

Abstract
Vesicular stomatitis virus (VSV) matrix protein (MP) can directly induce apoptosis via the mitochondrial pathway due to the inhibition of host gene expression. Our previous studies have demonstrated that MP gene therapy efficiently suppressed the growth of malignant tumor in vitro and in vivo. The present study was designed to determine the possibility that the combination of MP gene therapy with low‐dose cisplatin would improve therapeutic efficacy against murine melanoma. Immunocompetent C57BL/6 mice bearing B16‐F10 melanoma were established. Mice were treated once every 5 days with i.v. administration of 10 μg pVAX‐MP/30 μg liposome complex per mouse for 16 days and i.p. delivery of cisplatin at 4 mg/kg/mouse on days 6 and 12 after the initiation of MP treatment. We found that MP + cisplatin treatment resulted in significant inhibition of tumor growth and improved the survival time of melanoma‐bearing mice. MP successfully inhibited angiogenesis as assessed by CD31. Histological examination revealed that the combination therapy led to significant increased induction of apoptosis, tumor necrosis, and elevated CD8+ lymphocyte infiltration. Furthermore, the induction efficacy of the CTL response was dramatically enhanced by the combination therapy. Our findings may prove useful in further explorations of the application of these combinational approaches to the treatment of malignant melanoma.
(Cancer Sci 2010; 101: 1219–1225)
Cytotoxic chemotherapy has been one of the major medical approaches commonly used for the treatment of solid cancers. Cisplatin (CDDP), a platinum‐containing anticancer drug, remains the most widely used first‐line element of cytotoxic agents for the treatment of solid malignant tumors.( 1 , 2 ) However, clinical use of cisplatin in the treatment of human cancer is often limited by its acute and cumulative toxicities to normal tissues.( 3 ) One promising strategy to reduce the toxicity of chemotherapy while maintaining its therapeutic effects is the combination of low‐dose conventional chemotherapeutic drugs with agents that would increase cellular chemosensitivity.( 4 , 5 , 6 )
Vesicular stomatitis virus (VSV), the prototypic member of the Rhabdoviridae family, is an enveloped, negative‐sense RNA virus.( 7 ) VSV can selectively replicate in interferon (IFN)‐resistant tumor cells and induce apoptosis of host cells via signaling through double‐stranded RNA‐dependant serine/threonine protein kinase. Fas, and Daxx.( 8 ) Previous studies have demonstrated that VSV can efficiently suppress the growth of syngeneic tumors in immunocompetent mice or human tumor xenografts in nude mice and prolong the survival time of tumor‐bearing animals.( 7 , 9 , 10 , 11 , 12 , 13 , 14 ) However, severe adverse effects, including flu‐like symptoms, oral vesicles, encephalitis, ventriculitis or cervical lymphadenopathy, limit the clinical applications of replication‐competent VSV.( 15 , 16 , 17 , 18 )
Matrix protein (MP), one of the products of VSV, is a structural component of the virion and has multiple roles in viral assembly and cytopathogenesis. Expression of M protein alone causes many of the same cellular effects as infection with VSV by inhibiting host gene transcription and nucleocytoplasmic transport of host RNAs and proteins.( 19 , 20 , 21 , 22 ) We have reported previously that gene therapy with MP alone or combined with radiation would efficiently inhibit the growth of solid tumor and significantly prolong the survival time.( 23 , 24 , 25 , 26 ) These studies suggest that MP is a potent agent for the treatment of tumor and may act as an enhancer of other anticancer drugs.
In the present study, the possibility of MP gene therapy increasing the therapeutic efficacy of low‐dose cisplatin in melanoma‐bearing mice was assessed. Our results suggest that MP may be a beneficial supplement during cisplatin chemotherapy by increasing the induction of apoptosis and immune response, resulting in significant reduction in tumor volume and obviously improved survival time of melanoma‐bearing mice.
Materials and Methods
Cell lines. Murine melanoma B16‐F10 cells, human melanoma A375 cells, murine fibroblast NIH3T3 cells, and SV40 T‐antigen immortalized murine endothelial MS1 cells were obtained from the American Type Culture Collection (ATCC; Rockville, MD, USA). They were cultured in RPMI‐1640 supplemented with 10% FBS, and maintained in a 37°C incubator with a humidified 5% CO2 atmosphere.
Plasmid construction, preparation of cationic liposome, and liposome‐DNA complex. pVAX plasmid (Invitrogen, San Diego, CA, USA) expressing wild‐type VSV M protein, named MP, was constructed in our laboratory previously.( 26 ) As a control, pure pVAX plasmid was used as empty vector (null). The preparation of plasmid and liposome was done as described previously.( 26 , 27 ) DNA:liposome mixtures were prepared 30 min before use. DNA and stock liposome were diluted in 5% dextrose in water (D5W) or RPMI‐1640 medium without serum and mixed in equal volumes, with a DNA/liposome ratio of 1:3 (μg/μg). All reagents were diluted and mixed at room temperature.
