

Abstract
Endostatin (ED) is a carboxyl‐terminal fragment of collagen XVIII with strong antiangiogenic activity. ED has been considered as a highly specific inhibitor of endothelial cell proliferation and migration through interaction with its receptor on the surface of endothelial cells. Recently, direct antitumor effects of ED in colon cancer cells and head and neck squamous cell carcinoma cells has been reported. However, its effect on lung cancer cells has not been clarified. The purpose of the present study was to determine the effect of ED on in vitro lung cancer cell function and to identify its receptor on lung cancer cells. We revealed that α5 integrin is capable of being a functional ED receptor among several integrins that are expressed on murine lung cancer (Lewis lung cancer [LLC]) cells. We further demonstrated that the ED–integrin interaction modulates various in vitro biological functions of LLC cells as we revealed that immobilized ED helps in LLC cell adhesion and migration in an integrin‐dependent manner. Furthermore, ED inhibited LLC cell proliferation and induced apoptosis. Interestingly, ED did not demonstrate any antiproliferative activity against the other murine lung cancer cell line, KLN205, that lacks α5 integrin but binds to immobilized ED through the β1 integrin. In addition, the binding of ED to α5 integrin on LLC cells induced phosphorylation of focal adhesion kinase. Taken together, these results suggest that the interaction between ED and α5 integrin may play an important role in lung cancer cell function. (Cancer Sci 2007; 98: 830–837)
Tumor growth and metastasis depend on blood supply and vessel formation.( 1 ) Specifically, the expansion of solid tumors is critically dependent on angiogenesis, thus making cancer a potential clinical target for antiangiogenic therapy.( 2 ) In contrast to conventional chemotherapy and radiotherapy, which eradicate tumor cells,( 3 ) antiangiogenic therapies are designed to abrogate both nutritionally dependent tumor growth and cancer cell dissemination.( 4 , 5 , 6 )
Endostatin (ED) is a 22‐kDa polypeptide derived from a carboxyl‐terminal fragment of type XVIII collagen.( 7 , 8 , 9 ) Recombinant ED has been reported to inhibit the growth of a wide variety of tumors in mice with no known adverse effects. Tumors treated with several cycles of ED do not develop drug resistance and become dormant through inhibition of endothelial cell function.( 7 , 10 , 11 ) In fact, the antiangiogenic activity of ED is speculated to be specifically mediated by the inhibition of endothelial cell adhesion, migration, proliferation and induction of apoptosis.( 12 ) It has been previously reported that ED binds specifically to the cell surface receptor on endothelial cells and that the receptor–ligand complex is internalized into the cytoplasm.( 13 ) The integrins αv, α5 and β1 have been identified as functional ED receptors on endothelial cells, as binding of ED to these integrins induces intracellular signaling resulting in the promotion of cell adhesion and migration.( 14 ) In addition, cell surface glypicans have been identified as low‐affinity ED receptors, as antisense experiments suggest the critical importance of glypicans in mediating ED activity.( 15 )
Wilson et al. recently reported that ED directly inhibits migration and invasion of head and neck squamous cell carcinoma cells, which are essential for tumor progression.( 16 ) Dkhissi et al. revealed that ED directly inhibits in vitro cell growth of C51 murine colon cancer cells and HT29 human colon cancer cells and induces apoptosis of both of these colon cancer cells through its accumulation at the G1 phase.( 17 ) These results suggest that functional ED receptors, such as integrins, exist not only on endothelial cells, but also on cancer cells, and ED binding to its receptor expressed on cancer cells directly induces antitumor activity by suppressing in vitro cell migration and proliferation. However, it is not clear whether ED directly influences lung cancer cell functions such as migration, proliferation and apoptosis or whether it possesses antitumor activity in lung cancer.
In the present study, we identified α5 integrin as a functional ED receptor on murine lung cancer cells (Lewis lung cancer [LLC] cells). Moreover, we demonstrated that ED binding to its receptor inhibits in vitro cell proliferation, and promotes LLC cell adhesion and migration. We also observed that LLC cell binding to immobilized ED induces phosphorylation of focal adhesion kinase (FAK), resulting in cell apoptosis. Our study is the first report revealing that ED directly influences lung cancer cell function. The potential mechanisms are also discussed.
Materials and Methods
Cell lines and reagents. The murine lung cancer cell line LLC was purchased from the Riken gene bank (Ibaraki, Japan), and cultured in Iscove's Modified Dulbecco's medium containing 10% (v/v) fetal calf serum. The murine lung cancer cell line KLN205 was purchased from American Type Cell Culture Collection (Manassas, VA, USa) and maintained in Eagle's Minimal Essential medium with Eagle's BSS and 2 mM l‐glutamine containing 10% (v/v) fetal calf serum. Recombinant mouse ED protein used in this study was purchased from Calbiochem (San Diego, CA, USA). The biological activity of ED and its endotoxin‐free status were confirmed in accordance with the manufacturer's instructions. Antibodies used in this study were antimouse α5 antibody (HMα5), antimouse β1 antibody (HMβ1) and antimouse αv antibody (RMV‐7). All antibodies were kindly provided by Professor Okumura (Department of Immunology, Juntendo University, Tokyo, Japan), and have been proven to inhibit ligand binding.( 18 , 19 , 20 ) The anti‐FAK polyclonal antibody was purchased from Upstate Biotechnology (Lake Placid, NY, USA). The antiphosphotyrosine py‐69 antibody was purchased from BD Transduction Laboratories (Tokyo, Japan).
