Google Scholar]
- 10. Pastor N, Cortés F. Bufalin influences the repair of X‐ray‐induced DNA breaks in Chinese hamster cells. DNA Repair 2003; 2: 1353–60. [DOI] [PubMed] [Google Scholar]
- 11. Zhang L, Nakaya K, Yoshida T, Kuroiwa T. Induction by bufalin of differentiation of human leukemia cells HL60, U937, and ML1 toward macrophage/monocyte‐like cells and its potent synergistic effect on the differentiation of human leukemia cells in combination with other inducers. Cancer Res 1992; 52: 4634–41. [PubMed] [Google Scholar]
- 12. Watabe M, Masuda Y, Nakajo S, Yoshida T, Kuroiwa Y, Nakaya K. The cooperative interaction of two different signaling pathways in response to bufalin induces apoptosis in human leukemia U937 cells. J Biol Chem 1996; 271: 14 067–72. [DOI] [PubMed] [Google Scholar]
- 13. Han KQ, Huang G, Gu W, Su YH, Huang XQ, Ling CQ. Anti‐tumor activities and apoptosis‐regulated mechanisms of bufalin on the orthotopic transplantation tumor model of human hepatocellular carcinoma in nude mice. World J Gastroenterol 2007; 13: 3374–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee DY, Yasuda M, Yamamoto T, Yoshida T, Kuroiwa Y. Bufalin inhibits endothelial cell proliferation and angiogenesis in vitro . Life Sci 1997; 60: 127–34. [DOI] [PubMed] [Google Scholar]
- 15. Shi Y. A structural view of mitochondria‐mediated apoptosis. Nat Struct Biol 2001; 8: 394–401. [DOI] [PubMed] [Google Scholar]
- 16. Bristow RG, Hill RP. Hypoxia, DNA repair and genetic instability. Nat Rev Cancer 2008; 8: 180–92. [DOI] [PubMed] [Google Scholar]
- 17. Chinnaiyan AM, O’Rourke K, Tewari M, Dixit VM. FADD, a novel death domain‐containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 1995; 81: 505–12. [DOI] [PubMed] [Google Scholar]
- 18. Ranger AM, Malynn BA, Korsmeyer SJ. Mouse models of cell death. Nat Genet 2001; 28: 113–18. [DOI] [PubMed] [Google Scholar]
- 19. Zhuang SH, Burnstein KL. Antiproliferative effects of 1α,25‐dihydroxyvitamin D3 in human prostate cancer cell line LNCaP involves reduction of cyclin‐dependent kinase 2 activity and persistent G1 accumulation. Endocrinology 1998; 139: 1197–207. [DOI] [PubMed] [Google Scholar]
- 20. Kan SF, Huang WJ, Lin LC, Wang PS. Inhibitory effects of evodiamine on the growth of human prostate cancer cell line LNCaP. Int J Cancer 2004; 110: 641–51. [DOI] [PubMed] [Google Scholar]
- 21. Gottlieb RA, Granville DJ. Analyzing mitochondrial changes during apoptosis. Methods 2002; 26: 341–7. [DOI] [PubMed] [Google Scholar]
- 22. Shimada K, Nakamura M, Ishida E, Kishi M, Yonehara S, Konishi N. c‐Jun NH2‐terminal kinase‐dependent Fas activation contributes to etoposide‐induced apoptosis in p53‐mutated prostate cancer cells. Prostate 2003; 55: 265–80. [DOI] [PubMed] [Google Scholar]
- 23. Steel RGD, Torrie JH. Principles and Procedures of Statistics. New York: McGraw‐Hill, 1960. [Google Scholar]
- 24. Mesner PW Jr, Bible KC, Martins LM et al . Characterization of caspase processing and activation in HL‐60 cell cytosol under cell‐free conditions. Nucleotide requirement and inhibitor profile. J Biol Chem 1999; 274: 22 635–45. [DOI] [PubMed] [Google Scholar]
- 25. Watabe M, Ito K, Masuda Y, Nakajo S, Nakaya K. Activation of AP‐1 is required for bufalin‐induced apoptosis in human leukemia U937 cells. Oncogene 1998; 16: 779–87. [DOI] [PubMed] [Google Scholar]
- 26. Hong C, Shibayama‐Imazu T, Masuda Y, Shinki T, Nakajo S, Nakaya K. Involvement of Tiam1 in apoptosis induced by bufalin in HeLa cells. Anticancer Res 2007; 27: 245–9. [PubMed] [Google Scholar]
- 27. McConkey DJ, Lin Y, Nutt LK, Ozel HZ, Newman RA. Cardiac glycosides stimulate Ca2+ increases and apoptosis in androgen‐independent, metastatic human prostate adenocarcinoma cells. Cancer Res 2000; 60: 3807–12. [PubMed] [Google Scholar]
- 28. Trump BF, Berezesky IK. Calcium mediated cell injury and cell death. FASEB J 1995; 9: 219–28. [DOI] [PubMed] [Google Scholar]
- 29. Furuya Y, Lundmo P, Short AD, Gill DL, Isaacs JT. The role of calcium, pH, and cell proliferation in the programmed (apoptotic) death of androgen‐independent prostate cancer cells induced by thapsigargin. Cancer Res 1994; 54: 6167–75. [PubMed] [Google Scholar]