Detection of the expression of MP by reverse transcription‐PCR and western blotting. To test the expression of MP, B16‐F10 cells were transfected with pVAX‐MP or pVAX vector. Briefly, about 2 × 105 cells were plated in six‐well plates and treated with normal saline (NS), Lip‐null (2 μg pVAX/6 μg liposome mixtures), or Lip‐MP (2 μg pVAX‐MP/6 μg liposome mixtures). Forty‐eight hours after transfection, the total RNA was extracted with Trizol reagent (Invitrogen) and reverse transcription‐PCR was done using the One‐Step Reverse Transcription‐PCR Kit (Takara). Western blot analysis was performed with total protein from cell lysate homogenates using the antibody as prepared previously.( 26 )
MTT assay. The cytotoxicity of pVAX‐MP:liposome mixtures on murine melanoma B16‐F10 and murine fibroblast NIH3T3 cells was determined by the 3‐(4,5‐dimethylthiazol‐2‐ yl)‐2,5‐diphenyl tetrazolium bromide (MTT; Sigma, St. Louis, MO, USA) colorimetric assay. Briefly, about 5000 cells were seeded in a 96‐well plate at a volume of 100 μL of medium/well and incubated over night. The cells were treated with NS, Lip‐null (0.2 μg pVAX/0.6 μg liposome mixtures), Lip‐MP (0.2 μg pVAX‐MP/0.6 μg liposome mixtures), Lip‐null + CDDP (0.5 μg/mL), and Lip‐MP + CDDP. Six wells were included in each group. After 48 h of incubation, the medium was aspirated and 20 μL of 5 mg/mL MTT was added per well and incubated at 37°C for 4 h; then supernatant fluid was removed, and 150 μL of dimethyl sulfoxide (DMSO) was added per well. Spectrometric absorbance at 540 nm was measured using a microplate reader. The cell survival rate was assessed as percent cell viability in terms of non‐treated control cells.
Assessment of apoptosis in vitro. Cell apoptosis was analyzed by Hoechst 33258 staining (Apoptosis‐Hoechst staining kit; Beyotime, Jiangsu, China) and flow cytometry. About 2 × 105 cells were plated in six‐well plates and treated with NS, Lip‐null (2 μg pVAX/6 μg liposome mixtures), Lip‐MP (2 μg pVAX‐MP/6 μg liposome mixtures), Lip‐null + CDDP (0.5 μg/mL), and Lip‐MP + CDDP. After 48 h of incubation, cells were stained with Hoechst 33258 staining solution according to the manufacturer’s instructions and were analyzed by fluorescence microscopy (Leica, Benshaim, Germany). The apoptotic cells were featured as pyknotic and fragmented nuclei emitting intense fluorescence.( 28 ) Flow cytometric analysis was performed as previously described.( 24 , 25 ) Briefly, cells were collected and resuspended in 1 mL hypotonic fluorochrome solution containing 50 μg/mL propidium iodide (PI) in 0.1% sodium citrate plus 0.1% Triton X‐100, and analyzed by flow cytometry. Cells appearing in the sub‐G1 stage were considered as apoptotic cells.
Murine tumor model and treatment. Female C57BL/6 mice aged 6–8 weeks were purchased from the Laboratory Animal Center of Sichuan University and allowed to acclimate for 1 week before use. All the studies involving mice were performed in accordance with institutional guidelines concerning animal use and care.
About 5 × 105 B16‐F10 melanoma cells were inoculated subcutaneously in the right flank of each mouse. Mice were randomly divided into five groups when the size of tumors reached approximately 30 mm3 (6 days after tumor cell inoculation), and received the corresponding treatment: NS (100 μL), Lip‐null (10 μg pVAX/30 μg liposome mixtures, 100 μL), Lip‐MP (10 μg pVAX‐MP/30 μg liposome mixtures, 100 μL), Lip‐null + CDDP (10 μg pVAX/30 μg liposome mixtures, 100 μL; cisplatin, 4 mg/kg, 100 μL), and Lip‐MP + CDDP (10 μg pVAX‐MP/30 μg liposome mixtures, 100 μL; cisplatin, 4 mg/kg, 100 μL). NS and the liposome mixtures were injected into mice via the tail vein on days 6, 11, 16, and 21 after tumor cell inoculation. The regime of cisplatin (Qilu Pharmaceutical, Shandong, China) was cycled twice on days 6 and 12 after the initiation of liposome mixture treatment. The experiment for the observation of tumor volume and survival advantage included 10 mice per group.
Tumor dimensions were measured with a caliper every 3 days, and tumor volumes were calculated as π/6 × length (mm) × width (mm) × width (mm), where length is the largest superficial diameter and width is the smallest superficial diameter. All of the data are represented as means ± SE. On day 4 after the completion of treatment as described above, the mice were killed by cervical dislocation. Mice tissues of interest were excised and fixed in 10% neutral buffered formalin solution or frozen at −80°C.( 29 )
Histological analysis. To evaluate the potential side effects in the treated mice, the tissues from each group were embedded in paraffin. Sections of 3–5 μm were stained with hematoxylin–eosin (H&E). Immunostaining for VSV‐MP expression and neovascularization in tumors was done as described previously.( 4 ) Briefly, sections were deparaffinized in xylol and rehydrated through graded alcohol series. Antigen retrieval was done by autoclaving sections in 10 mm EDTA (pH 6.0) and incubating them with rabbit immunoserum at 1:100 dilution( 26 ) and a polyclonal antibody rabbit antimouse CD31 (platelet/endothelial cell adhesion molecule 1) at a 1:200 dilution (Labvision, Freemont, CA, USA), followed by incubating with biotinylated rat antirabbit antibody and then streptavidin biotin reagents. Vessel density was determined by counting the number of microvessels per high‐power field in the sections, as described.( 4 )
Immunofluorescence staining was used to determine the infiltration of cytotoxic T lymphocytes (CTLs) in the tumor tissue. The frozen sections were blocked (10% FBS, 3% BSA) for 30 min before staining with anti‐CD8 monoclonal antibody (Cy5PE conjugate; eBioscience, San Diego, CA, USA). Fluorescence was visualized, and images were captured with fluorescence microscopy (Leica). For flow cytometry analysis of CD8+ lymphocyte infiltration, the tumor tissues were digested with type IV collagenase (Sigma) and passed through a 70‐μm‐pore‐size nylon mesh. Samples were stained with Cy5PE‐conjugated antibody to CD8 and then analyzed by flow cytometry.
