

Abstract
Tamoxifen is an antagonist of estrogen receptor, which is used widely as an estrogen receptor‐positive breast cancer drug that blocks growth signals and provokes apoptosis. However, recent studies have revealed that tamoxifen induces apoptosis even in estrogen receptor‐negative cells. In the present study, we synthesized several tamoxifen derivatives to augment the apoptosis‐inducing effect of tamoxifen and evaluated the apoptosis‐inducing pathway. The estrogen receptor‐positive human leukemia cell line HL‐60 and estrogen receptor‐negative human leukemia cell line Jurkat were treated with tamoxifen and synthesized tamoxifen derivatives, and thereafter subjected to cell viability‐detection assays. Tamoxifen derivatives, as well as the lead compound tamoxifen, decreased the cell viability despite the expression of estrogen receptor. Among all of the synthesized tamoxifen derivatives, ridaifen‐B had more potent cancer cell‐damaging activity than tamoxifen. Ridaifen‐B fragmented Jurkat cell DNA and activated caspases, suggesting that the ridaifen‐B‐induced apoptosis pathway is estrogen receptor independent. Moreover, mitochondrial involvement during ridaifen‐B‐induced apoptosis was estimated. Ridaifen‐B significantly reduced mitochondrial membrane potential, and overexpression of Bcl‐2 inhibited ridaifen‐B‐induced apoptosis. These results suggest that the induction of apoptosis by ridaifen‐B, a novel tamoxifen derivative, is dependent on mitochondrial perturbation without estrogen receptor involvement. (Cancer Sci 2008; 99: 608–614)
Abbreviations:
- ΔΨm
mitochondrial membrane potential
- Ac
acetyl
- CHO
aldehyde
- DHR
dihydrorhodamine
- EDTA,
ethylenediaminetetraacetic acid
- ER
estrogen receptor
- IC50
median inhibitory concentration
- JC‐1
5,5′,6,6′‐tetrachloro‐1,1′,3,3′‐tetraethylbenzimidazolylcarbocyanine iodide
- PBS
phosphate‐buffered saline
- pNA
p‐nitroaniline
- RID
ridaifen
- ROS
reactive oxygen species
- SDS
sodium dodecylsulfate
- SERM
selective estrogen receptor modulator
- TAM
tamoxifen
- TMPO
3,3,5,5‐tetramethyl‐1‐pyrolline‐N‐oxide.
One way of enhancing the ability of cancer cells to multiply is by utilizing growth factor receptors, as well as growth hormone receptors, to effectively receive cellular growth signals. One of the hormone receptors, ER, is expressed in most breast cancer cells. ER has two subtypes, ER‐a and ER‐b. ER‐a is a transcription factor that induces not only growth‐promoting genes but also differentiation‐related genes,( 1 , 2 ) resulting in the mediation of reproduction, metabolism, and cancer‐cell growth. Another ER subtype, ER‐b, was cloned in 1996,( 3 ) and has a ligand‐binding domain identical to that of ER‐a. However, the physiological effect of ER‐b seems to be the opposite of that of ER‐a: it inhibits proliferation.( 4 ) The non‐steroidal SERM TAM binds to ER instead of the endogenous growth hormone estrogen 17b‐estradiol, leading to inhibition of breast cancer cell proliferation and resulting in apoptosis.( 5 ) Therefore, TAM is now used widely as an ER‐positive breast cancer drug. However, how TAM induces apoptosis, which is the pharmacological effect of TAM, is obscure as several reports have demonstrated that a high dose and long time treatment with TAM is able to induce apoptosis even in ER‐negative cancer cells.( 6 , 7 ) TAM therefore seems to induce apoptosis in both ER‐positive and ‐negative cells, suggesting that ER is not necessary for TAM to provoke a cancer‐cell decrease.
Mitochondria play an important role in apoptosis. With multiple cytotoxic stimuli, including UV, X‐ray, and many chemical drugs, mitochondrial perturbation occurs by DYm reduction and cytochrome c release into the cytosol.( 8 , 9 ) Bcl‐2 family proteins are involved in the stability of the mitochondrial membrane by promoting or preventing such mitochondrial perturbation.( 10 , 11 , 12 ) When mitochondrial stability is attenuated, released cytochrome c binds to the cytosolic adaptor protein Apaf‐1, and the complex activates one of the initiator caspases, caspase‐9.( 13 ) Activated caspase‐9 in turn activates the effector caspases (caspase‐3, ‐6, and ‐7) to execute apoptosis by means of chromatin DNA fragmentation, morphological changes, and cell‐volume loss. Overall, mitochondrial perturbation tends to induce apoptosis. However, involvement of the mitochondria in TAM‐induced apoptosis is not clear. Mandlekar et al. discovered that caspase‐9 is slightly activated during TAM‐induced apoptosis, suggesting mitochondrial participation.( 14 ) In contrast, Ferlini et al. have reported that mitochondria‐localized anti‐apoptotic protein Bcl‐2 expression and functional inactivation by phosphorylation is unchanged during TAM‐induced apoptosis, assuming that TAM‐induced apoptosis is independent of mitochondria.( 6 )
As TAM acts as a SERM, it is also a partial agonist of ER‐a and is associated with an increased incidence of endometrial cancer.( 15 ) For the purpose of using a more effective SERM for reducing side effects and promoting antiproliferative effects, many TAM derivatives have since been synthesized. For example, idoxifene has greater binding ability to ER than TAM, exerting inhibition of cell proliferation.( 16 )
In the present study, we synthesized novel TAM derivatives to strengthen the apoptotic effect of TAM with the hope of creating promising drugs and determining the apoptotic pathway, especially in terms of the involvement of ER and mitochondria. We examined the effects of TAM derivatives on an ER‐negative helper T leukemia cell line, Jurkat.( 6 ) In lymphoid cell lines, promyelotic and cytotoxic T cells bear ER, whereas others, including helper T cell lines, do not.( 17 ) Using helper T cell line Jurkat cells, we revealed that TAM derivatives, as well as TAM itself, induced apoptosis independently of ER. Moreover, we discovered that the TAM derivative RID‐B had a potent apoptosis‐inducing effect compared with TAM. RID‐B induced mitochondria‐involved apoptosis, suggesting that mitochondria play a pivotal role in RID‐B‐induced apoptosis.