Flow cytometric analysis. Cells were detached from plates with 5 mM ethylenediaminetetracetic acid (EDTA) in phosphate‐buffered saline (PBS), washed with PBS, and then incubated with HMα5‐2, RMV‐7 or HMβ1 in serum‐free RPMI‐1640 medium at 10 µg/mL for 30 min at 4°C. Cells were washed with PBS and then incubated with either fluorescein‐labeled antihamster IgG (Santa Cruz Biotechnology, Santa Cruz, CA, USA) for HMα5‐2 and HMβ1 analysis, or fluorescein‐labeled antirat IgG (Santa Cruz Biotechnology) for RMV‐7 analysis at 4°C for 30 min. Cells were washed twice with PBS, and resuspended in PBS. Then, propidium iodide (Sigma, St Louis, MO, USA) was added to a final concentration of 10 µg/mL. Samples were analyzed with FACscan (Becton Dickinson, Mountain View, CA, USA).
Cell adhesion assay. Plastic 96‐well flat bottom assay plates (Corning Inc., Corning, NY, USA) were coated with recombinant mouse ED (20 µg/mL) or bovine serum albumin (BSA) (10 mg/mL) in PBS and incubated overnight at 4°C. The plates were washed with PBS and non‐specific adhesion sites were blocked with 1% BSA in PBS for 2 h at 37°C. After washing the wells with PBS, 5 × 104 cells in 100 µL of serum‐free RPMI‐1640 medium were seeded in each well. For some experiments, the cell suspensions were treated with HMα5‐2, HMβ1 or RMV‐7 antibodies at a concentration of 10 µg/mL where indicated. Adhesion was allowed to proceed for 2 h at 37°C. The plates were inverted and centrifuged at 150 g for 3 min, and unattached cells were aspirated. The adherent cells were then placed in RPMI‐1640 medium containing 10% (v/v) 2‐(2‐methoxy‐4‐nitrophenyl)‐3‐(4‐nitrophenyl)‐5‐(2,4‐disulfophenyl)‐2H‐tetrazolium monosodium salt (WST‐8; Dojindo, Kumamoto, Japan) at 37°C for 4 h for color development. Absorbance was determined at 450 nm on a microplate reader with microplate manager (Bio‐Rad, Richmond, CA, USA). All experiments were carried out in triplicate.
In vitro cell proliferation assay. Two thousand cells were seeded in 96‐well culture plates, maintained with RPMI‐1640 medium containing 1% (v/v) insulin‐transferrin‐selenium (ITS) medium (Invitrogen, Auckland, New Zealand). After 1 h incubation, various concentrations of mouse ED (25 or 50 µg/mL) were added to the cells and cultured at 37°C with 5% CO2 for 5 days. After incubation, the culture mediums were replaced with RPMI‐1640 without phenol red containing 0.5 mg/mL methylthiazoletetrazolium (MTT; Sigma) and reincubated for 2 h at 37°C. The culture media was then removed and formazan crystals were dissolved with 200 µL dimethylsulfoxide. After vigorous shaking of the plate, the optical density of each well was determined using a microplate reader with microplate manager at 560–640‐nm wavelength. All experiments were conducted in triplicate.
In vitro cell migration assay. In vitro cell migration was analyzed using a cell culture insert with 8‐µm micropore membrane (Becton Dickinson, Franklin Lake, NJ, USA) as described previously.( 21 ) Briefly, the reverse side of the membrane was coated with mouse ED at the various concentrations ranging from 10 to 100 µg/mL and BSA (10 mg/mL). After 15 min incubation, the excess substrate was removed by washing twice with PBS. The cells were resuspended in 0.1% BSA in Iscove's Modified Dulbecco's medium (IMDM) and seeded on the upper chamber at a concentration of 5 × 104/200 µL. Five hundred microliters of 0.1% BSA in IMDM was added to the lower chamber. After incubation for 6 h at 37°C, the filters were fixed with 10% formalin and stained with 0.2% crystal violet. The cells on the upper surface of the filters were removed by swabbing with a cotton swab and the cells that had migrated to the reverse side were counted in 10 random fields under a microscope at ×400 magnification. We also conducted additional experiments by treating cells with mouse ED protein (50 and 100 µg/mL) or antibodies against integrins α5, β1 or αv at a concentration of 10 µg/mL to confirm that cell migration were mediated by the interaction between ED and its receptors.
Immunocytochemical staining for transferase‐mediated dUTP nick‐end labeling and single‐stranded DNA. One × 104 cells were cultured on a four‐well glass slide Lab Tek chamber (Nunc, Naperville, IL, USA) in RPMI‐1640 medium containing 10% fetal calf serum overnight. The cells were washed in PBS, and 500 µL of RPMI‐1640 medium containing 1% (v/v) ITS medium was added to each chamber. After 1 h incubation, mouse ED was added at a final concentration of 25 µg/mL and cultured for 48 h at 37°C. The cells were then fixed with 4% paraformaldehyde at room temperature for 30 min, and apoptotic cells were detected with an in situ Cell Death Detection kit (Roche, Indianapolis, IN, USA) according to the manufacturer's instructions. Briefly, each experiment conducted with transferase‐mediated dUTP nick‐end labeling (TUNEL) reaction mixture without terminal transferase served as the negative control. The fluorescent images were obtained using an epifluorescence microscope.
To identify apoptotic cells, we used another method. Antibody specific for single‐stranded DNA (ssDNA) (Dako, Kyoto, Japan) was used to identify cells with DNA fragmentation. In brief, cells pretreated with ED (25 µg/mL) at 37°C for 48 h were harvested and fixed on slides using the cytospin method. The slides were fixed with 4% PFA, washed with PBS, and incubated for 30 min in 10% normal goat serum. Then the anti‐ssDNA polyclonal antibody was applied at a dilution of 1:400 and incubated overnight at 4°C. Specific binding was detected through avidin–biotin–peroxidase complex formation with a biotin‐conjugated goat antirabbit IgG (Vectastain ABC kit; Vector Laboratories, Burlingame, CA, USA) and diaminobendizine (DAB) (Sigma) as the substrate. Staining was absent when isotype‐matched immunoglobulin was used as the control.