Quantitative assessment of apoptosis. Tumor species were prepared as described above. The presence of apoptotic cells within the tumor sections was determined using the In Situ Cell Death Detection Kit (DeadEnd Fluorometric TUNEL System; Promega, Madison, WI, USA), according to the manufacturer’s instructions. In the tissue sections, four equal‐sized fields were randomly chosen and analyzed. The apoptotic index (AI) was defined as follows: AI (%) = 100 × apoptotic cells/total tumor cells.
Cytotoxicity assay. A 4‐h 51Cr release assay was performed to evaluate the adaptive cellular immune response as described previously.( 5 , 24 ) Briefly, spleen T lymphocytes obtained from the mice 4 days after cessation of treatment were isolated from single‐cell suspensions with Nylon Fiber Column T (LType; Wako, Osaka, Japan) as CTL effector cells. B16‐F10 murine melanoma cells served as target cells after incubation at 37°C with 51Cr for 2 h. A total of 200 μL of effector and target cells were seeded into the 96‐well microtiter plate at different effector/target ratios, and incubated for 4 h at 37°C. The supernatant was harvested, and the CTL activity was calculated by the formula: % lysis = [(experimental release − spontaneous release)/(maximum release − spontaneous release)] × 100.
Evaluation of potential adverse effects. To evaluate the potential side effects or toxicity on mice during the treatment, gross measures such as weight loss, ruffling of fur, lifespan, behavior, and feeding were investigated. The tissue from the heart, liver, spleen, lung, kidney, and brain were fixed in 10% neutral buffered formalin solution, embedded in paraffin, and then stained with H&E.
Statistical analysis. Data were assayed by anova and an unpaired Student’s t‐test.( 30 ) Kaplan–Meier curves were established for each group of animals, and survival curves were compared by the log‐rank test.( 31 ) Differences between means or ranks as appropriate were considered significant when yielding a P < 0.05.
Results
In vitro expression of MP and therapeutic efficacy of the combination treatment. B16‐F10 melonoma cells were transfected with MP‐expressing plasmid pVAX‐MP and its expression was confirmed by reverse transcription‐PCR and western blotting (Fig. 1A,B) In order to test the combined effect of M‐protein gene therapy and cisplatin in vitro, the B16‐F10 melanoma and murine fibroblast NIH3T3 cells were treated with NS, Lip‐null, Lip‐MP, Lip‐null + CDDP, and Lip‐MP + CDDP. According to the results of the MTT assays, the combined treatment more effectively inhibited the growth of tumor cells (B16‐F10 melanoma) compared with the normal cells (murine fibroblast NIH3T3) (Fig. 1C).
Figure 1.
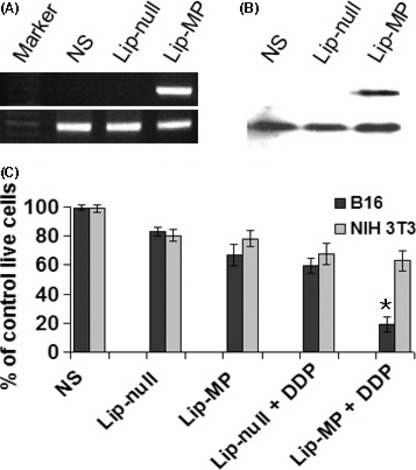
In vitro expression of matrix protein (MP) and viability of cancer cells by MTT assay. (A) Vesicular stomatitis virus (VSV)‐MP was expressed at the RNA level in B16‐F10 cells. (B) Western blot analysis of VSV‐MP expression after transfection of B16‐F10 cells. (C) B16‐F10 and NIH3T3 cells were incubated in 96‐well plates and then treated with normal saline (NS), Lip‐null (0.2 μg pVAX/0.6 μg liposome mixtures), Lip‐MP (0.2 μg pVAX‐MP/0.6 μg liposome mixtures), Lip‐null + CDDP (cisplatin; 0.5 μg/mL), and Lip‐MP + CDDP. The cells were cultured for 48 h and MTT assay was performed for observing the viability of cells as described in the Materials and Methods. The proportion of surviving cells was calculated as a percentage of the control. Data are represented as the mean ± SE of three independent experiments. (*P < 0.01, compared with the NS and Lip‐null groups; P < 0.05, compared with the Lip‐MP and Lip‐null + CDDP groups).