Materials and Methods
Cells. The human lymphoid helper T‐cell line Jurkat, transfected stably with a human bcl‐2‐expression plasmid (bcl‐2) or neomycin‐resistance plasmid (neo), was provided by Dr T. Miyashita of the National Research Institute for Child Health and Development (Tokyo, Japan). The human promyelocytic lymphoma HL‐60 and human endocervical carcinoma cell line MCF‐7 were supplied by the Cell Resource Center for Biomedical Research, Tohoku University (Sendai, Japan).
Reagents. Tamoxifen and its derivatives (Fig. 1) were synthesized chemically in our laboratory based on the methods described by Shiina et al.( 18 , 19 , 20 , 21 )
Figure 1.
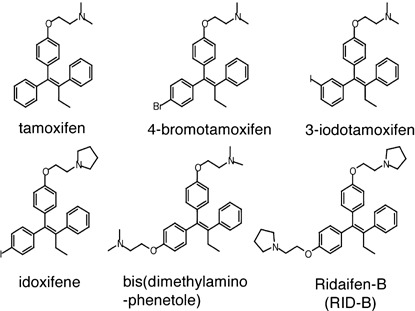
Chemical structures of tamoxifen and its derivatives.
Medium and cell cultures. Cells were cultured in RPMI‐1640 medium supplemented with 10% fetal bovine serum and 75 mg/L kanamycin sulfate, and maintained at 37°C in a humidified chamber under an atmosphere of 95% air and 5% CO2.
Western blotting. Cells (4 × 106) were collected, washed with PBS, and placed on ice for 20 min in lysis buffer (50 mM HEPES‐NaOH, 10% glycerol, 150 mM NaCl, 1% Triton X‐100, 1 mM ethylene glycol bis(βaminoethyl ether)‐N,N,N′,N′‐tetra‐acetic acid (EGTA), 1.5 mM MgCl2, 1% proteinase inhibitor cocktail [Sigma], pH 7.5). Cell lysates were centrifuged at 4°C for 20 min at 15 000g. The protein concentrations of the supernatant were determined using a bicinchoninic acid protein assay (Pierce, Rockford, IL, USA). Cell lysates were mixed with an equivalent volume of sample application buffer (4% SDS, 125 mM Tris‐HCl, 10% glycerol, 0.02 mg/mL bromophenol blue, and 10% 2‐mercaptoethanol, pH 6.8). The mixture was placed in boiling water for 3 min. Proteins (20 µg/lane) were separated by SDS‐polyacrylamide gel electrophoresis with 12% gel and then transferred to polyvinylidine difluoride membrane (Millipore, Bedford, MA, USA). After blocking, the membrane was incubated with anti‐ER‐α and anti‐ER‐β antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Thereafter, horseradish peroxidase‐conjugated antirabbit IgG was applied as a secondary antibody, and positive bands were detected using enhanced chemiluminescence (GE Healthcare, Buckinghamshire, UK).
WST‐8 assay. Cells (2 × 104) were incubated in 96‐well plates with or without drugs at 37°C for 4 h. Two hours prior to the end of incubation, 10 µL of WST‐8 solution (Dojindo Laboratories, Kumamoto, Japan) was added to each well, and the plates were incubated further for 2 h. Cell viability was determined by measuring the created formazan at 570 nm for each well using a microplate reader (Bio‐Rad, Hercules, CA, USA).
Agarose gel electrophoresis. Apoptosis was determined by DNA fragmentation, which was assessed by agarose‐gel electrophoresis. Cells (1 × 106) were rinsed once with 10 mM Tris‐HCl buffer, pH 8.7, containing 3 mM MgCl2 and 2 mM 2‐mercaptoethanol and were dissolved in 50 mM Tris‐HCl buffer, pH 7.8, containing 10 mM EDTA, 0.5% sodium lauryl sarcocinate, and 1 mg/mL proteinase K. After incubation at 50°C for 30 min, RNase A was added at a concentration of 0.5 mg/mL and the samples were further incubated at 50°C for 15 min. Lysates were mixed with an equal volume of loading buffer containing TBE buffer (89 mM Tris, pH 8.4, 2.5 mM EDTA, 89 mM boric acid), 20% glycerol, and 0.01% bromophenol blue. Samples were electrophoresed on 1.8% agarose gels in TBE containing 0.5 mg/mL ethidium bromide.