Immunoprecipitation and immunoblot analysis. Polystyrene dishes (Corning) were coated with mouse ED (50 µg/mL), vitronectin (20 µg/mL) or BSA (10 mg/mL) in PBS and incubated overnight at 4°C. The dishes were then washed three times with PBS and blocked with Iscove's Modified Dulbecco's medium containing 1% (v/v) ITS (IMDM/ITS+) at 37°C for 1 h. LLC or KLN205 cells were harvested after a brief incubation in 0.05% trypsin–EDTA solution (Invitrogen, consisting of 0.05% trypsin and 0.02% EDTA) and washed twice with PBS containing 0.5 mg/mL soybean trypsin inhibitor. This procedure dephosphorylates FAK.( 22 ) Cells were resuspended in IMDM/ITS+, and 1 × 106 cells were added to the dishes and incubated at 37°C for 45 min in the absence or presence of anti‐α5 antibody (5 µg/mL). The cells were then homogenized with lysis buffer (1% Triton X‐100 in PBS, 1.5 mM MgCl2, 1 mM sodium fluoride, 10 mM sodium PPi, 0.2 mM sodium orthovanadate, 20 µg/mL phenylmethylsulfonyl fluoride, 1 µg/mL aprotinin, 1 µg/mL leupeptin). Nuclei were removed by centrifugation at 12 000 g for 30 min at 4°C and the lysate was precleared with protein G‐sepharose beads (Amersham Biosciences, Uppsala, Sweden). Next, the cell lysates were incubated with protein G‐sepharose beads conjugated with anti‐FAK antibody for 1 h at 4°C. The beads were washed three times and boiled in one volume of 2× sodium dodecylsulfate (SDS) sample buffer. Immunoprecipitates were analyzed with sodium dodecylsulfate–polyacrylamide gel electrophoresis under reducing conditions, and electroblotted. After blocking the filters, specific proteins were detected using horseradish peroxidase‐linked antirabbit antibody (Amersham Biosciences) for anti‐FAK polyclonal antibody or antimouse antibody (Amersham Biosciences), or for antiphosphotyrosine py 69 antibody using an enhanced chemiluminescence system (Amersham Bioscience).
RNA interference assay. LLC cells were transfected with 5 nM small interfering RNA (siRNA) using Hiperfect Transfection Reagent (Qiagen, Hilden, Germany) according to the manufacturer's instructions. Briefly, the day before transfection, 3 × 105/4 mL LLC cells were seeded in 6‐cm dishes. After incubation for 24 h at 37°C, siRNA‐Hiperfect Transfection Reagent complexes were added to each dish and they were incubated for 24 h. Knockdown efficacy was evaluated using quantitative real‐time polymerase chain reaction (PCR) and flow cytometric analysis. siRNA directed against α5 integrin (Mm_Itga5–1_HP siRNA) and negative control (Allstars Negative Control siRNA) were purchased from Qiagen. The LLC cells transfected with siRNA for α5 integrin and negative control siRNA were designated LLC/α5 siRNA and LLC/control, respectively. Subsequently, the cells were subjected to in vitro cell proliferation assays for investigating the effect of ED on the proliferation of siRNA‐transfected LLC cells.
Quantitative real‐time PCR. Total RNA was extracted from siRNA‐transfected cells using TRIzol reagent (Invitrogen). First‐strand cDNA was synthesized from 2 µg of total RNA of each cell sample using a Gene Amp RNA PCR Kit (Applied Biosystems, Branchburg, NJ, USA) according to the manufacturer's instructions. The cDNA was then used as templates for individual PCR using specific Quantitect primer assays (Qiagen). PCR were carried out using the Quantitect SYBR Green PCR Kit (Qiagen). The quantitative PCR analysis was carried out using the Applied Biosystem 7500 (Applied Biosystems). Glyceraldehyde‐3‐phosphate dehydrogenase was used for normalizing the expression data.
Statistics. Statistical analysis was carried out with analysis of variance (ANOVA). All data are presented as mean ± SD. Differences between means were considered statistically significant at P < 0.05.
Results
Integrin expression on murine lung cancer cells. Because αvβ1, αvβ3 and α5β1 integrins have been reported as ED receptors, we investigated what receptors are expressed on the surface of LLC and KLN205 cells using flow cytometric analysis. As shown in Fig. 1a, α5, β1 and αv integrins were shown to be expressed on LLC cells, whereas β3 integrin was not expressed on LLC cells (data not shown). Interestingly, only β1 integrin was expressed on the KLN205 cells (Fig. 1b).
Figure 1.
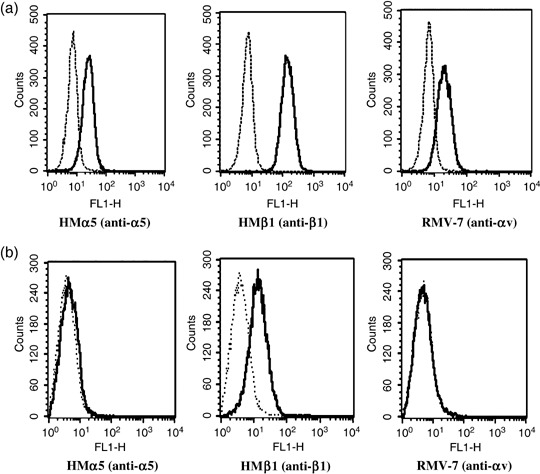
Expression of α5, β1 and αv integrins on (a) Lewis lung cancer (LLC) and (b) KLN205 cells. Integrin expression was assessed with flow cytometric analysis. The dotted lines indicate background fluorescence intensity. The solid lines indicate the fluorescence intensity of α5 (HMα5: left), β1 (HMβ1: middle) and αv (RMV‐7: right) integrin, respectively.