Hoechst staining demonstrated that the apoptotic rate of tumor cells was affected by Lip‐MP alone treatment, whereas the combination treatment resulted in a significant increase of apoptotic tumor cells (Fig. 2A). The apoptotic effect of the combined treatment on B16‐F10 and A375 cells was further quantitatively evaluated by observing sub‐G1 cells using flow cytometry. The combined treatment significantly increased the number of apoptotic cells compared with the other treatments (Fig. 2B).
Figure 2.
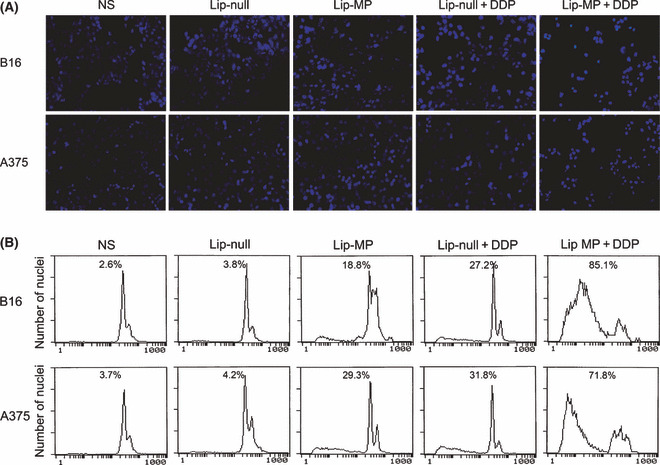
Induction of apoptosis in B16‐F10 and A375 cells by treatment with matrix protein (MP) and cisplatin (CDDP). (A) Tumor cells were treated with normal saline (NS), Lip‐null, Lip‐MP, Lip‐null + CDDP, and Lip‐MP + CDDP. Forty‐eight hours after treatment, cells were stained with Hoechst 33258. Lip‐MP + CDDP treatment resulted in significant increment of morphological changes characteristic of apoptosis (×200). (B) A percentage of sub‐G1 cells (apoptotic cells) identified by observing propidium iodide (PI)‐stained nuclei of tumor cells was assessed using flow cytometry. The results revealed that Lip‐MP + CDDP significantly increased the number of apoptotic tumor cells as compared with NS, Lip‐null, Lip‐MP, or Lip‐null + CDDP. Data are represented as the mean ± SE of three independent experiments.
Tumor growth inhibition. Tumor volume of mice assay showed that both Lip‐MP and Lip‐null + CDDP resulted in effective suppression of tumor growth. However, the combined treatment of Lip‐MP + CDDP had a superior antitumor effect, resulting in more than 65% inhibition in tumor volume compared with the NS group (P < 0.05, 4 days after the completion of treatment). No significant difference of tumor volumes was observed between the NS and Lip‐null groups (Fig. 3A). Moreover, a significant increase was observed in the survival rate of Lip‐MP + CDDP treated mice (P < 0.05, by log‐rank test; Fig. 3B). Although Lip‐null + CDDP therapy did not significantly prolong the lifespan of the animals, this treatment efficiently inhibited tumor growth.
Figure 3.
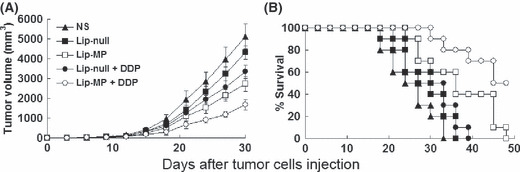
Tumor suppression and survival advantage in mice. Tumor‐bearing mice (10 mice per group) were treated with i.v. administration of normal saline (NS) (100 μL), Lip‐null (10 μg pVAX/30 μg liposome mixtures, 100 μL), or Lip‐MP (10 μg pVAX‐MP/30 μg liposome mixtures, 100 μL) on days 6, 12, 18, and 24 after tumor cell inoculation and administration of cisplatin (CDDP) cycled twice (4 mg/kg, on days 6 and 12 after the initiation of liposome mixtures treatment). (A) Suppression of s.c. tumor growth in mice. Lip‐MP + CDDP treatment resulted in significant tumor growth inhibition versus NS controls (P < 0.01), Lip‐null (P < 0.01), Lip‐null + CDDP (P < 0.05), or Lip‐MP (P < 0.05) from day 20 after initiation of treatment. Points, average tumor volume; bars, ± SD. (B) A significant increase was observed in the survival rates of Lip‐MP + CDDP treatment mice compared with the other groups (P < 0.05, by log‐rank test).
In vivo expression of MP and therapeutic effect on apoptosis. The expression of the VSV‐MP gene in tumor tissue was successfully detected by immunohistochemical analyses (Fig. 4A). In order to test whether apoptosis was indeed the mechanism by which Lip‐MP enhanced the antitumor effects of cisplatin in vivo, TUNEL assay was carried out. Tumors resected 4 days after the completion of treatment were subjected to TUNEL assays. Lip‐MP or Lip‐null + CDDP treatment affected the apoptotic rate of tumor cells, whereas an apparent increase in the number of apoptotic cells was observed within the tumors of the Lip‐MP + CDDP‐treated group (Fig. 4B). Data represent the mean apoptotic index ± SDs of cancer cells as percent normalized to apoptotic index of cancer cells (Fig. 4B).
Figure 4.