Detection of caspase activities. Activation of caspases was determined by measuring the hydrolyzing activities of the caspase‐3 substrate Ac‐DEVD‐pNA (Alexis, San Diego, CA, USA), the caspase‐8 substrate Ac‐IETD‐pNA (Alexis), or the caspase‐9 substrate Ac‐LEHD‐pNA (Alexis). Cells (2.0 × 106) were washed with PBS. The pellets were lysed in RIPA buffer (25 mM Tris‐HCl, 150 mM KCl, 5 mM EDTA, 1% Nonidet P‐40, 0.5% sodium deoxycholate, 0.1% SDS, pH 7.4), and cell extracts were obtained by centrifugation at 10 000g for 5 min at 4°C. Protein concentrations were determined using the bicinchoninic acid protein assay. Cell lysates were incubated in caspase buffer (100 mM HEPES, pH 7.4, 0.5 mM EDTA, 20% glycerol, 5 mM dithiothreitol) containing 100 µM of the substrate Ac‐DEVD‐pNA, Ac‐IETD‐pNA, or Ac‐LEHD‐pNA for 4 h at 37°C. Caspase activities were assayed by measuring the released pNA at 405 nm using a microplate reader.
Determination of apoptosis morphology. Cells (2 × 105) were fixed with Carnoy fluid (acetic acid : methanol = 1:3) for 5 min, and thereafter stained with 2% Giemsa solution for 30 min. Stained nuclei were observed with a phase‐contrast microscope.
Detection of ΔΨm. Cells (1 × 106) were incubated for 15 min with 2.5 µg/mL of the cation dye JC‐1 (Sigma). Thereafter, cells were collected, washed, and resuspended in PBS. The fluorescence of mitochondria localized in the aggregated form (red) (excitation 585 nm/emission 590 nm) and in the cell‐incorporated monomer form (green) (excitation 510 nm/emission 527 nm) was measured by spectrofluorometer. The ratio of red fluorescence/green fluorescence resembled the ΔΨm of each cell sample.
Detection of mitochondrial ROS. Cells (1 × 106) were incubated for 10 min with 5 µg/mL DHR123 (Cayman Chemical, Ann Arbor, MI, USA). Thereafter, cells were collected, washed, and resuspended in PBS. The fluorescence of rhodamine (excitation 500 nm/emission 530 nm) was measured by spectrofluorometer.
Results
Tamoxifen derivatives induced cell injury in both ER‐positive and ER‐negative cells. The effects of TAM and their derivatives on leukemia cells were determined. Western blot analysis indicated that the promyelocytic leukemia cell line HL‐60 expressed ER‐α and ‐β, whereas ER‐α and ‐β were not observed in T‐cell lymphoma Jurkat cells (Fig. 2). Indeed, the breast cancer cell line MCF‐7 expressed both types of ER. To evaluate whether the sensitivity of TAM and its derivatives to leukemia cells was ER dependent, we carried out a WST‐8 assay to determine cellular NADH levels for estimating cell viability. Similar to other ER‐positive cells (e.g. MCF‐7), the ER‐positive leukemia cell HL‐60 had markedly decreased viability with a 4‐h incubation with TAM (Fig. 3a). The synthesized TAM derivatives 4‐bromoTAM and idoxifene also decreased cell viability similarly to TAM. However, 3‐iodoTAM had less cell‐damage activity than TAM. Among all of the synthesized TAM derivatives, bis(dimethylaminophenetole) and RID‐B were found to be the most effective damaging agents to HL‐60. Next, TAM and their derivatives were used to treat the ER‐negative leukemia cell line Jurkat. Surprisingly, TAM derivatives as well as TAM induced cell damage to Jurkat identically to HL‐60 (Fig. 3b). The TAM derivative RID‐B significantly decreased cell viability even without the presence of ER. A 4‐h treatment with TAM did not damage Jurkat cells at a dose of 10 µM, whereas RID‐B impaired almost all of the cells at the same dose. The estimated IC50 doses of TAM and RID‐B were approximately 30 and 4 µM, respectively. Moreover, prolonged treatment of Jurkat cells with RID‐B led to a marked decrease in cell viability with lesser IC50 doses (approximately 0.1 µM in 48 h) compared with short‐time treatment (approximately 4 µM in 4 h), indicating that RID‐B affected the dose‐dependent viability decrease as well as time‐dependent decrease to ER‐negative Jurkat cells (Fig. 3c).
Figure 2.
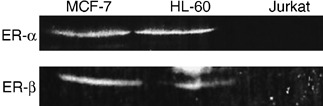
Estrogen receptor (ER) expression in leukemia cell lines. HL‐60 and Jurkat cells were lysed and subjected to western blot analysis using anti‐ER‐α and anti‐ER‐β antibodies. MCF‐7 cells were used as a positive control.
Figure 3.
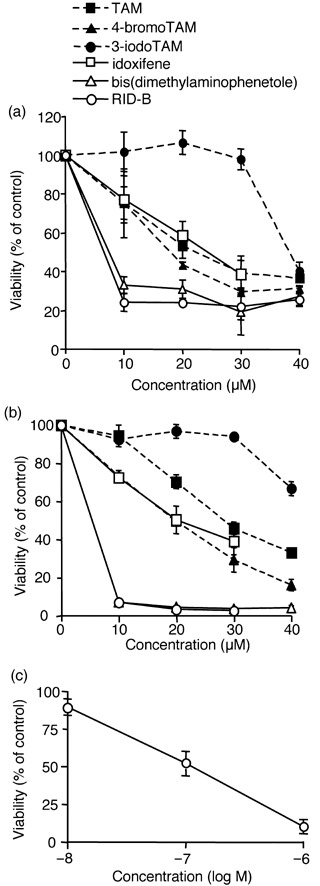
Tamoxifen (TAM) derivatives induced cell damage to estrogen receptor (ER)‐positive and ‐negative cells. (a) ER‐positive HL‐60 cells or (b) ER‐negative Jurkat cells were incubated with the indicated doses of TAM derivatives for 4 h. (c) Jurkat cells were incubated with the indicated doses of ridaifen (RID)‐B for 48 h. Cell viability was estimated by WST‐8 assay. Each bar denotes the standard deviation (n = 3).
