

Abstract
Cell transformation arises from activation of oncoproteins and/or inactivation of tumor suppressor proteins. During the initial stage of carcinogenesis, transformation occurs in a single cell within an epithelial monolayer. However, it is not known what happens at the interface between normal and transformed cells once the initial transformation has occurred. Using elaborate cell culture systems, recent reports have shown that interactions between normal and transformed epithelial cells can induce various phenomena. For example, when Ras‐ or Src‐transformed cells are surrounded by normal epithelial cells, multiple signaling pathways are activated and the transformed cells are apically extruded from the epithelium. In addition, normal and certain types of transformed cells compete with each other for cell survival, and the transformed cells undergo apoptosis. Importantly, when transformed cells alone are present, neither apoptosis nor elimination from epithelia occurs, indicating that the presence of surrounding normal cells influences the signaling pathways and fate of transformed cells. Comparable phenomena are also observed in zebrafish and mice in vivo model systems. In this review, I will introduce this newly emerging research field and discuss how these studies can potentially lead to establishment of novel types of cancer prevention and treatment. (Cancer Sci 2011; 102: 1749–1755)
In our human society, to lead a peaceful life, each of us communicates with each other and properly behaves by responding to a variety of information. However, there often exist those who disturb our harmonious society with disorderly conduct. Therefore, we have created social and cultural systems to cope with the bad guys. For extremely harmful individuals (e.g. terrorists, serial murderers), a professional group of people such as police or army will handle them. In contrast, for those who are slightly disturbing (e.g. a lazy student in a lab or a noisy neighborhood), surrounding ordinary people will try to deal with their problems that are too minor for police or army to take care of. Similarly, in our body, each cell receives various signals or inputs from the surrounding environment and responds to them accordingly. However, mutations in oncogenes or tumor suppressor genes produce cells that have different properties from neighboring cells and become harmful for the society of cells. It is well known that for highly malignant cancer cells, specialized immune cells (police or army in the cell society) will detect and eliminate them. Then, for less transformed cells at the earlier stage of oncogenesis, such as cells with a single oncogenic mutation, do surrounding normal cells (ordinary people in the cell society) recognize their presence and take some action?
In humans, more than 80% of cancers are derived from epithelial tissues including lung, stomach, colon, and mammary glands. Therefore, it is important to investigate the interface between transformed and non‐transformed epithelial cells in order to understand phenomena occurring at the initial stage of oncogenesis. However, it is technically difficult to study the interaction between different types of epithelial cells using conventional co‐culture assays because mixing different epithelial cell populations often leads to cell sorting, a process in which cells with different cell–cell adhesion strength or with unequal membrane surface tensions segregate from each other.( 1 , 2 , 3 ) This cell sorting process would minimize the interface between normal and transformed epithelial cells and hinder thorough analyses on the interaction between them. To circumvent this problem, recently published studies have used cell lines expressing oncoproteins or shRNA of tumor suppressor proteins in an inducible manner.( 4 , 5 , 6 ) In those studies, Madin–Darby canine kidney (MDCK) cells that form a polarized epithelial monolayer were used, providing a useful tool to analyze the physiological interaction between normal and transformed epithelial cells. With these newly established cell culture systems, it has been shown that the presence of surrounding normal cells affects the signaling pathways and behavior of transformed cells (Table 1). In addition, competition for cell survival has been shown to occur between normal and transformed cells in cultured cells and mice (Table 1). In this review article, I will introduce this newly emerging research field: interactions between normal and transformed epithelial cells. Interactions between transformed epithelial cells and underlying stromal cells (such as fibroblasts, endothelial cells, and immune cells) have also been shown to play vital roles in tumorigenesis and tumor maintenance. However, this topic is not included in this review, so please refer to other excellent review articles.( 7 , 8 , 9 , 10 , 11 )
Table 1.