Cell adhesion to immobilized ED is mediated by integrins. To confirm whether LLC or KLN205 cells bind to immobilized ED, cell adhesion assays were carried out. As shown in Fig. 2, LLC and KLN205 cells bound significantly to immobilized ED compared to immobilized BSA. To identify the integrin receptors that mediate cell adhesion to ED, anti‐integrin antibodies (HMα5, HMβ1 and RMV‐7) were preincubated with cell suspension prior to conducting the adhesion assay. Our results showed that HMα5 significantly inhibited LLC cell adhesion to ED, whereas RMV‐7 and HMβ1 did not influence LLC cell adhesion (Fig. 2a). In contrast, KLN205 cell adhesion to ED was significantly inhibited only with the addition of HMβ1 antibody (Fig. 2b). These results suggest that α5 and β1 integrin may serve as functional ED receptors in LLC and KLN205 cells, respectively.
Figure 2.
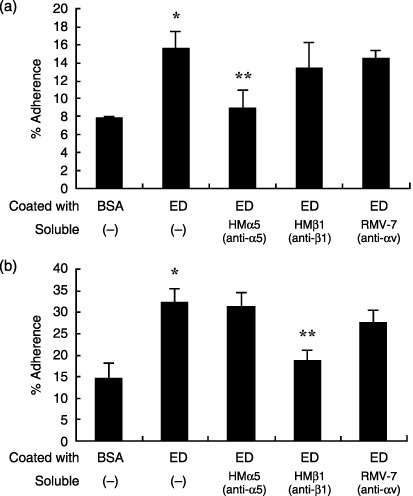
Effect of anti‐integrin antibodies on endostatin (ED) binding to Lewis lung cancer (LLC) and KLN205 cells. Cells were allowed to adhere for 1 h at 37°C to 96‐well plates coated with ED (20 µg/mL) or bovine serum albumin (BSA; 10 mg/mL) in the absence or presence of anti‐α5 antibody (HMα5), anti‐β1 antibody (HMβ1) or anti‐αv antibody (RMV‐7) at 10 µg/mL. The bound cells were quantitated using the colorimetric WST‐8 agent. Note that HMα5 significantly reduced ED binding to LLC cells (a), but did not influence ED binding to KLN205 cells (b). In contrast, HMβ1 markedly reduced ED binding to KLN205 cells. Data are presented as mean ± SD. *P < 0.01 versus BSA; **P < 0.05 versus coated with ED, soluble (–).
Effect of soluble ED on the in vitro proliferation of murine lung cancer cell lines. To investigate whether soluble ED influences LLC or KLN205 cell growth in vitro, various concentrations of ED (25 and 50 µg/mL) were added to the cell suspension and incubated for 5 days at 37°C in RPMI‐1640 medium containing 1% (v/v) ITS. As shown in Table 1, soluble ED inhibited in vitro LLC cell proliferation in a concentration‐dependent manner. In contrast, the inhibitory effect of ED on in vitro cell proliferation was not confirmed in KLN205 cells.
Table 1.
Effect of soluble endostatin (ED) on the proliferation of cells
| Cell line | Soluble ED (µg/mL) | Inhibition ratio (%) |
|---|---|---|
| LLC | 0 | 0 |
| 25 | 12.1 ± 4.4* | |
| 50 | 21.0 ± 2.4* | |
| KLN205 | 0 | 0 |
| 25 | 3.6 ± 3.4 | |
| 50 | 4.0 ± 3.2 | |
| LLC/control | 0 | 0 |
| 50 | 24.1 ± 3.4 | |
| LLC/α5 siRNA | 0 | 0 |
| 50 | 11.4 ± 5.6** |
The cells were incubated in the absence (control) or the presence (samples) of soluble ED (25 or 50 µg/mL) for 5 days. Cell proliferation was assessed by MTT assay. Inhibition ratio (%) was calculated as follows: inhibition ratio (%) = (optical density at 560 nm of control – optical density at 560 nm of sample)/optical density at 560 nm of control ×100. Data are presented as mean ± SD. *P < 0.05 versus control; **P < 0.05 versus Lewis lung cancer (LLC)/control, soluble ED 50 µg/mL. siRNA, small interfering RNA.
Cell migration mediated by immobilized ED. To assess the potential migration of LLC cells toward immobilized ED, a Boyden chamber modified method was used. The membrane was coated with various concentrations of ED. The number of cells that migrated through the membrane was counted. As expected, LLC cells significantly migrated toward ED to a greater extent than toward BSA in a dose‐dependent manner (Fig. 3a). In contrast, immobilized ED did not induce KLN205 cell migration (Fig. 3b). Furthermore, LLC cell migration toward ED was completely inhibited with the addition of soluble ED or antimouse α5 antibody (HMα5‐2) to the upper chambers (Fig. 3c). Interestingly, HMβ1 and RMV‐7 did not influence LLC cell migration toward ED. These results are consistent with our finding that attachment of LLC cell to ED was mediated by α5 integrin as shown in Fig. 2a.
Figure 3.