Tamoxifen derivative RID‐B‐induced apoptosis of ER‐negative cells. As the TAM derivative RID‐B rapidly and effectively decreased the cell viability of ER‐negative Jurkat cells, we further determined whether RID‐B is able to induce apoptosis despite the absence of ER. DNA fragmentation and condensation of chromatin is a hallmark of apoptosis events. As shown in Fig. 4a, doses of RID‐B greater than 2 µM fragmented DNA in a 4‐h treatment (Fig. 4a). Similarly, 4‐h treatment with 4 µM RID‐B changed Jurkat nuclei to a ‘bubbled’ form, which resembled condensed chromatin (Fig. 4b). Moreover, RID‐B activated numerous caspases, caspase‐3, ‐8, and ‐9, time dependently (Fig. 5a). The caspase‐3‐specific inhibitor Ac‐DEVD‐CHO completely inhibited RID‐B‐induced caspase‐3 activity, and caused a significant caspase‐8 activity reduction. However, Ac‐DEVD‐CHO did not inhibit RID‐B‐induced caspase‐9 activation, assuming that caspase‐9 activation is prior to caspase‐3 activation, and caspase‐8 tended to be activated after caspase‐3 activation (Fig. 5b). Furthermore, RID‐B‐induced DNA fragmentation was inhibited by Ac‐DEVD‐CHO, suggesting that RID‐B‐induced DNA fragmentation is caspase dependent (Fig. 5c).
Figure 4.

Ridaifen (RID)‐B induced apoptosis. (a) Jurkat cells were incubated with the indicated doses of RID‐B for 4 h. The cells were lysed, and DNA was prepared. DNA fragmentation was analyzed by agarose gel electrophoresis. (b) Jurkat cells were incubated with 4 µM RID‐B for 4 h. Cells were stained with Giemsa solution. Cellular morphology was observed by microscopy. The arrow indicates a chromatin‐condensed cell.
Figure 5.
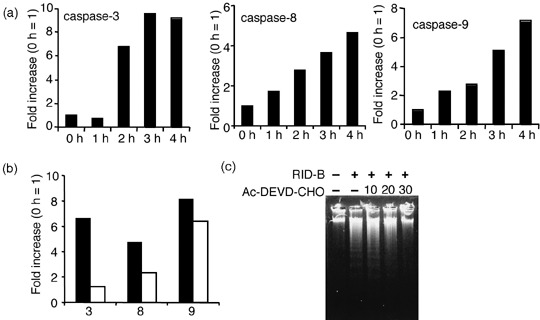
Ridaifen (RID)‐B‐induced apoptosis is caspase dependent. (a) Jurkat cells were incubated with 4 µM RID‐B for the indicated times. The cells were lysed and subjected to caspase‐3, caspase‐8, and caspase‐9 assay. The results are presented as a comparison with the 0‐h treatment. Data are representative of three independent experiments. (b) Jurkat cells were incubated with or without the indicated doses of acetyl (Ac)‐DEVD‐CHO for 1 h, and thereafter incubated with or without 4 µM RID‐B for 4 h. The cells were lysed and subjected to caspase‐3, caspase‐8, and caspase‐9 assay. The results are presented as a comparison with the 0‐h treatment. Data are representative of three independent experiments. (c) Jurkat cells were incubated with or without the indicated doses of Ac‐DEVD‐CHO for 1 h, and thereafter incubated with or without 4 µM RID‐B for 4 h. DNA fragmentation assay was carried out by agarose gel electrophoresis.
Ridaifen‐B perturbed mitochondria to induce apoptosis. As caspase‐9 activation was involved in RID‐B‐induced apoptosis (Fig. 5b), we determined the mitochondrial involvement to clarify the mechanism of RID‐B‐induced apoptosis. The mitochondria‐localized antiapoptotic protein Bcl‐2 was overexpressed in Jurkat. As shown in Fig. 6a, overexpression of Bcl‐2 clearly inhibited the decrease in RID‐B‐induced cell viability. At doses of 4 µM, the viability of Jurkat cells dropped to 50% as Bcl‐2‐overexpressed cells were not damaged at all. Moreover, overexpression of Bcl‐2 blocked RID‐B‐induced DNA fragmentation (Fig. 6b) as well as caspase activation (Fig. 6c), suggesting that mitochondrial perturbation is crucial to RID‐B‐induced apoptosis. During apoptosis, mitochondria perturbation results in ΔΨm loss and release of cytochrome c into the cytosol. The cation fluorescence dye JC‐1 aggregates to mitochondria depending on the ΔΨm. In highly energized mitochondria, JC‐1 aggregates to the mitochondria and shows red fluorescence. However, depolarized apoptotic mitochondria cannot accumulate JC‐1 and exist as a single monomer form with green fluorescence that is only incorporated in cells. We estimated the ΔΨm during Jurkat treatment with RID‐B. Cells were treated with RID‐B and thereafter stained with JC‐1. Both the aggregated form (red fluorescence) and the monomer form (green fluorescence) were measured, and the ratio of red fluorescence/green fluorescence was calculated to determine ΔΨm at a constant cell density. As shown in Fig. 6d, ΔΨm decreased significantly over time with RID‐B treatment, suggesting that RID‐B perturbed mitochondria. However, this ΔΨm decrease was inhibited by Bcl‐2 overexpression (Fig. 6e). Furthermore, caspase‐3 inhibition by Ac‐DEVD‐CHO did not inhibit RID‐B‐induced ΔΨm reduction (Fig. 6f), suggesting that RID‐B‐induced caspase‐3 activation is a postmitochondrial event.