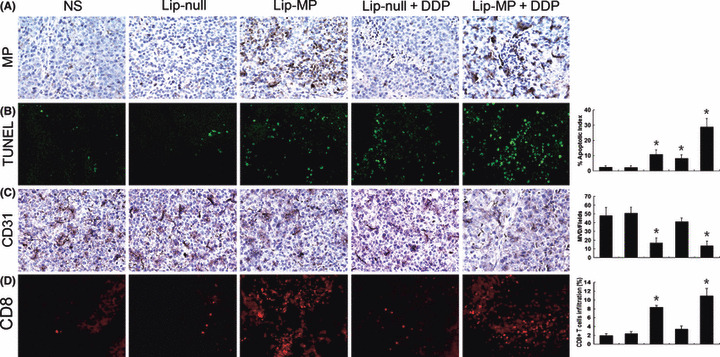
Histochemical analysis of tumors. (A) Immuohistochemical analysis of vesicular stomatitis virus matrix protein (VSV‐MP) expression in tumor tissue. VSV‐MP‐expressing cells could be detected in both Lip‐MP and Lip‐MP + CDDP groups after intravenous administration of pVAX‐MP:lipo complexes versus the other groups. (B) Apoptotic tumor cells within tumor tissues were evaluated by TUNEL assays. An apparent increase in the number of apoptotic cells and apoptotic index was observed within the tumor tissues treated with Lip‐MP + CDDP compared with the control groups (P < 0.05). Data represent the mean apoptotic index ± SDs of cancer cells. (C) Inhibition of angiogenesis assayed by immunohistochemistry with CD31. The number of vessels per ×200 field was counted as described in the Materials and Methods. The Lip‐MP + CDDP‐ and Lip‐MP‐treated groups displayed decreased microvessel density as compared with the control groups (P < 0.05). (D) Fluorescence staining of infiltrated lymphocytes. The frozen sections were stained with anti‐CD8+ monoclonal antibodies as described in the Materials and Methods. CD8+ cytotoxic lymphocyte infiltration was significantly enhanced in the tumor tissues of Lip‐MP + CDDP‐ or Lip‐MP‐treated groups. The number of infiltrating CD8+ cytotoxic lymphocyte was further quantified by flow cytometry. Original magnification, ×200. The columns in all graphs correspond to the labeled columns in the picture. * P < 0.05, significantly different from the normal saline (NS) or Lip‐null groups.
Inhibition of angiogenesis. Angiogenesis within tumor tissue was assessed by CD31 immunohistochemical vessel staining. The most highly vascularized area of each tumor was identified on low power and five high‐powered fields were counted in this area of greatest vessel density. Lip‐MP + CDDP therapy resulted in an apparent inhibition of tumor angiogenesis compared with the control groups, including NS, Lip‐null or Lip‐null + CDDP (P < 0.05, Fig. 4C). Our results suggested that the antitumor effects of the combined treatment acted partly via an anti‐angiogenic mechanism to promote tumor regression.
Fluorescence analysis of intratumoral T‐cell infiltration. To determine whether the therapy improved the infiltration of CD8+ lymphocytes into the tumors, anti‐CD8 monoclonal antibodies were used in immunofluorescence staining. The results showed that Lip‐MP alone or combined with cisplatin would significantly increase the infiltration of CD8+ CTLs (Fig. 4D).
Apoptosis induction of the combined treatment on MS1 murine endothelial cells in vitro. In order to find out the possible mechanism by which the combined treatment inhibited angiogenesis, SV40 T‐antigen immortalized murine endothelial cells were treated with NS, Lip‐null, Lip‐MP, Lip‐null + CDDP, and Lip‐MP + CDDP. Lip‐MP + CDDP treatment resulted in morphological changes characterized as apoptosis. Flow cytometric analysis revealed that there were a larger amount of apoptotic cells in the Lip‐MP + CDDP‐treated group compared with the other groups (Fig. 5).
Figure 5.

Combined treatment induced apoptosis of SV40 T‐antigen immortalized murine endothelial cells in vitro. MS1 cells were treated with normal saline (NS), Lip‐null, Lip‐MP, Lip‐null + CDDP, and Lip‐MP + CDDP. (A) Forty‐eight hours after treatment, cells were stained with Hoechst 33258. Significant morphological changes of cells could be observed in the combination‐treated group. (B) A percentage of sub‐G1 cells (apoptotic cells) identified by observing propidium iodide (PI)‐stained nuclei of tumor cells was assessed using flow cytometry. Representative flow cytograph from different groups are shown. Data are represented as the mean ± SE of three independent experiments.
Effect of combination therapy on CTL activities. Determination of CTL activity provided evidence for the involvement of an immunological mechanism in the antitumor effects of the treatments. Spleen T lymphocytes isolated from mice treated with Lip‐MP or Lip‐MP + CDDP showed higher cytotoxicity against B16‐F10 melanoma cells than those from the other groups (Fig. 6).
Figure 6.
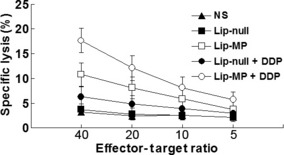
Cytotoxicity assay. The cytotoxic activity of spleen lymphocytes was measured by 4‐h 51Cr‐release assay. The purified T lymphocytes were added to 51Cr‐labeled B16‐F10 melanoma cells immediately after isolation from spleens. The activation of CTLs was induced by Lip‐MP gene therapy and enhanced by cisplatin (CDDP) chemotherapy.