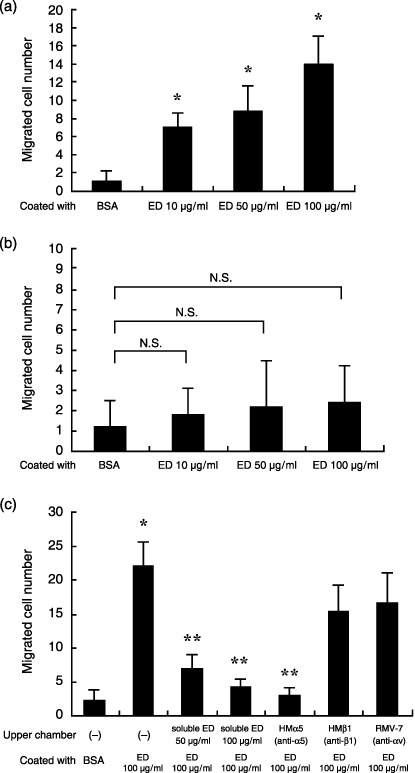
Cell migration toward endostatin (ED). A cell culture chamber separated by a membrane into an upper and lower chamber was utilized where the membrane surface facing the lower chamber was coated with ED at concentration ranging from 10 to 100 µg/mL or 10 mg/mL bovine serum albumin (BSA). The Lewis lung cancer (LLC) cells were placed in the upper chambers and after 6 h of incubation, cells that migrated through the porous filter were counted at ×400 magnification. (a) LLC cells that migrated toward immobilized ED were observed to a much greater extent than cells that migrated toward immobilized BSA. Data are presented as mean ± SD. *P < 0.001 versus BSA. (b) In contrast to LLC cells, immobilized ED did not induce KLN205 cell migration. (c) Enhanced migration of LLC cells toward ED was abrogated with addition of either ED (50 or 100 µg/mL) or HMα5 (10 µg/mL) to the upper chambers. Data are presented as mean ± SD. *P < 0.0001 versus BSA; **P < 0.0001 versus upper chamber (–), coated with ED 100 µg/mL.
Examination for DNA fragmentation. To investigate whether apoptosis is induced with soluble ED treatment of LLC or KLN205 cells, we carried out both TUNEL and ssDNA staining. TUNEL‐positive cells were scattered throughout the colonies of both the control and ED‐treated LLC cells (LLC + ED). TUNEL‐positive cells in LLC + ED (Fig. 4a, right) were more numerous than those in the control (Fig. 4a, left). As shown in Fig. 4b, greater than three‐fold TUNEL‐positive cells were confirmed in the LLC + ED group in comparison to the control. In contrast, the number of TUNEL‐positive KLN205 cells was not increased by treatment with ED (Fig. 4c). In the same way, the number of ssDNA‐positive cells was markedly increased in the LLC + ED group (Fig. 5a, right) in comparison with that of the controls (Fig. 5a, left). As shown in Fig. 5b, greater than 10‐fold ssDNA‐positive cells were identified in the LLC + ED group compared with the control. As expected, there was no significant difference in the number of ssDNA‐positive cells between control and ED‐treated KLN205 cells (Fig. 5c). These results suggest that ED binding to its receptor on LLC cells is capable of directly inducing apoptosis.
Figure 4.
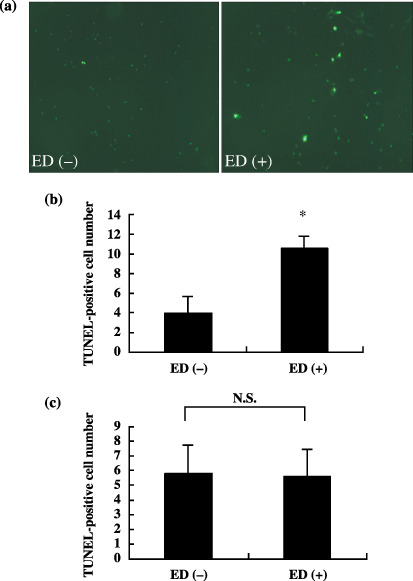
Immunofluorescence analysis for transferase‐mediated dUTP nick‐end labeling (TUNEL) staining of the cells treated with or without soluble endostatin (ED). (a) TUNEL‐positive cells were rarely observed in the untreated Lews lung cancer (LLC) cells (left), whereas there were several positive cells in LLC cells treated with ED (right). Quantification of TUNEL‐positive cells in control and ED‐treated LLC cells are shown in (b). (c) Quantification of TUNEL‐positive cells in control and ED‐treated KLN205 cells. There was no significant difference in the number of apoptotic cells between control and ED‐treated KLN205 cells. The number of TUNEL‐positive cells in five fields was counted at ×200 and presented as the mean ± SD. *P < 0.001 versus ED (–).
Figure 5.
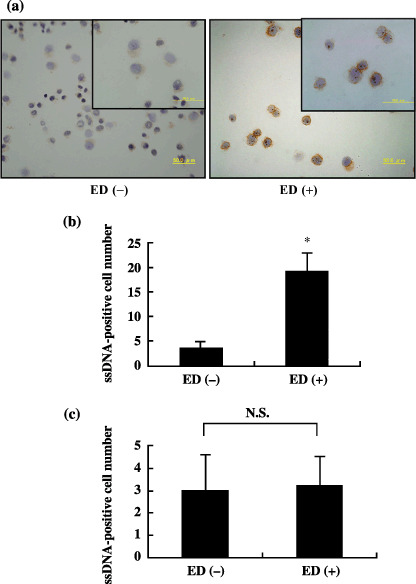
Immunocytochemistry for single‐stranded DNA (ssDNA) in control and endostatin (ED)‐treated Lewis lung cancer (LLC) cells. (a) ssDNA‐positive cells were rarely identified in the control cells (left), whereas there were several positive cells in ED‐treated LLC cells (right) (original magnification ×400). Insets are magnified views (×1000). (b) Quantification of ssDNA‐positive cells in control cells and ED‐treated LLC cells. (c) The number of ssDNA‐positive cells in ED‐treated KLN205 cells was not significantly different from that of control cells. The number of ssDNA‐positive cells in five fields of cytospin was counted at ×400 and presented as mean ± SD. *P < 0.001 versus ED (–).