Figure 6.
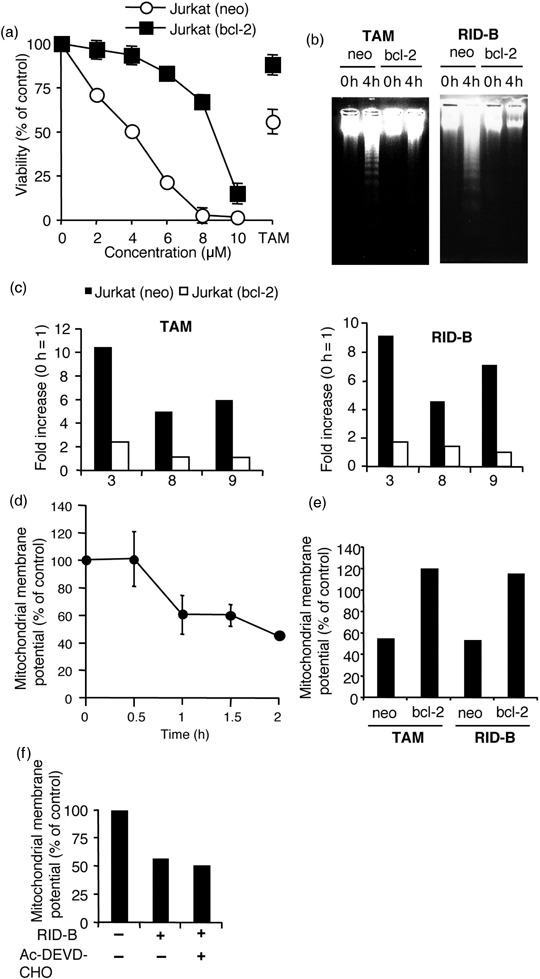
Mitochondria are involved in ridaifen (RID)‐B‐induced apoptosis. (a) Jurkat (neo) and Jurkat (bcl‐2) cells were incubated with the indicated doses of RID‐B or IC50 value (30 µM) of tamoxifen (TAM) (control) for 4 h. Cell viability was observed with a WST‐8 assay. The results are presented as a comparison with the non‐additive control. Each bar denotes the standard deviation (n = 3). (b) Jurkat (neo) and Jurkat (bcl‐2) cells were incubated with or without 4 µM RID‐B or IC50 value of TAM (for control) for 4 h. DNA fragmentation was carried out by agarose gel electrophoresis. (c) Jurkat (neo) and Jurkat (bcl‐2) cells were incubated with 4 µM RID‐B or IC50 value of TAM (for control) for 4 h. Caspase‐3, ‐8, and ‐9 assays were carried out. The results are presented as a comparison with the non‐additive control for each of the cells and assayed caspase numbers only are indicated in the graph. Data are representative of three independent experiments. (d) The mitochondrial membrane potential was observed by 5,5′,6,6′‐tetrachloro‐1,1′,3,3′‐tetraethylbenzimidazolylcarbocyanine iodide (JC‐1). Jurkat (neo) cells were incubated with 4 µM RID‐B for the indicated times and stained with JC‐1. The results are presented as a comparison with the non‐additive control. Each bar denotes the standard deviation (n = 3). (e) Jurkat (neo) and Jurkat (bcl‐2) cells were incubated with 4 µM RID‐B or IC50 value of TAM (for control) for 2.5 h and stained with JC‐1. The results are presented as a comparison with the non‐additive control. Data are representative of three independent experiments. (f) Jurkat cells were incubated with or without 30 µM acteyl (Ac)‐DEVD‐CHO for 1 h, and thereafter incubated with or without 4 µM RID‐B for 4 h. Mitochondrial membrane potential was observed by JC‐1. The results are presented as a comparison with the non‐additive control. Data are representative of three independent experiments.
Mitochondria‐involved RID‐B‐induced apoptosis is not related to ROS generation. Further experiments were carried out to observe how RID‐B induces mitochondrial perturbation. The production of ROS is correlated with many types of apoptosis, especially in mitochondria‐involved apoptosis, as mitochondria are a major site of ROS production.( 22 ) In the present study, DHR123 was used to determine the generation of mitochondrial ROS. DHR123 is a membrane‐permeable dye that is oxidized and converted to rhodamine123, and subsequently localized to the mitochondria.( 23 ) As shown in Fig. 7a, cells treated with RID‐B accumulated significant amounts of rhodamine123, indicating that RID‐B induced mitochondrial ROS production. Next, RID‐B‐induced ROS production was inhibited by prior treatment with the spin‐trap reagent TMPO.( 23 ) However, as TMPO completely blocked RID‐B‐induced ROS production (Fig. 6a), TMPO did not inhibit the decrease in RID‐B‐induced cell viability (Fig. 7b), DNA fragmentation (Fig. 7c), or caspase‐3 activation (Fig. 7d). Moreover, Bcl‐2 overexpression as well as caspase‐3 inactivation still accumulated ROS (Fig. 7e,f), suggesting that RID‐B has the ability to produce mitochondrial ROS, but this ROS production is not related to RID‐B‐induced apoptosis.