Interactions between normal and transformed epithelial cells in vertebrates, except where hematopoietic stem and precursor cells were used for analyses (†)
| Mutations | Phenomena (reference) | Signaling pathways involved | |
|---|---|---|---|
| Cell culture | Ras | Apical extrusion or basal protrusion formation of Ras‐transformed cells (4) | MAPK, Myosin‐II, Cdc42, ROCK |
| Src | Apical extrusion of Src‐transformed cells (5) | MAPK, Myosin‐II, FAK | |
| Mahjong | Apoptosis of Mahjong‐knockdown cells (6) | JNK | |
| Scribble | Apoptosis of Scribble‐knockdown cells (Norman et al., unpublished data; Medical Research Council, Laboratory for Molecular Cell Biology, London, UK) | p38MAPK | |
| In vivo | |||
| Zebrafish | Src | Apical extrusion of Src‐transformed cells (5) | Unknown |
| Mice | Minute | Elimination of Minute‐knockout cells (37) | Unknown |
| p53† | Cell competition between wild‐type and p53 mutant cells (30) | Unknown | |
Interface between normal and Ras‐transformed epithelial cells
Ras is one of the small GTPase superfamily that regulates multiple cellular processes including cell proliferation, differentiation, and motility.( 12 ) Mutant Ras alleles have been frequently found in various types of human cancers, such as pancreatic and colonic carcinomas.( 12 ) In order to examine the interaction between normal and Ras‐transformed epithelial cells, Hogan et al. ( 4 ) established MDCK epithelial cells expressing constitutively active, oncogenic Ras (RasV12) in a tetracycline‐inducible manner (MDCK‐pTR GFP‐RasV12 cells, hereafter referred as RasV12 cells). The authors first labeled RasV12 cells with a plasma membrane‐permeable fluorescent dye and mixed them with non‐transfected normal MDCK cells at a ratio of 1:100 (RasV12 cells:normal cells). The mixture of cells was cultured on a collagen gel in the absence of tetracycline until they formed an epithelial monolayer. Tetracycline was then added in the culture medium to induce RasV12 expression. Under this condition, the authors observed that 80% of RasV12 cells were apically extruded from the epithelial monolayer (Fig. 1a). This apical extrusion of RasV12 cells occurred independently of cell death, and the extruded cells continued to proliferate and formed multicellular aggregates. Importantly, when RasV12 cells alone were cultured, no apical extrusion was observed (Fig. 1b), indicating that activation of cell‐autonomous Ras signaling pathways per se is not sufficient to induce apical extrusion of RasV12 cells, but that interaction with surrounding normal cells is also required. What is the physiological significance of apical extrusion of Ras‐transformed cells? Generally, to metastasize into distant tissues, transformed epithelial cells have to migrate basally through the underlying matrix. Thus, apical extrusion can be considered to be an antimetastatic cellular process, in which cells move into the direction opposite to that required for metastasis. To understand the molecular mechanism of apical extrusion, the authors analyzed RasV12 cells that were not yet extruded and remained within an epithelial monolayer, just prior to apical extrusion. They found that the height of RasV12 cells along the apicobasal axis was significantly higher than that of surrounding normal cells. In addition, F‐actin accumulated at cell–cell contacts between RasV12 cells and phosphorylation of myosin light chain of myosin‐II was enhanced in RasV12 cells. These changes were not observed when RasV12 cells alone were cultured, suggesting that RasV12 cells recognize that they are surrounded by normal cells and modulate their cell shape and cytoskeleton accordingly. Furthermore, Cdc42, a member of Rho GTPases, was activated in RasV12 cells surrounded by normal cells. Because apical extrusion did not occur in 20% of RasV12 cells surrounded by normal cells, the authors next examined the fate of the non‐extruded RasV12 cells and found that they formed large protrusions underneath surrounding normal cells (Fig. 2). When expression of RasV12 was induced in a group of cells within a monolayer of normal cells, large protrusions were frequently found at the interface between normal and RasV12 cells, but rarely observed between RasV12 cells, suggesting that protrusion formation also occurs specifically at the boundary between normal and RasV12 cells. Experiments with inhibitors for various signaling pathways revealed that apical extrusion and basal protrusion formation are regulated, at least partially, by distinct molecular mechanisms. Finally, the authors investigated the molecular mechanisms of how the fate of RasV12 cells is determined, either being apically extruded or forming basal protrusions. They found that expression of constitutively inactive Cdc42 or dominant negative ROCK (also known as Rho kinase) in RasV12 cells strongly suppressed their apical extrusion while promoting basal protrusion formation. In addition, when surrounded by E‐cadherin‐deficient cells, RasV12 cells produced large basal protrusions more frequently and were more often basally delaminated from the monolayer than when surrounded by normal cells. These data suggest that the fate of RasV12 cells is influenced by the activity of Cdc42 and ROCK in RasV12 cells and E‐cadherin‐based intercellular adhesions between surrounding normal cells (Fig. 2). Thus, RasV12 cells leave epithelial sheets either apically or basally in a cell context‐dependent manner, and most importantly these phenomena occur only when they are surrounded by normal epithelial cells.
Figure 1.