Immobilized ED promotes FAK phosphorylation in LLC cells but not in KLN205 cells. Integrin‐mediated cell attachment on cognate integrin ligands, such as extracellular matrix proteins, results in dissemination of focal adhesion formation and induction of protein tyrosine phosphorylation.( 23 ) To examine whether immobilized ED is capable of inducing FAK phosphorylation, LLC cells were incubated on dishes that had been coated with ED under IMDM/ITS+ conditions. As shown in Fig. 6a, immobilized ED, similar to vitronectin, induced a high level of tyrosine phosphorylation of FAK in LLC cells. In contrast, immobilized ED did not influence tyrosine phosphorylation of FAK in KLN205 cells, which do not express α5 integrin (Fig. 6b). As expected, induced phosphorylation of FAK was abrogated with antiα5 antibody (data not shown). No changes were observed in the total amounts of FAK, indicating that the enhanced signals were attributable to tyrosine phosphorylation of FAK. These results suggest that immobilized ED serves as an adhesive substrate for LLC cells, possibly by interacting with integrins on the cell surface.
Figure 6.
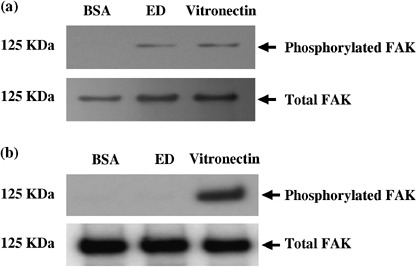
Tyrosine phosphorylation of focal adhesion kinase (FAK). Lewis lung cancer (LLC) and KLN205 cells were placed for 45 min at 37°C on dishes that had been coated with endostatin (ED; 50 µg/mL), vitronectin (20 µg/mL) or bovine serum albumin (BSA; 10 mg/mL). Cell lysates containing equal amounts of protein were immunoprecipitated with anti‐FAK antibody, and one‐half of the precipitates was analyzed by immunoblotting with antiphosphotyrosine antibodies (top panel). The other half was probed with anti‐FAK antibody to confirm loading (bottom panel). Note that high levels of phosphorylated FAK in LLC cells plated on ED and vitronectin were observed (a). In contrast, phosphorylation of FAK was not observed in KLN205 cells cultured on ED (b).
Downregulation of α5 integrin diminishes the inhibitory effect of ED on LLC cell proliferation. To prepare α5 integrin‐knockdown LLC cells, we transfected 5 nM siRNA for α5 integrin in 20 µL of Hiperfect Transfection Reagent into LLC cells and estimated the knockdown efficacy with quantitative real‐time PCR and flow cytometric analysis after 24 h transfection. As shown in Fig. 7a, transfection of LLC cells with siRNA resulted in a significant decrease in α5 integrin mRNA levels. As expected, expression of α5 integrin was markedly reduced in the LLC/α5 siRNA (Fig. 7b, right) compared with LLC/control (Fig. 7b, left). To investigate whether antiproliferative signal of ED to LLC cells was mediated by α5 integrin on LLC cells, α5 integrin knockdown LLC cells were subjected to in vitro cell proliferation assays. As shown in Table 1, LLC/α5 siRNA significantly attenuated the inhibitory effect of ED on LLC cell proliferation. These results indicate that the antiproliferative signal is mediated by ED binding to lung cancer cells through the α5 integrin receptor expressed on LLC cells.
Figure 7.
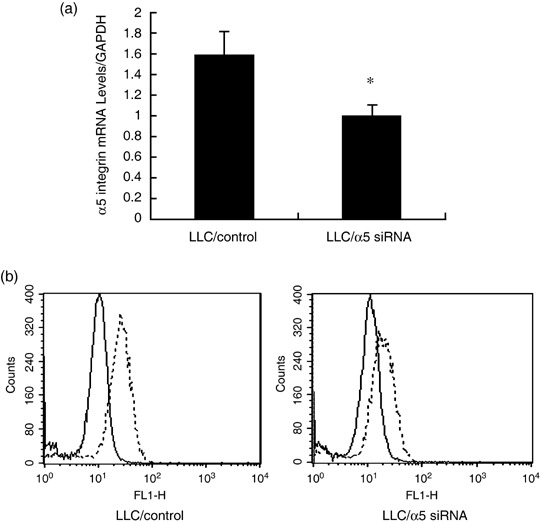
Small interfering RNA (siRNA)‐mediated suppression of α5 integrin. (a) α5 integrin mRNA expression levels in Lewis lung cancer (LLC)/control and LLC/α5 siRNA were determined with quantitative real‐time polymerase chain reaction using specific primer sets. The result is expressed as mean ± SD. P < 0.05 versus LLC/control. (b) The expression of α5 integrin in LLC/control and LLC/α5 siRNA were assessed with flow cytometric analysis. The dotted lines indicate the fluorescence intensity of α5 integrin.
Discussion
It has previously been reported that ED possesses antiangiogenic activity, but does not regulate cancer cell functions.( 7 ) Recently, ED has been studied not only for its inhibitory effect on vascular endothelial cell function but also its direct antitumor effect on cancer cell migration and proliferation of head and neck squamous cell carcinoma cells.( 16 ) In the present study, we demonstrated that soluble ED directly inhibits the proliferation and migration of mouse lung cancer cells (LLC). Furthermore, we showed that ED induces LLC cell apoptosis using TUNEL assay and ssDNA staining. These findings are inconsistent with those previously reported by Dkhissi et al.( 17 ) Their study examined direct antitumor effects of ED using several cancer cells, including LLC cells, and demonstrated that ED inhibits the growth of colon carcinoma cell lines, C51 and HT29, but did not influence in vitro LLC cell growth. Where does this difference come from? First, this could be explained by differences in the experimental methods, in which complete medium with fetal calf serum, which might have contained various growth factors, was used in their study. These various growth factors might have interfered with ED's in vitro functions in LLC cells. Second, we used higher concentrations of ED (50 µg/mL) than in their study (5 µg/mL). These differences in the experimental protocol may account for this difference between results.