Observation of potential toxicity. To evaluate the possible adverse effects of the treatments, the weight of the mice was monitored every 3 days throughout the whole experiment and considered as a variable for evaluation of systemic well‐being or cachexia. No significant differences in weights were found among the five groups. No adverse consequences in other gross measures such as ruffling of fur, behavior, feeding, or toxic death were observed in Lip‐MP + CDDP group. However, the chemotherapy may not be entirely innocuous. In the histopathological examination of the kidney, destroyed renal tubular cells could be observed in the Lip‐null + CDDP and Lip‐MP + CDDP groups; interestingly, reduced toxicity could be observed in the Lip‐MP + CDDP group (Fig. 7). The nephrotoxicity may be the reason that Lip‐null + CDDP did not efficiently prolong the lifespan of the animals, although the treatment could obviously inhibit tumor growth.
Figure 7.
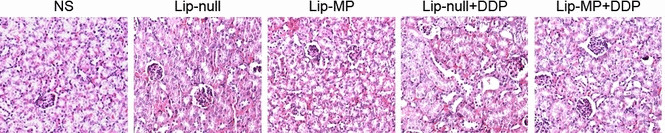
Histopathological examination of kidneys by hematoxylin–eosin (H&E) staining. Kidneys from mice in each group on day 4 after the completion of treatment were removed and embedded in paraffin. The sections were stained with H&E for histopathological examination and observed under light microscope. The result shown here is the representative cortex from each group. The destruction of renal tubular cells could be detected in the Lip‐null + CDDP and Lip‐MP + CDDP groups.
Discussion
Cytotoxic chemotherapy has been the mainstay of medical approaches to the treatment of a variety of tumors. However, dose‐dependent toxicity and drug resistance greatly limit the clinical applications of chemotherapy. Previous studies have demonstrated that drug resistance to chemotherapy is thought to be partly mediated by the ability to circumvent apoptosis.( 31 , 32 ) As increasingly evident, therapies based on the combination of agents impairing different molecular pathways that regulate tumor cell survival can result in an improvement of current cancer management.( 33 , 34 )
It has been reported that the vesicular stomatitis virus matrix protein (VSV‐MP), one structural protein of the virus, can cause the apoptosis of tumor cells by disorganizating cytoskeletal elements and inhibiting host gene expression,( 19 , 20 , 21 , 22 ) which is totally different from the mechanisms of cisplatin.( 35 ) In this regard, we wonder whether gene therapy with MP, which is capable of directly inducing cell death, could enhance the antitumor effects of cisplatin.
In the present study, we show that combined therapy with Lip‐MP and low‐dose cisplatin not only specifically and significantly reduced the growth of the highly tumorigenic and poorly immunogenic B16 melanoma but also efficiently improved the survival of tumor‐bearing animals, without obvious systemic toxicity. Although the exact mechanism by which the combination of Lip‐MP with cisplatin can enhance the therapeutic effects remained to be determined, the improved antitumor efficacy may in part result from the increased induction of the apoptosis. This suggestion is supported by the present findings. In vitro combination treatment with Lip‐MP and cisplatin significantly reduced the viability (Fig. 1) and increased the apoptosis of tumor cells compared with other therapies (Fig. 2). The more apparent apoptotic cells were observed in the tumor tissues of the Lip‐MP + CDDP combination‐treated group (Fig. 4B). The mechanism by which Lip‐MP increased cisplatin‐induced apoptosis may be related to decreasing the expression of Delta‐NP63alpha and increasing the expression of BAX.( 36 )
Previous studies in our laboratory have shown that Lip‐MP could efficiently inhibit tumor angiogenesis.( 23 ) Our findings that Lip‐MP alone or combined with cisplatin significantly decreased microvessel density, leading to the observed increase in tumor cell apoptosis, coincide with previous reports.( 23 ) It has been stated that anti‐angiogenic agents maybe helpful for inhibiting the metastasis of tumor. In the current study, significant survival advantage was observed in the Lip‐MP alone‐ or combination‐treated groups which may in part result from the anti‐angiogenic activity of Lip‐MP, resulting in less dissemination of tumor cells. Thus, we speculate that the anti‐angiogenesis activity of Lip‐MP may play an important role in reducing tumor growth and metastasis, resulting in tumor growth stasis and prolonged survival.
Breaking of immune tolerance against self‐tumor antigen and induction of auto‐immunity against tumors should be a useful approach for the treatment of tumors. It has been reported that introduction of new antigens into tumor cells would stimulate immune responses against autologous malignant cells.( 37 , 38 , 39 ) In the present study, we found that systemic delivery of Lip‐MP could significantly increase the infiltration of CD8+ T lymphocytes (Fig. 4D) and induce tumor‐specific CTL activity in vivo (Fig. 6). Moreover, low‐dose chemotherapeutics not only reduce their toxic effects against normal tissues and avoid the immunosuppressive effects of these drugs, but also improve antitumor immunity through the acquisition of tumor‐associated antigens from apoptotic tumor cells.( 6 ) This may in part explain the result that combined treatment with Lip‐MP and low‐dose cisplatin induced more potent CTL activity in homogeneous tumor cells as compared with the other treatments.