Figure 7.
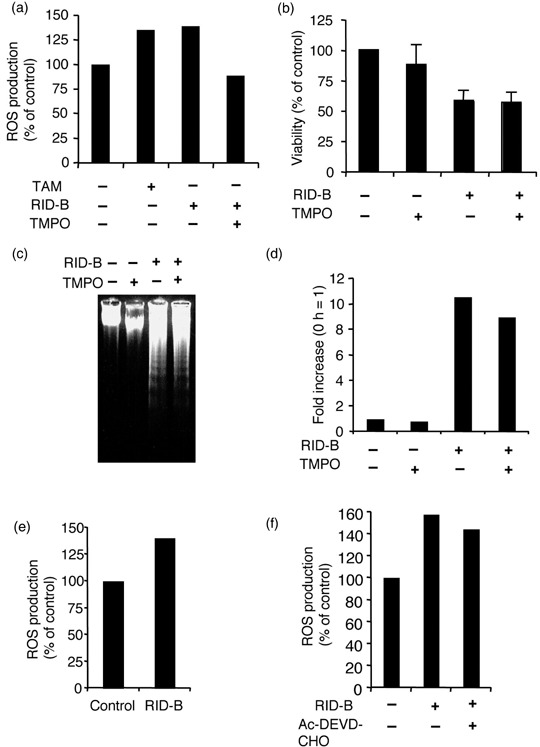
Ridaifen (RID)‐B‐induced apoptosis is not related to reactive oxygen species (ROS). (a) Jurkat cells were incubated with or without 10 mM 3,3,5,5‐tetramethyl‐1‐pyrolline‐N‐oxide (TMPO) for 2 h, and subsequently incubated with or without 4 µM RID‐B or IC50 value of tamoxifen (TAM) (control) for 4 h. Ten minutes prior to the end of incubation, 5 µg/mL DHR123 was added. Rhodamine fluorescence was measured by spectrofluorometer, and the results are presented as a comparison to the non‐additive control. Data are representative of three independent experiments. (b) Jurkat cells were incubated with or without 10 mM TMPO for 2 h, and thereafter incubated with or without 4 µM RID‐B for 4 h. Cell viability was estimated by WST‐8 assay. Each bar denotes the standard deviation (n = 3). (c) Jurkat cells were incubated with or without 10 mM TMPO for 2 h, and thereafter incubated with or without 4 µM RID‐B for 4 h. The cells were lysed, and DNA was prepared. DNA fragmentation was analyzed by agarose gel electrophoresis. (d) Jurkat cells were incubated with or without 10 mM TMPO for 2 h, and thereafter incubated with or without 4 µM RID‐B for 4 h. The caspase‐3 assay was then carried out. The results are presented as a comparison with the non‐additive control for each of the cells. Data are representative of three independent experiments. (e) Jurkat (bcl‐2) cells were incubated with or without 4 µM RID‐B for 4 h. Ten minutes prior to the end of incubation, 5 µg/mL DHR123 was added. Rhodamine fluorescence was measured by spectrofluorometer, and the results are presented as a comparison with the non‐additive control. Data are representative of three independent experiments. (f) Jurkat cells were incubated with or without 30 µM Ac‐DEVD‐CHO for 1 h, and thereafter incubated with or without 4 µM RID‐B for 4 h. Ten minutes prior to the end of incubation, 5 µg/mL DHR123 was added. Rhodamine fluorescence was measured by spectrofluorometer, and the results are presented as a comparison to the non‐additive control. Data are representative of three independent experiments.
Discussion
In the present study, we synthesized several TAM derivatives in order to strengthen the pharmacological effects of TAM, and discovered that RID‐B is a powerful TAM derivative exerting apoptotic induction. RID‐B was found to induce apoptosis in an ER‐negative leukemia cell line, suggesting that the RID‐B apoptosis pathway does not require ER. Moreover, mitochondria are indispensable for the RID‐B‐induced apoptosis pathway, such as downstream caspase activation.
In the present study, we focused on two human leukemia cell lines, ER‐negative Jurkat and ER‐positive HL‐60, to determine whether ER is dependent on RID‐B‐induced apoptosis and discovered that RID‐B can induce apoptosis in spite of ER expression in leukemia cell lines. In our recent study, we treated 39 human cancer cell lines with RID‐B to determine organ‐specific reactions and the cell specificity of RID‐B, including the breast cancer cell lines MCF‐7 (ER positive) and MDA‐MB‐231 (ER negative), the ovary cell lines SK‐OV‐3 (ER positive) and OVCAR‐3 (ER negative), and the prostate cell lines PC‐3 (ER positive) and DU‐145 (ER negative).( 21 ) The cell growth rates of all of the cell lines used in the study were inhibited by half at similar doses of RID‐B (approximately 1 µM for a 48‐h treatment) regardless of the presence of ER. Taken together, ER seems not to be correlated with RID‐B‐induced apoptosis, which seems to be different from the mechanism of TAM‐induced apoptosis.