The αv, α5 and β1 integrins have been identified as ED receptors expressed on endothelial cells.( 14 ) Sudhakar et al. reported that ED binding to α5β1 integrins on endothelial cells promotes antiangiogenic activities.( 24 ) Karumanchi et al. identified glypicans as low‐affinity ED receptors on both endothelial and epithelial cells.( 15 ) In the present study, we used LLC and KLN205 cells to investigate whether mouse ED is capable of binding to integrin receptors on the surface of lung cancer cells and alter lung cancer cell function. We first demonstrated that α5 integrin on LLC cells and β1 integrin on KLN205 cells mediate ED binding. Although the β1 integrin is expressed on LLC cells, HMβ1 did not inhibit binding of ED to LLC cells. This discrepancy may be attributed to the difference in glycosylation levels of integrins, which could alter their affinity to ligands. As expected, β1 integrins on LLC and KLN205 cells showed different molecular weights by western blot analysis using anti‐β1 integrin antibody (data not shown). In fact, Isaji et al. reported that N‐glycosylation of β1 integrin influences its functions, such as cell migration, ligand binding and dimer formation.( 25 ) Interestingly, ED showed strong antiproliferative activity in LLC cells in vitro, but this activity was not observed in KLN205 cells even though these cells are capable of binding to ED through the β1 integrin receptor. These results suggest that α5 integrin expressed on LLC cells is a functional receptor, whereas β1 integrin on KLN205 cells is not. In fact, the immobilized ED significantly induced LLC cell migration, which was completely inhibited with the addition of anti‐α5 antibody and soluble ED. Interestingly, immobilized ED did not induce the migration of KLN205 cells lacking α5 integrin. To the best our knowledge, there have been no studies that determined the expression of ED receptors on lung cancer cells. Therefore our report could be the first study that identified and characterized the functional ED receptor on lung cancer cells.
It has been shown by Kim et al. that ED inhibits tumor cellular invasion by blocking the activation and catalytic activity of matrix metalloproteinase 2.( 26 ) Hanai et al. have reported that ED can repress β‐catenin‐dependent cyclin D1 promoter activity, which is thought to be linked to the inhibition of endothelial cell proliferation. Moreover, ED is capable of inhibiting the Wnt‐dependent signaling pathway by stimulating the degradation of β‐catenin both in endothelial and DLD‐1 colon cancer cells.( 27 , 28 ) In the present study, we conducted in vitro cell proliferation assays to investigate whether ED influences in vitro LLC cell growth. To avoid the influence of various growth factors in serum, LLC cells were resuspended in RPMI‐1640 medium with 1% (v/v) ITS, which does not contain serum. A large amount of ED (25 and 50 µg/mL) significantly inhibited in vitro LLC cell growth in a concentration‐dependent manner, but did not affect in vitro KLN205 cell growth. The finding that high concentrations of ED (50 µg/mL) did not affect in vitro cell proliferation of KLN205 cells excludes the possibility that ED was contaminated with endotoxin. Furthermore, downregulation of α5 integrin on LLC cells with the siRNA technique significantly attenuated the inhibitory effect of ED on LLC cell growth in vitro. These results suggest that the antiproliferative signal of ED to LLC cells is mediated by α5 integrin receptor on LLC cells. To determine the cell signaling pathway via the ED receptor, we conducted immunoprecipitation–western blot analysis using FAK antibody. As expected, immobilized ED induced FAK phosphorylation in LLC cells, but not in KLN205 cells. Moreover, FAK phosphorylation in LLC cells was inhibited by anti‐α5 integrin antibody (data not shown). These results suggest that ED serves as an adhesive substrate for LLC cells and induces FAK phosphorylation by interacting with α5‐integrin on the surface of LLC cells.
ED has been reported to inhibit tumor vascularization and to directly suppress the growth of a wide variety of tumors in the systems of murine models.( 7 , 29 ) However, the overall therapeutic efficacy of recombinant ED has been generally moderate in several human clinical studies, especially in patients with lung cancer.( 30 ) We do not know the exact reason why ED has not been useful in these clinical studies. In the present study, we showed that ED binding to α5‐integrin induces significant anticancer activity in LLC cells. In contrast, the same results were not observed in the KLN205 cells that lack α5‐integrin expression. These results indicate that the anticancer efficiency of ED against lung cancer could depend on the presence of α5 integrin expression. One possibility to explain the low therapeutic efficacy of recombinant ED would be the low expression of α5 integrin on cancer tissues in these clinical studies, although it has not been mentioned.
In the present study, we showed the α5 integrin is a functional ED receptor expressed on LLC cells, and high dose of ED binding to its receptor directly suppresses LLC cell functions such as migration and proliferation. The efficiency of ED therapy for lung cancer could be predicted by exploration of the α5 integrin expression on tumor cells. The high dose of ED may also enhance ED's antitumor activity. Further investigation is needed to confirm these hypotheses.