Taken together, our data in the present study suggest that the combination of Lip‐MP gene therapy and low‐dose cisplatin significantly enhanced antitumor efficacy compared with either treatment alone. This synergistic antitumor effect may result from triggering the apoptosis of cancer cells and enhancing the immune response specifically targeting the tumor cells. The present findings may be of importance to the further exploration of the potential application of this combined approach in the treatment of malignant melanoma.
Acknowledgments
This work was supported by National Natural Science Foundation of China (30973452), the National Key Basic Research Program of China (973 project 2010CB529900) and the Hi‐tech Research and Development Program of China (863 project 2007AA021106).
Shan Luo and Ping Chen contributed equally to this work.
References
- 1. Arriagada R, Bergman B, Dunant A et al. Cisplatin‐based adjuvant chemotherapy in patients with completely resected non‐small‐cell lung cancer. N Engl J Med 2004; 350: 351–60. [DOI] [PubMed] [Google Scholar]
- 2. Olaussen KA, Dunant A, Fouret P et al. DNA repair by ERCC1 in non‐small‐cell lung cancer and cisplatin‐based adjuvant chemotherapy. N Engl J Med 2006; 355: 983–91. [DOI] [PubMed] [Google Scholar]
- 3. Chirino YI, Hernández‐Pando R, Pedraza‐Chaverrí J. Peroxynitrite decomposition catalyst ameliorates renal damage and protein nitration in cisplatin‐induced nephrotoxicity in rats. BMC Pharmacol 2004; 4: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li G, Tian L, Hou JM et al. Improved therapeutic effectiveness by combining recombinant CXC chemokine ligand 10 with cisplatin in solid tumors. Clin Cancer Res 2005; 11: 4217–24. [DOI] [PubMed] [Google Scholar]
- 5. Zhang R, Tian L, Chen LJ et al. Combination of MIG (CXCL9) chemokine gene therapy with low‐dose cisplatin improves therapeutic efficacy against murine carcinoma. Gene Ther 2006; 13: 1263–71. [DOI] [PubMed] [Google Scholar]
- 6. Perrotta C, Bizzozero L, Falcone S et al. Nitric oxide boosts chemo‐immunotherapy via inhibition of acid sphingomyelinase in a mouse model of melanoma. Cancer Res 2007; 67: 7559–64. [DOI] [PubMed] [Google Scholar]
- 7. Li Q, Wei YQ, Wen YJ et al. Induction of apoptosis and tumor regression by vesicular stomatitis virus in the presence of gemcitabine in lung cancer. Int J Cancer 2004; 112: 143–9. [DOI] [PubMed] [Google Scholar]
- 8. Gaddy DF, Lyles DS. Oncolytic vesicular stomatitis virus induces apoptosis via signaling through PKR, Fas, and Daxx. J Virol 2007; 81: 2792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Balachandran S, Barber GN. Vesicular stomatitis virus (VSV) therapy of tumors. IUBMB Life 2000; 50: 135–8. [DOI] [PubMed] [Google Scholar]
- 10. Balachandran S, Porosnicu M, Barber GN. Oncolytic activity of vesicular stomatitis virus is effective against tumors exhibiting aberrant p53, Ras, or myc function and involves the induction of apoptosis. J Virol 2001; 75: 3474–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stojdl DF, Lichty BD, TenOever BR et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti‐cancer agents. Cancer Cell 2003; 4: 263–75. [DOI] [PubMed] [Google Scholar]
- 12. Shinozaki K, Ebert O, Kournioti C, Tai YS, Woo SL. Oncolysis of multifocal hepatocellular carcinoma in the rat liver by hepatic artery infusion of vesicular stomatitis virus. Mol Ther 2004; 9: 368–76. [DOI] [PubMed] [Google Scholar]
- 13. Ahmed M, Cramer SD, Lyles DS. Sensitivity of prostate tumors to wild type and M protein mutant vesicular stomatitis viruses. Virology 2004; 330: 34–49. [DOI] [PubMed] [Google Scholar]
- 14. Ebert O, Harbaran S, Shinozaki K, Woo SL. Systemic therapy of experimental breast cancer metastases by mutant vesicular stomatitis virus in immune‐competent mice. Cancer Gene Ther 2005; 12: 350–8. [DOI] [PubMed] [Google Scholar]
- 15. Letchworth GJ, Rodriguez LL, Del cbarrera J. Vesicular stomatitis. Vet J 1999; 157: 239–60. [DOI] [PubMed] [Google Scholar]
- 16. Forger JM 3rd, Bronson RT, Huang AS, Reiss CS. Murine infection by vesicular stomatitis virus: initial characterization of the H‐2d system. J Virol 1991; 65: 4950–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huneycutt BS, Bi Z, Aoki CJ, Reiss CS. Central neuropathogenesis of vesicular stomatitis virus infection of immunodeficient mice. J Virol 1993; 67: 6698–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sur JH, Allende R, Doster AR. Vesicular stomatitis virus infection and neuropathogenesis in the murine model are associated with apoptosis. Vet Pathol 2003; 40: 512–20. [DOI] [PubMed] [Google Scholar]