- 30. Lin H, Juang JL, Wang PS. Involvement of Cdk5/p25 in digoxin‐triggered prostate cancer cell apoptosis. J Biol Chem 2004; 279: 29 302–7. [DOI] [PubMed] [Google Scholar]
- 31. Yeh JY, Huang WJ, Kan SF, Wang PS. Inhibitory effects of digitalis on the proliferation of androgen dependent and independent prostate cancer cells. J Urol 2001; 166: 1937–42. [PubMed] [Google Scholar]
- 32. Chen AY, Lin LF. DNA topoisomerase: essential enzymes and lethal targets. Annu Rev Pharmacol Toxicol 1994; 34: 191–218. [DOI] [PubMed] [Google Scholar]
- 33. Panesar NS. Bufalin and unidentified substances in traditional Chinese medicine cross react in commercial digoxin assay. Clin Chem 1992; 38: 2155–6. [PubMed] [Google Scholar]
- 34. Brubacher JR, Lachmanen D, Ravikumar PR, Hoffman RS. Efficacy of digoxin specific Fab fragments (Digibind) in the treatment of toad venom poisoning. Toxicon 1999; 37: 931–42. [DOI] [PubMed] [Google Scholar]
- 35. Jing Y, Ohizumi H, Kawazoe N et al . Selective inhibitory effect of bufalin on growth of human tumor cells in vitro: association with the induction of apoptosis in leukemia HL‐60 cells. Jpn J Cancer Res 1994; 85: 645–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nasu K, Nishida M, Ueda T et al . Bufalin induces apoptosis and the G0/G1 cell cycle arrest of endometriotic stromal cells: a promising agent for the treatment of endometriosis. Mol Hum Reprod 2005; 11: 817–23. [DOI] [PubMed] [Google Scholar]
- 37. Li Y, Raffo AJ, Drew L et al . Fas‐mediated apoptosis is dependent on wild‐type p53 status in human cancer cells expressing a temperature‐sensitive p53 mutant alanine‐143. Cancer Res 2003; 63: 1527–33. [PubMed] [Google Scholar]
- 38. Van Brussel JP, Van Steenbrugge GJ, Romijn JC, Schroder FH, Mickisch GH. Chemosensitivity of prostate cancer cell lines and expression of multidrug resistance‐related proteins. Eur J Cancer 1999; 35: 664–71. [DOI] [PubMed] [Google Scholar]
- 39. Goping IS, Gross A, Lavoie JN et al . Regulated targeting of BAX to mitochondria. J Cell Biol 1998; 143: 207–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim MK, Oh HL, Choi BY, Lim H, Cho YH, Lee CH. CR229, a novel derivative of β‐carbolin‐1‐one, induces cell cycle arrest and apoptosis in HeLa cells via p53 activation. Cancer Sci 2007; 98: 1402–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Carroll AG, Voeller HJ, Sugars L, Gelmann EP. P53 oncogene mutations in three human prostate cancer cell lines. Prostate 1993; 23: 123–34. [DOI] [PubMed] [Google Scholar]
- 42. Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998; 94: 491–501. [DOI] [PubMed] [Google Scholar]
- 43. Fukazawa T, Fujiwara T, Morimoto Y et al . Differential involvement of the CD95 (Fas/APO‐1) receptor/ligand system on apoptosis induced by the wild‐type p53 gene transfer in human cancer cells. Oncogene 1999; 18: 2189–99. [DOI] [PubMed] [Google Scholar]
- 44. Toyoshima F, Moriguchi T, Nishida E. Fas induces cytoplasmic apoptotic responses and activation of the MKK7‐JNK/SAPK and MKK6‐p38 pathways independent of CPP32‐like proteases. J Cell Biol 1997; 139: 1005–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen JJ, Sun Y, Nable GJ. Regulation of proinflammatory effects of Fas ligand (CD95L). Science 1998; 282: 1714–17. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. The percentage of the prostate cancer cell population in sub‐G0–G1 after treatment with bufalin and cinobufagin. Cancer cells were treated with bufalin and cinobufagin at a concentration of 0.1 µmol/L for 2–4 days. The cell cycle distribution was examined by flow cytometry. The percentage of the population in sub‐G0–G1 is indicated at the upper‐right corner of each figure. The percentage increased after 2–4 days of treatment in three cancer cell lines. Although treatment with 0.1 µmol/L for 18 and 24 h didn’t induce significant apoptotic effects (Fig. 3), the results of S1 suggest that the concentration of 0.1 µmol/L used in Figure 1 could induce cancer cell apoptosis after a longer treatment time.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item