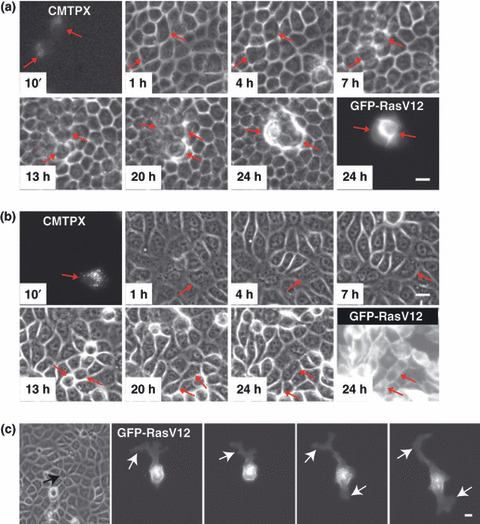
Apical extrusion and basal protrusion formation of RasV12‐expressing cells surrounded by normal epithelial cells. Fluorescently labeled MDCK‐pTR GFP‐RasV12 cells (CMTPX) are mixed with non‐transformed MDCK cells (a,c) or MDCK‐pTR GFP‐RasV12 cells (b). Images are extracted from a representative time‐lapse analysis. Scale bar = 20 μm. (a,b) Red arrows indicate fluorescently labeled RasV12 cells. (c) Images are captured at 50‐min intervals. Black arrow and white arrows indicate RasV12 cell surrounded by non‐transformed cells and protrusions, respectively. (Adapted from Hogan et al., 4 with permission, 1, 3.)
Figure 2.
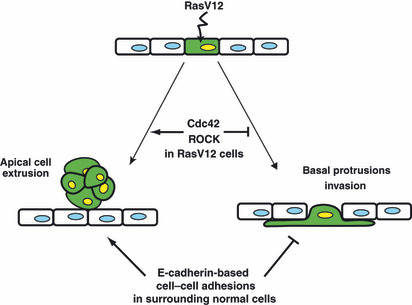
Schematic model for the fate of RasV12 cells surrounded by normal epithelial cells. When expression of RasV12 is induced in a single epithelial cell (green) within a monolayer of normal cells, two independent phenomena can occur in a non‐cell‐autonomous fashion. RasV12‐expressing cells are either apically extruded from the monolayer or form basal protrusions beneath the surrounding neighbors. The fate of RasV12 cells is influenced by the activity of Cdc42 and ROCK in RasV12 cells and by E‐cadherin‐based cell–cell adhesions in the surrounding normal cells. (Adapted from Hogan et al., 4 with permission, fig. 5.)
Interface between normal and Src‐transformed epithelial cells
The Rous sarcoma virus src gene (v‐src) was the first identified oncogene.( 13 ) v‐Src and its cellular counterpart c‐Src are non‐receptor tyrosine kinases that phosphorylate various substrate proteins and regulate cell proliferation, cell adhesions, and other cellular processes.( 14 , 15 ) Increased Src expression and/or activity have been described in many different tumor types including breast and colon cancer.( 15 ) To investigate the phenomena occurring at the interface between normal and Src‐transformed cells, Kajita et al. ( 5 ) used MDCK cells transformed with a temperature‐sensitive v‐Src mutant (ts‐Src MDCK cells). In this cell line, activity of ts‐Src is controlled by temperature shifts between 40.5°C (the non‐permissive temperature for ts‐Src activity) and 35°C (the permissive temperature for ts‐Src activity).( 16 , 17 ) As described above for RasV12 cells, ts‐Src MDCK cells (hereafter referred as Src cells) were first stained with a fluorescent dye and mixed with untransfected (normal) MDCK cells at a ratio of 1:100. The mixture of cells was cultured at 40.5°C until they formed a monolayer of epithelial sheets. Subsequently, activation of ts‐Src was induced by a temperature shift to 35°C, and the fate of Src cells surrounded by normal cells was examined. The authors found that the majority of Src cells were extruded from the apical surface of the monolayer. During the apical extrusion, activity of myosin‐II and focal adhesion kinase was increased in Src cells, leading to activation of downstream MAPK. They also showed that activation of these molecules plays a crucial role in apical extrusion of Src cells. When Src cells alone were cultured, neither apical extrusion nor activation of these molecules was observed, suggesting that the presence of surrounding normal cells influences the fate and signaling pathways of Src cells.