- 19. Kopecky SA, Lyles DS. The cell‐rounding activity of the vesicular stomatitis virus matrix protein is due to the induction of cell death. J Virol 2003; 77: 5524–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kopecky SA, Lyles DS. Contrasting effects of matrix protein on apoptosis in HeLa and BHK cells infected with vesicular stomatitis virus are due to inhibition of host gene expression. J Virol 2003; 77: 4658–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gaddy DF, Lyles DS. Vesicular stomatitis viruses expressing wild‐type or mutant M proteins activate apoptosis through distinct pathways. J Virol 2005; 79: 4170–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ahmed M, McKenzie MO, Puckett S, Hojnacki M, Poliquin L, Lyles DS. Ability of the matrix protein of vesicular stomatitis virus to suppress beta interferon gene expression is genetically correlated with the inhibition of host RNA and protein synthesis. J Virol 2003; 77: 4646–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhong Q, Wen YJ, Yang HS et al. Efficient inhibition of cisplatin‐resistant human ovarian cancer growth and prolonged survival by gene transferred vesicular stomatitis virus matrix protein in nude mice. Ann Oncol 2008; 19: 1584–91. [DOI] [PubMed] [Google Scholar]
- 24. Zhao JM, Wen YJ, Li Q et al. A promising cancer gene therapy agent based on the matrix protein of vesicular stomatitis virus. FASEB J 2008; 22: 4272–80. [DOI] [PubMed] [Google Scholar]
- 25. Du XB, Lang JY, Xu JR et al. Vesicular stomatitis virus matrix protein gene enhances the antitumor effects of radiation via induction of apoptosis. Apoptosis 2008; 13: 1205–14. [DOI] [PubMed] [Google Scholar]
- 26. Shi W, Tang Q, Chen X et al. Antitumor and antimetastatic activities of vesicular stomatitis virus matrix protein in a murine model of breast cancer. J Mol Med 2009; 87: 493–506. [DOI] [PubMed] [Google Scholar]
- 27. Templeton NS, Lasic DD, Frederik PM, Strey HH, Roberts DD, Pavlakis GN. Improved DNA: liposome complexes for increased systemic delivery and gene expression. Nat Biotechnol 1997; 15: 647–52. [DOI] [PubMed] [Google Scholar]
- 28. Yang F, Li Z, Deng H et al. Efficient inhibition of ovarian cancer growth and prolonged survival by transfection with a novel pro‐apoptotic gene, hPNAS‐4, in a mouse model. In vivo and in vitro results. Oncology 2008; 75: 137–44. [DOI] [PubMed] [Google Scholar]
- 29. Sauter BV, Martinet O, Zhang WJ, Mandeli J, Woo SL. Adenovirus‐mediated gene transfer of endostatin in vivo results in high level of transgene expression and inhibition of tumor growth and metastases. Proc Natl Acad Sci U S A 2000; 97: 4802–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Peto R, Peto J. Asymptotically efficient rank invariant test procedures. J R Stat Soc Ser A 1972; 135: 185–207. [Google Scholar]
- 31. Malaguarnera L. Implications of apoptosis regulators in tumorigenesis. Cancer Metastasis Rev 2004; 23: 367–87. [DOI] [PubMed] [Google Scholar]
- 32. Ferreira CG, Epping M, Kruyt FA, Giaccone G. Apoptosis: target of cancer therapy. Clin Cancer Res 2002; 8: 2024–34. [PubMed] [Google Scholar]
- 33. Zupi G, Scarsella M, Semple SC, Mottolese M, Natali PG, Leonetti C. Antitumor efficacy of bcl‐2 and c‐myc antisense oligonucleotides in combination with cisplatin in human melanoma xenografts: relevance of the administration sequence. Clin Cancer Res 2005; 11: 1990–8. [DOI] [PubMed] [Google Scholar]
- 34. Bourhis J, Rivera F, Mesia R et al. Phase I/II study of cetuximab in combination with cisplatin or carboplatin and fluorouracil in patients with recurrent or metastatic squamous cell carcinoma of the head and neck. J Clin Oncol 2006; 24: 2866–72. [DOI] [PubMed] [Google Scholar]
- 35. Sasaki H, Sheng Y, Kotsuji F, Tsang BK. Down‐regulation of X‐linked inhibitor of apoptosis protein induces apoptosis in chemoresistant human ovarian cancer cells. Cancer Res 2000; 60: 5659–66. [PubMed] [Google Scholar]
- 36. Megyeri K, Orosz L, Kemény L. Vesicular stomatitis virus infection triggers apoptosis associated with decreased DeltaNp63alpha and increased Bax levels in the immortalized HaCaT keratinocyte cell line. Biomed Pharmacother 2007; 61: 254–60. [DOI] [PubMed] [Google Scholar]
- 37. Schirrmacher V, Haas C, Bonifer R, Ahlert T, Gerhards R, Ertel C. Human tumor cell modification by virus infection: an efficient and safe way to produce cancer vaccine with pleiotropic immune stimulatory properties when using Newcastle disease virus. Gene Ther 1999; 6: 63–73. [DOI] [PubMed] [Google Scholar]
- 38. Rosenberg SA. Progress in human tumour immunology and immunotherapy. Nature 2001; 411: 380–4. [DOI] [PubMed] [Google Scholar]
- 39. Plautz GE, Yang ZY, Wu BY, Gao X, Huang L, Nabel GJ. Immunotherapy of malignancy by in vivo gene transfer into tumors. Proc Natl Acad Sci U S A 1993; 90: 4645–9. [DOI] [PMC free article] [PubMed] [Google Scholar]