A family of aspartate‐specific cysteine proteases, caspases, is pivotally involved in the initiation and execution of apoptotic cell death. Usually, caspases exist as proforms in the cytosol, and these proforms are activated by cleavage induced by two major upstream pathways. One of the initiation‐phase caspases, caspase‐8, is cleaved by death ligand‐mediated apoptotic molecules,( 24 ) whereas another initiation‐phase caspase, caspase‐9, is cleaved by mitochondria‐mediated apoptotic molecules.( 13 ) Activated initiation caspases cleave the execution‐phase caspases caspase‐3 and ‐7 to undergo apoptosis. In the present study, the executor caspase caspase‐3, as well as the initiator caspases caspase‐8 and ‐9 were all activated by RID‐B treatment. As the caspase‐3 inhibitor Ac‐DEVD‐CHO clearly blocked RID‐B‐induced DNA fragmentation, caspase‐3 activation would be a key regulator of RID‐B‐induced apoptosis (Fig. 5c). Moreover, RID‐B activated two different initiator caspases, caspase‐8 and ‐9, thereby posing the question of what is the upstream pathway for cleaving initiator caspases; in other words, neither death ligands nor mitochondria are important to RID‐B‐induced apoptosis. We used mitochondria‐localized antiapoptotic protein Bcl‐2‐overexpressed cells to inhibit the postmitochondrial apoptotic pathway to determine whether RID‐B apoptosis is dependent on mitochondria. Bcl‐2 overexpression completely inhibited the mitochondrial downstream caspases caspase‐9 and ‐3. Surprisingly, Bcl‐2 overexpression also blocked caspase‐8 activation. These results suggest that the activation of caspase‐8 by RID‐B is a postmitochondrial apoptotic event that is not under death ligand regulation. Similar results were revealed by Slee et al., in which activated caspase‐3 cleaves caspase‐2 and ‐6, and in turn activates the death ligand‐mediated initiator caspases caspase‐8 and ‐10.( 25 ) In agreement with Slee et al., caspase‐3 inhibition attenuated RID‐B‐induced caspase‐8 activation. Taken together, these results suggest that RID‐B‐induced apoptosis depends on caspase activation and that this activation is a postmitochondrial event.
Because mitochondria are essential for the induction of apoptosis by RID‐B, how RID‐B perturbs mitochondria to exert an apoptotic signal is an important issue in determining the mechanism of RID‐B‐induced apoptosis. One possibility is that RID‐B promotes the production of mitochondrial ROS, followed by mitochondrial perturbation, as the lead compound of RID‐B, TAM, is reported to be induced during oxidative stress treatment.( 14 ) The mitochondrial respiratory chain is the major site for the generation of superoxide, from which the mitochondrial superoxide dismutase produces hydrogen peroxide. Similar to TAM, RID‐B induced mitochondrial ROS production (Fig. 6a). However, the radical spin scavenger TMPO blocked RID‐B‐produced ROS, but not RID‐B‐induced apoptosis, assuming that RID‐B‐induced ROS production is not a fundamental phenomenon to perturb mitochondria and subsequent apoptosis. Another mitochondrial upstream candidate was reported by Kallio et al. They reported that TAM‐induced mitochondria‐involved apoptosis is ER dependent, assuming that the TAM derivative RID‐B might have a similar effect.( 26 ) However, we discovered RID‐B‐induced apoptosis in ER‐negative Jurkat cells and that this phenomenon was blocked by overexpression of the mitochondrial antiapoptotic protein Bcl‐2. These results suggest that RID‐B‐induced mitochondria‐involved apoptotic pathway is independent of ER. We are currently seeking the binding protein of RID‐B that exerts mitochondrial perturbation and apoptosis.
Overall, these results suggest that a novel TAM derivative, RID‐B, initially perturbs mitochondria to produce ROS and decrease ΔΨm. However, mitochondrial ROS production is not related to RID‐B‐induced apoptosis. RID‐B‐induced mitochondrial perturbation induces caspase activation, followed by DNA fragmentation to execute apoptosis. RID‐B may strongly and rapidly induce apoptosis independently of the presence of ER, and RID‐B may therefore be a promising drug for the treatment of various cancers.
Acknowledgments
We are thankful to Mr Yuki Sano, Mr Hironobu Horibe, Mr Akihiko Kikuchi, Mr Taichi Sato, and Mr Youhei Noguchi for technical assistance.