Apical extrusion of Src and RasV12 cells shares several common features. First, apical extrusion occurs only when transformed cells are surrounded by normal cells. Second, apical extrusion occurs independently of apoptosis of the transformed cells. Third, the height of the transformed cells increases when they are surrounded by normal cells. Fourth, activity of myosin‐II is enhanced in the transformed cells, and this activation is involved in apical extrusion. Finally, the MAPK pathway is involved in apical extrusion. It is intriguing that activation of Src and Ras, each of which activates distinct signaling pathways, induces similar phenomena. However, basal protrusion formation, which is observed for RasV12 cells, does not occur in Src cells, indicating that different signaling pathways are also regulated in the respective transformed cells.
Furthermore, the authors examined whether apical extrusion of Src‐transformed cells also occurs in vivo using zebrafish embryos.( 5 ) They induced v‐Src expression in a mosaic manner within the outer epithelial layer (enveloping layer) of zebrafish gastrula and showed that v‐Src‐expressing cells were frequently extruded from the apical surface of the epithelium (Fig. 3). This result indicates that apical extrusion of Src‐transformed cells indeed occurs in vivo.
Figure 3.
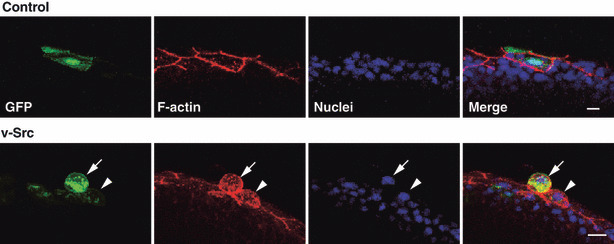
Apical extrusion of a v‐Src‐expressing cell from a monolayer of the enveloping layer in a zebrafish embryo. Immunofluorescence images (semilateral view) of zebrafish embryos (at 8–9 h postfertilization), injected with the GFP‐ (control) or v‐Src‐expressing vector at the 1‐ to 2‐cell stage. Embryos are stained with phalloidin (red) and Hoechst (blue). Arrowhead and arrow indicate the v‐Src‐expressing cell with increased cell height and extruded v‐Src‐expressing cell, respectively. Scale bar = 10 μm. (Adapted from Kajita et al., 5 with permission, fig. 3.)
Cell competition between normal and Mahjong‐transformed epithelial cells
In Drosophila, it has been reported that normal and transformed epithelial cells compete with each other for cell survival.( 18 , 19 , 20 ) For example, when Drosophila Myc (dMyc)‐overexpressing cells coexist with wild‐type cells in the wing disc epithelium, wild‐type cells neighboring dMyc‐overexpressing cells undergo apoptosis and are eliminated from the epithelium.( 21 , 22 ) In contrast, when cells deficient for tumor suppressor protein Scribble coexist with wild‐type cells, Scribble‐deficient cells die by apoptosis.( 23 ) These phenomena are called “cell competition” and are currently intensively studied in Drosophila. However, the underlying molecular mechanisms of cell competition remain largely unknown, and it was not clearly understood whether comparable phenomena also occur in mammals.
Lethal giant larvae (Lgl) was originally identified as a tumor suppressor protein in Drosophila.( 24 ) In Drosophila imaginal discs, mutations of lgl cause loss of apicobasal polarity and uncontrolled proliferation, leading to neoplastic tumor formation.( 24 , 25 ) These data indicate that Lgl plays an important role in cell polarity and cell proliferation. In addition, Lgl is also involved in cell competition; in Drosophila eye discs or wing discs bearing lgl mutant clones in a mosaic manner, some lgl mutant cells undergo apoptosis and are eliminated from the epithelia.( 6 , 26 ) In order to understand the molecular mechanisms of Lgl‐induced cell competition, Tamori et al. ( 6 ) carried out immunoprecipitation using mammalian epithelial cell lines expressing GFP‐tagged mammalian Lgl. They identified a novel Lgl‐binding protein and named it Mahjong, the name of a table game in which winners and losers are determined through strong competition. Mahjong is a cytosolic protein that contains LisH and WD40‐like domains. In Drosophila, there exists one Mahjong homologue protein, and the authors confirmed that Drosophila Mahjong protein interacts with Lgl, suggesting that Mahjong is an evolutionally conserved Lgl‐binding protein. Furthermore, they showed that Mahjong is involved in cell competition in Drosophila; mahj −/− clones adjacent to wild‐type cells frequently underwent apoptosis and were eliminated from the epithelium, whereas mahj −/− clones that were not adjacent to wild‐type cells rarely died. These data suggest that neighboring wild‐type cells trigger mahj −/− cells to undergo apoptosis. The authors also investigated the epistatic relationship between lgl and Mahjong and found that Mahjong acts downstream of Lgl and is involved in Lgl‐mediated cell competition.