References
- 1. Folkman J. Seminars in Medicine of the Beth Israel Hospital, Boston. Clinical applications of research on angiogenesis. N Engl J Med 1995; 333: 1757–63. [DOI] [PubMed] [Google Scholar]
- 2. Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996; 86: 353–64. [DOI] [PubMed] [Google Scholar]
- 3. Vogel CA, Galmiche MC, Buchegger F. Radioimmunotherapy and fractionated radiotherapy of human colon cancer liver metastases in nude mice. Cancer Res 1997; 57: 447–53. [PubMed] [Google Scholar]
- 4. Bergers G, Javaherian K, Lo KM, Folkman J, Hanahan D. Effects of angiogenesis inhibitors on multistage carcinogenesis in mice. Science 1999; 284: 808–12. [DOI] [PubMed] [Google Scholar]
- 5. Deplanque G, Harris AL. Anti‐angiogenic agents: clinical trial design and therapies in development. Eur J Cancer 2000; 36: 1713–24. [DOI] [PubMed] [Google Scholar]
- 6. Gradishar WJ. An overview of clinical trials involving inhibitors of angiogenesis and their mechanism of action. Invest New Drugs 1997; 15: 49–59. [DOI] [PubMed] [Google Scholar]
- 7. O’Reilly MS, Boehm T, Shing Y et al . Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell 1997; 88: 277–85. [DOI] [PubMed] [Google Scholar]
- 8. Wen W, Moses MA, Wiederschain D, Arbiser JL, Folkman J. The generation of endostatin is mediated by elastase. Cancer Res 1999; 59: 6052–6. [PubMed] [Google Scholar]
- 9. Zatterstrom UK, Felbor U, Fukai N, Olsen BR. Collagen XVIII/endostatin structure and functional role in angiogenesis. Cell Struct Funct 2000; 25: 97–101. [DOI] [PubMed] [Google Scholar]
- 10. Boehm T, Folkman J, Browder T, O’Reilly MS. Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature 1997; 390: 404–7. [DOI] [PubMed] [Google Scholar]
- 11. Dhanabal M, Ramchandran R, Volk R et al . Endostatin: yeast production, mutants, and antitumor effect in renal cell carcinoma. Cancer Res 1999; 59: 189–97. [PubMed] [Google Scholar]
- 12. Dhanabal M, Ramchandran R, Waterman MJ et al. Endostatin induces endothelial cell apoptosis. J Biol Chem 1999; 274: 11 721–6. [DOI] [PubMed] [Google Scholar]
- 13. Dixelius J, Larsson H, Sasaki T et al. Endostatin‐induced tyrosine kinase signaling through the Shb adaptor protein regulates endothelial cell apoptosis. Blood 2000; 95: 3403–11. [PubMed] [Google Scholar]
- 14. Rehn M, Veikkola T, Kukk‐Valdre E et al. Interaction of endostatin with integrins implicated in angiogenesis. Proc Natl Acad Sci USA 2001; 98: 1024–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Karumanchi SA, Jha V, Ramchandran R et al. Cell surface glypicans are low‐affinity endostatin receptors. Mol Cell 2001; 7: 811–22. [DOI] [PubMed] [Google Scholar]
- 16. Wilson RF, Morse MA, Pei P et al. Endostatin inhibits migration and invasion of head and neck squamous cell carcinoma cells. Anticancer Res 2003; 23: 1289–95. [PubMed] [Google Scholar]
- 17. Dkhissi F, Lu H, Soria C et al. Endostatin exhibits a direct antitumor effect in addition to its antiangiogenic activity in colon cancer cells. Hum Gene Ther 2003; 14: 997–1008. [DOI] [PubMed] [Google Scholar]
- 18. Noto K, Kato K, Okumura K, Yagita H. Identification and functional characterization of mouse CD29 with a mAb. Int Immunol 1995; 7: 835–42. [DOI] [PubMed] [Google Scholar]
- 19. Takahashi K, Nakamura T, Koyanagi M et al. A murine very late activation antigen‐like extracellular matrix receptor involved in CD2‐ and lymphocyte function‐associated antigen‐1‐independent killer–target cell interaction. J Immunol 1990; 145: 4371–9. [PubMed] [Google Scholar]
- 20. Yasuda M, Hasunuma Y, Adachi H et al. Expression and function of fibronectin binding integrins on rat mast cells. Int Immunol 1995; 7: 251–8. [DOI] [PubMed] [Google Scholar]
- 21. Takahashi F, Takahashi K, Okazaki T et al. Role of osteopontin in the pathogenesis of bleomycin‐induced pulmonary fibrosis. Am J Respir Cell Mol Biol 2001; 24: 264–71. [DOI] [PubMed] [Google Scholar]
- 22. Zagzag D, Shiff B, Jallo GI et al. Tenascin‐C promotes microvascular cell migration and phosphorylation of focal adhesion kinase. Cancer Res 2002; 62: 2660–8. [PubMed] [Google Scholar]
- 23. Schwartz MA, Schaller MD, Ginsberg MH. Integrins: emerging paradigms of signal transduction. Annu Rev Cell Dev Biol 1995; 11: 549–99. [DOI] [PubMed] [Google Scholar]
- 24. Sudhakar A, Sugimoto H, Yang C, Lively J, Zeisberg M, Kalluri R. Human tumstatin and human endostatin exhibit distinct antiangiogenic activities mediated by αvβ3 and α5β1 integrins. Proc Natl Acad Sci USA 2003; 100: 4766–71. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 25. Isaji T, Sato Y, Zhao Y et al. N‐glycosylation of the β‐propeller domain of the integrin α5 subunit is essential for α5β1 heterodimerization, expression on the cell surface, and its biological function. J Biol Chem 2006; 281: 33 258–67. [DOI] [PubMed] [Google Scholar]
- 26. Kim YM, Jang JW, Lee OH et al. Endostatin inhibits endothelial and tumor cellular invasion by blocking the activation and catalytic activity of matrix metalloproteinase. Cancer Res 2000; 60: 5410–13. [PubMed] [Google Scholar]
- 27. Hanai J, Dhanabal M, Karumanchi SA et al. Endostatin causes G1 arrest of endothelial cells through inhibition of cyclin D1. J Biol Chem 2002; 277: 16 464–9. [DOI] [PubMed] [Google Scholar]
- 28. Hanai J, Gloy J, Karumanchi SA et al. Endostatin is a potential inhibitor of Wnt signaling. J Cell Biol 2002; 158: 529–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yoon SS, Eto H, Lin CM et al. Mouse endostatin inhibits the formation of lung and liver metastases. Cancer Res 1999; 59: 6251–6. [PubMed] [Google Scholar]
- 30. Herbst RS, Hess KR, Tran HT et al. Phase I study of recombinant human endostatin in patients with advanced solid tumors. J Clin Oncol 2002; 20: 3792–803. [DOI] [PubMed] [Google Scholar]