References
- 1. Katzenellenbogen BS, Katzenellenbogen JA. Estrogen receptor transcription and transactivation. Estrogen receptor α and estrogen receptor β: regulation by selective estrogen receptor modulators and importance in breast cancer. Breast Cancer Res 2000; 2: 335–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shiau AK, Barstad D, Loria PM et al . The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 1998; 95: 927–37. [DOI] [PubMed] [Google Scholar]
- 3. Mosselman S, Polman J, Dijkema R. ER β: identification and characterization of a novel human estrogen receptor. FEBS Lett 1996; 392: 49–53. [DOI] [PubMed] [Google Scholar]
- 4. Imamov O, Shim GJ, Warner M, Gustafsson JA. Estrogen receptor β in health and disease. Biol Reprod 2005; 73: 866–71. [DOI] [PubMed] [Google Scholar]
- 5. Mandlekar S, Kong AN. Mechanisms of tamoxifen‐induced apoptosis. Apoptosis 2001; 6: 469–77. [DOI] [PubMed] [Google Scholar]
- 6. Ferlini C, Scambia G, Marone M et al . Tamoxifen induces oxidative stress and apoptosis in oestrogen receptor‐negative human cancer cell lines. Br J Cancer 1999; 79: 257–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kang Y, Cortina R, Perry RR. Role of c‐myc in tamoxifen‐induced apoptosis estrogen‐independent breast cancer cells. J Natl Cancer Inst 1996; 88: 279–84. [DOI] [PubMed] [Google Scholar]
- 8. Zamzami N, Marzo I, Susin SA et al . The thiol crosslinking agent diamide overcomes the apoptosis‐inhibitory effect of Bcl‐2 by enforcing mitochondrial permeability transition. Oncogene 1998; 16: 1055–63. [DOI] [PubMed] [Google Scholar]
- 9. Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell‐free extracts: requirement for dATP and cytochrome c . Cell 1996; 86: 147–57. [DOI] [PubMed] [Google Scholar]
- 10. Narita M, Shimizu S, Ito T et al . Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proc Natl Acad Sci USA 1998; 95: 14 681–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zha J, Harada H, Osipov K, Jockel J, Waksman G, Korsmeyer SJ. BH3 domain of BAD is required for heterodimerization with BCL‐XL and pro‐apoptotic activity. J Biol Chem 1997; 272: 24 101–4. [DOI] [PubMed] [Google Scholar]
- 12. Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 1998; 94: 481–90. [DOI] [PubMed] [Google Scholar]
- 13. Li P, Nijhawan D, Budihardjo I et al . Cytochrome c and dATP‐dependent formation of Apaf‐1/caspase‐9 complex initiates an apoptotic protease cascade. Cell 1997; 91: 479–89. [DOI] [PubMed] [Google Scholar]
- 14. Mandlekar S, Yu R, Tan TH, Kong AN. Activation of caspase‐3 and c‐Jun NH2‐terminal kinase‐1 signaling pathways in tamoxifen‐induced apoptosis of homan breast cancer cells. Cancer Res. 2000; 60: 5995–6000. [PubMed] [Google Scholar]
- 15. Dhingra K. Antiestrogens – tamoxifen, SERMs and beyond. Invest New Drugs 1999; 17: 285–311. [DOI] [PubMed] [Google Scholar]
- 16. Nuttall ME, Bradbeer JN, Stroup GB et al . Idoxifene: a novel selective estrogen receptor modulator prevents bone loss and lowers cholesterol levels in ovariectomized rats and decreases uterine weight in intact rats. Endocrinology 1998; 139: 5224–34. [DOI] [PubMed] [Google Scholar]
- 17. Danel L, Menouni M, Cohen JH et al . Distribution of androgen and estrogen receptors among lymphoid and haemopoietic cell lines. Leuk Res 1985; 9: 1373–8. [DOI] [PubMed] [Google Scholar]
- 18. Shiina I, Suzuki M, Yokoyama K. Short‐step synthesis of tamoxifen and its derivatives via the three‐component coupling reaction and migration of the double bond. Tetrahedron Lett 2004; 45: 965–7. [Google Scholar]
- 19. Shiina I, Sano Y, Nakata K et al . Synthesis of the new pseudo‐symmetrical tamoxifen derivatives and their anti‐tumor activity. Bioorg Med Chem Lett 2007; 17: 2421–4. [DOI] [PubMed] [Google Scholar]
- 20. Shiina I, Sano Y, Nakata K et al . An expeditious synthesis of tamoxifen, a representative SERM (selective estrogen receptor modulator), via the three‐component coupling reaction among aromatic aldehyde, cinnamyltrimethylsilane, and β‐chlorophenetole. Bioorg Med Chem 2007; 15: 7599–617. [DOI] [PubMed] [Google Scholar]
- 21. Shiina I, Sano Y, Nakata K et al . Synthesis and pharmacological evaluation of the novel pseudo‐symmetrical tamoxifen derivatives as anti‐tumor agents. Biochem Pharm 2007; Epub 22 Nov 2007: doi: 10.1016/j.bcp.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 22. Cai J, Jones DP. Superoxide in apoptosis. Mitochondrial generation triggered by cytochrome c loss. J Biol Chem 1998; 273: 11 401–4. [DOI] [PubMed] [Google Scholar]
- 23. Cheng J, Park TS, Chio LC, Fischl AS, Ye XS. Induction of apoptosis by sphingoid long‐chain bases in Aspergillus nidulans . Mol Cell Biol 2003; 23: 163–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Muzio M, Chinnaiyan AM, Kischkel FC et al . FLICE, a novel FADD‐homologous ICE/CED‐3‐like protease, is recruited to the CD95 (Fas/APO‐1) death‐inducing signaling complex. Cell 1996; 85: 817–27. [DOI] [PubMed] [Google Scholar]
- 25. Slee EA, Harte MT, Kluck RM et al . Ordering the cytochrome c‐initiated caspase cascade: hierarchical activation of caspases‐2‐3‐6‐7‐8, and ‐10 in a caspase‐9‐dependent manner. J Cell Biol 1999; 144: 281–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kallio A, Zheng A, Dahllund J, Heiskanen KM, Harkonen P. Role of mitochondria in tamoxifen‐induced rapid death of MCF‐7 breast cancer cells. Apoptosis 2005; 10: 1395–410. [DOI] [PubMed] [Google Scholar]