To explore the involvement of Mahjong in cell competition in mammalian cells, the authors established MDCK cell lines that stably express Mahjong shRNA in a tetracycline‐inducible manner (MDCK‐pTR Mahjong shRNA cells, hereafter referred as Mahjong shRNA cells).( 6 ) Mahjong shRNA cells were first labeled with a fluorescent dye and mixed with normal MDCK cells at a ratio of 1:10. The mixture of cells was cultured in the absence of tetracycline until they formed a monolayer. The authors then induced expression of Mahjong shRNA with tetracycline and observed the fate of Mahjong‐knockdown cells surrounded by normal cells. They found that after 24–52 h of tetracycline addition, ∼45% of Mahjong shRNA cells underwent apoptosis and were apically extruded from the monolayer (Fig. 4). When Mahjong shRNA cells alone were incubated in the presence of tetracycline, neither apoptosis nor apical extrusion was observed, indicating that apoptosis of Mahjong‐knockdown cells depends on the presence of surrounding normal cells. Furthermore, the authors showed that the JNK signaling pathway is involved in apoptosis of Mahjong‐knockdown cells in both Drosophila and mammalian cells. This is the first evidence showing that cell competition can occur in a mammalian cell culture system.
Figure 4.
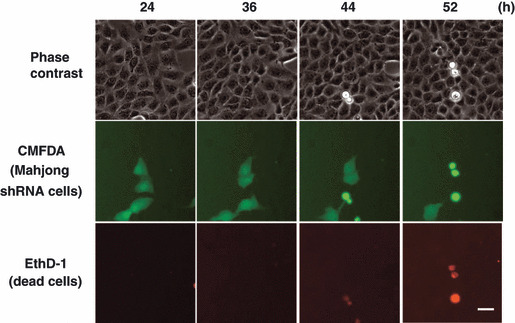
Cell competition induced by knockdown of Mahjong in MDCK epithelial cells. MDCK‐pTR Mahjong shRNA cells are fluorescently labeled with green fluorescent dye CMFDA (green) and mixed with normal MDCK cells at a ratio of 1:10, and cultured in the presence of tetracycline and EthD‐1 (red) for the indicated times. Scale bar = 30 μm. (Adapted from Tamori et al., 6 with permission, fig. 3.)
Cell competition between normal and p53‐transformed cells in mice
The transcription factor p53 has been most extensively studied in its capacity to mediate tumor suppression.( 27 , 28 , 29 ) Expression of p53 becomes upregulated upon various stress responses including irradiation‐mediated DNA damage. To investigate the involvement of p53 in cell competition in mammals, Bondar and Medzhitov( 30 ) used genetic mosaic mouse models and bone marrow chimeras with different levels of p53 activity. First, the authors examined the effect of stress on the competitive status of cells in the hematopoietic system using a low dose of ionizing radiation (IR). The bone marrow from irradiated mice was mixed with that from untreated mice, and the mixture of bone marrow was transferred into lethally irradiated recipient mice. Sixteen weeks later, the total number and the percentage of hematopoietic stem and progenitor cells (HSPCs) were analyzed, and the result showed that hematopoietic cells from untreated mice outcompeted those from irradiated mice. The authors further showed that HSPCs from the untreated mice replaced those from irradiated wild‐type mice, but not from irradiated p53+/− mice, suggesting that IR‐induced competition depends on the p53 level in the competing cells. To further characterize p53‐mediated cell competition, the authors created an inducible genetic mouse model expressing the oncogenic p53 mutant R172H (mp53) in a mosaic manner. The R172H mutant suppresses endogenous p53 activity in a dominant‐negative fashion by forming mixed hetero‐tetramers with wild‐type p53 and reducing p53 binding to the p53‐responsive element in its target genes.( 31 , 32 ) mp53 mice were crossed to Cre‐ER mice, in which Cre can be inducibly activated by tamoxifen injection. Because the Cre‐mediated recombination in the Cre‐ER mice occurred only in a fraction of cells, tamoxifen injection resulted in the generation of genetic mosaic. Analyses of the percentage of mp53‐expressing HSPCs showed that expression of mp53 does not confer a selective advantage under homeostatic conditions. In contrast, after IR, the percentage of mp53‐expressing HSPCs substantially increased. There was no significant difference in total HSPC numbers between wild‐type and mosaic mice after IR, suggesting that mp53‐expressing cells replaced wild‐type cells, rather than simply expanding. The authors also showed that this IR‐induced, p53‐mediated cell competition occurs predominantly in the HSPC compartment, not in differentiated lymphocyte compartments in the hematopoietic system. Interestingly, the classical p53‐mediated DNA damage response did not contribute to the cell competition, and apoptosis was not involved in the process, which is different from cell competition in Drosophila where apoptosis plays a crucial role. Instead, several positive markers of cell proliferation, such as Ki67, cyclin B1, and cyclin A2, were expressed at higher levels in mp53 cells than in wild‐type cells from irradiated mosaic mice. In contrast, genes encoding negative cell proliferation regulators, p57kip and necdin, displayed the opposite pattern of expression. Importantly, these gene expression changes were dependent on the presence of the competitor cells, suggesting that one of the underlying mechanisms on p53‐mediated cell competition is through the non‐cell‐autonomous effect of p53 on cell proliferation. In addition, the authors observed senescence‐like phenotype in outcompeted HSPCs. p16INK4a is a marker and mediator of senescence of hematopoietic stem cells.( 33 , 34 ) Expression of p16INK4a was higher in outcompeted wild‐type cells than in mp53 cells from irradiated mosaic mice. Similarly, P‐selectin and Sdpr, which are upregulated in aged hematopoietic stem cells,( 35 , 36 ) were upregulated in outcompeted wild‐type cells and decreased in mp53 cells. These data suggest that induction of senescence also plays a role in p53‐mediated cell competition.
In addition to this study, there are other reports showing the occurrence of cell competition in mice.( 37 , 38 ) Thus, taken together with data from cultured cells, it is likely that cell competition is a general cellular process in mammals. To examine whether cell competition occurs during various stages of carcinogenesis, further in vivo studies need to be carried out. In conventional mouse model systems, tissue‐specific promoters are used to knock‐in or knock‐out specific genes within the entire tissues. These methods are suitable to examine the effect of genetic changes on cell‐autonomous processes, but not to analyze interactions between normal and transformed cells. Therefore, novel mouse model systems need to be established to induce genetic changes in a mosaic manner within the epithelium. In addition, apical extrusion or cell death of transformed cells at the earlier stage of carcinogenesis might have been overlooked in previous studies, as cells extruded into the apical epithelial lumen encounter physically harsh environments such as flow of urine or stool and are likely to leave the epithelium shortly after extrusion. Thus, in vivo live image analyses are also required to carefully examine these phenomena.
Clinical applications and future perspective
Accumulating evidence has indicated that various phenomena can occur at the interface between normal and transformed cells in vertebrates, especially within the epithelium (Table 1).( 4 , 5 , 6 , 30 , 37 ) However, at present, the underlying molecular mechanisms are still largely unknown (Fig. 5). How do normal and transformed cells recognize the difference(s) between them? How does the cell‐recognition machinery cause activation of signaling pathways in transformed cells or surrounding normal cells, leading to cell death or apical extrusion of transformed cells? Recently, it was shown in Drosophila that a membrane protein Flower is involved in cell competition between normal and dMyc‐overexpressing cells.( 39 ) In Drosophila, the fwe locus produces three alternative splicing isoforms. During cell competition, expression of one of the Flower isoforms is upregulated in loser cells (normal cells that surround dMyc‐overexpressing cells), and cell–cell comparison of relative levels of Flower isoforms determines which cells undergo apoptosis. It remains to be studied whether a comparable molecular mechanism is also involved in cell competition in mammals. It is expected that future studies will reveal a number of molecules that play a crucial role in cell–cell recognition and downstream signaling pathways at the interface between normal and transformed epithelial cells.
Figure 5.
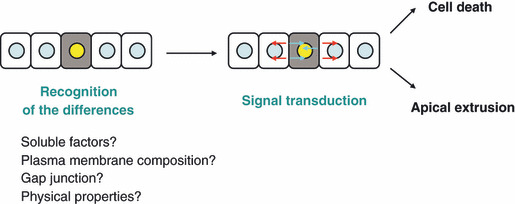
Schematic model for the interaction between normal and transformed epithelial cells.
Several reports have shown that transformed cells undergo apoptosis and/or are apically eliminated from the tissues when surrounded by normal cells, suggesting that normal cells have a capability to attack and remove transformed cells.( 4 , 5 , 6 , 37 ) This is an unexplored scheme of defensive forces against cancer, equipped by surrounding normal cells, not by specialized immune cells. Once molecules that specifically function at the boundary between normal and transformed cells are identified, they will become drug targets. By developing these studies, we may be able to establish novel types of cancer prevention and treatment: enhancing the ability of surrounding normal cells to fight against cancer cells or attenuating the defense of cancer cells against neighboring normal cells. In most of the previous cell competition studies, researchers have examined the interaction of normal cells with cells transformed with single oncogenic mutations. To apply cell competition research into the development of cancer treatment, the interaction between normal cells and malignant cells that contain multiple mutations also needs to be studied. Hence, from the data available at present, it is more realistic to consider a possibility that cell competition research may lead to cancer prevention or treatment of precancerous lesions rather than treatment of malignant cancers. In recent years, most of the efforts of oncologists and pharmaceutical companies have been directed toward treating malignant cancers. However, in most clinical trials, with very few exceptions, cancer patients at advanced stages are often resistant to treatment, and complete remission cannot be achieved because of recurrence. Thus, like other chronic diseases such as diabetes mellitus, hypertension, and rheumatoid arthritis, it might be sensible to put more effort into tackling the identification and treatment of cancer at earlier stages. The interaction between normal and transformed epithelial cells is a newly emerging research field, and the future studies of this topic will be of promising potential to provide us with a new weapon to fight against cancers.
Disclosure Statement
The author has no conflict of interest.
Acknowledgments
This work is supported by the Funding Program for Next Generation World‐Leading Researchers (NEXT Program). Yasuyuki Fujita is also supported by Takeda Science Foundation, the Naito Foundation, the Sagawa Foundation for promotion of Cancer Research, the Yasuda Medical Foundation, Ono Cancer Research Fund, the NOVARTIS Foundation (Japan) for the Promotion of Science, and the Ichiro Kanehara Foundation. I thank Mihoko Kajita for critical reading of the manuscript.
References
- 1. Foty RA, Steinberg MS. The differential adhesion hypothesis: a direct evaluation. Dev Biol 2005; 278: 255–63. [DOI] [PubMed] [Google Scholar]
- 2. Krieg M, Arboleda‐Estudillo Y, Puech PH et al. Tensile forces govern germ‐layer organization in zebrafish. Nat Cell Biol 2008; 10: 429–36. [DOI] [PubMed] [Google Scholar]
- 3. Steinberg MS, Takeichi M. Experimental specification of cell sorting, tissue spreading, and specific spatial patterning by quantitative differences in cadherin expression. Proc Natl Acad Sci USA 1994; 91: 206–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hogan C, Dupre‐Crochet S, Norman M et al. Characterization of the interface between normal and transformed epithelial cells. Nat Cell Biol 2009; 11: 460–7. [DOI] [PubMed] [Google Scholar]
- 5. Kajita M, Hogan C, Harris AR et al. Interaction with surrounding normal epithelial cells influences signalling pathways and behaviour of Src‐transformed cells. J Cell Sci 2010; 123: 171–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tamori Y, Bialucha CU, Tian AG et al. Involvement of Lgl and Mahjong/VprBP in cell competition. PLoS Biol 2010; 8: e1000422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bhowmick NA, Moses HL. Tumor‐stroma interactions. Curr Opin Genet Dev 2005; 15: 97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bissell MJ, Radisky D. Putting tumours in context. Nat Rev Cancer 2001; 1: 46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liotta LA, Kohn EC. The microenvironment of the tumour‐host interface. Nature 2001; 411: 375–9. [DOI] [PubMed] [Google Scholar]
- 10. Egeblad M, Nakasone ES, Werb Z. Tumors as organs: complex tissues that interface with the entire organism. Dev Cell 2010; 18: 884–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McAllister SS, Weinberg RA. Tumor‐host interactions: a far‐reaching relationship. J Clin Oncol 2010; 28: 4022–8. [DOI] [PubMed] [Google Scholar]
- 12. Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol 2008; 9: 517–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hunter T, Sefton BM. Transforming gene product of Rous sarcoma virus phosphorylates tyrosine. Proc Natl Acad Sci USA 1980; 77: 1311–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Frame MC. Src in cancer: deregulation and consequences for cell behaviour. Biochim Biophys Acta 2002; 1602: 114–30. [DOI] [PubMed] [Google Scholar]
- 15. Frame MC, Fincham VJ, Carragher NO, Wyke JA. v‐Src’s hold over actin and cell adhesions. Nat Rev Mol Cell Biol 2002; 3: 233–45. [DOI] [PubMed] [Google Scholar]
- 16. Behrens J, Vakaet L, Friis R et al. Loss of epithelial differentiation and gain of invasiveness correlates with tyrosine phosphorylation of the E‐cadherin/beta‐catenin complex in cells transformed with a temperature‐sensitive v‐SRC gene. J Cell Biol 1993; 120: 757–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fujita Y, Krause G, Scheffner M et al. Hakai, a c‐Cbl‐like protein, ubiquitinates and induces endocytosis of the E‐cadherin complex. Nat Cell Biol 2002; 4: 222–31. [DOI] [PubMed] [Google Scholar]
- 18. Baker NE, Li W. Cell competition and its possible relation to cancer. Cancer Res 2008; 68: 5505–7. [DOI] [PubMed] [Google Scholar]
- 19. Diaz B, Moreno E. The competitive nature of cells. Exp Cell Res 2005; 306: 317–22. [DOI] [PubMed] [Google Scholar]
- 20. Johnston LA. Competitive interactions between cells: death, growth, and geography. Science 2009; 324: 1679–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. de la Cova C, Abril M, Bellosta P, Gallant P, Johnston LA. Drosophila myc regulates organ size by inducing cell competition. Cell 2004; 117: 107–16. [DOI] [PubMed] [Google Scholar]
- 22. Moreno E, Basler K. dMyc transforms cells into super‐competitors. Cell 2004; 117: 117–29. [DOI] [PubMed] [Google Scholar]
- 23. Brumby AM, Richardson HE. Scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila . EMBO J 2003; 22: 5769–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mechler BM, McGinnis W, Gehring WJ. Molecular cloning of lethal(2)giant larvae, a recessive oncogene of Drosophila melanogaster . EMBO J 1985; 4: 1551–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bilder D, Li M, Perrimon N. Co‐operative regulation of cell polarity and growth by Drosophila tumor suppressors. Science 2000; 289: 113–6. [DOI] [PubMed] [Google Scholar]
- 26. Grzeschik NA, Amin N, Secombe J, Brumby AM, Richardson HE. Abnormalities in cell proliferation and apico‐basal cell polarity are separable in Drosophila lgl mutant clones in the developing eye. Dev Biol 2007; 311: 106–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature 2004; 432: 307–15. [DOI] [PubMed] [Google Scholar]
- 28. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature 2000; 408: 307–10. [DOI] [PubMed] [Google Scholar]
- 29. Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol 2007; 8: 275–83. [DOI] [PubMed] [Google Scholar]
- 30. Bondar T, Medzhitov R. p53‐mediated hematopoietic stem and progenitor cell competition. Cell Stem Cell 2010; 6: 309–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. de Vries A, Flores ER, Miranda B et al. Targeted point mutations of p53 lead to dominant‐negative inhibition of wild‐type p53 function. Proc Natl Acad Sci USA 2002; 99: 2948–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Willis A, Jung EJ, Wakefield T, Chen X. Mutant p53 exerts a dominant negative effect by preventing wild‐type p53 from binding to the promoter of its target genes. Oncogene 2004; 23: 2330–8. [DOI] [PubMed] [Google Scholar]
- 33. Janzen V, Forkert R, Fleming HE et al. Stem‐cell ageing modified by the cyclin‐dependent kinase inhibitor p16INK4a . Nature 2006; 443: 421–6. [DOI] [PubMed] [Google Scholar]
- 34. Sharpless NE, DePinho RA. How stem cells age and why this makes us grow old. Nat Rev Mol Cell Biol 2007; 8: 703–13. [DOI] [PubMed] [Google Scholar]
- 35. Chambers SM, Shaw CA, Gatza C, Fisk CJ, Donehower LA, Goodell MA. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol 2007; 5: e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rossi DJ, Bryder D, Seita J, Nussenzweig A, Hoeijmakers J, Weissman IL. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature 2007; 447: 725–9. [DOI] [PubMed] [Google Scholar]
- 37. Oliver ER, Saunders TL, Tarle SA, Glaser T. Ribosomal protein L24 defect in belly spot and tail (Bst), a mouse Minute. Development 2004; 131: 3907–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Oertel M, Menthena A, Dabeva MD, Shafritz DA. Cell competition leads to a high level of normal liver reconstitution by transplanted fetal liver stem/progenitor cells. Gastroenterology 2006; 130: 507–520. [DOI] [PubMed] [Google Scholar]
- 39. Rhiner C, Lopez‐Gay JM, Soldini D et al. Flower forms an extracellular code that reveals the fitness of a cell to its neighbors in Drosophila . Dev Cell 2010; 18: 985–98. [DOI] [PubMed] [Google Scholar]