

Abstract
To clarify the clinical presentation and outcome of nodal marginal zone B‐cell lymphoma (NMZBL), 65 Japanese patients with this disease were studied and compared with the published literature from western countries. The clinical findings of our 65 cases were similar to those of their cases in some aspects: (1) 58% of the patients were >60 years old (median age, 64 years); (2) there was a slight female predominance; (3) 90% of the patients exhibited asymptomatic lymphadenopathy in the head and neck area; (4) only a minority of patients had B symptoms (6%) and poor performance status (8%); and (5) only 5% of patients were positive for M‐protein. However, the 65 patients in this series exhibited relatively longer 5‐year overall survival (85%) and failure‐free survival (60%) than the NMZBL series published in western literature, suggesting that NMZBL should be classified as indolent lymphoma. Moreover, based on the histological findings, we further classified four histological subtypes as follows: (1) splenic type (n = 7); (2) floral type (n = 9); (3) mucosa‐associated lymphoid tissue (MALT) type (n = 29); and (4) diffuse large B‐cell lymphoma (DLBCL) + MALT type (n = 20). DLBCL + MALT type exhibited significantly poorer 5‐year overall survival than the splenic variant. The recognition of DLBCL + MALT type appears important. No API2–MALT1 fusion transcript was detected in any of the 14 cases examined. (Cancer Sci 2007; 98: 44–49)
Nodal marginal zone B‐cell lymphoma (NMZBL) is a rare and controversial entity, accounting for only 1.0% of all lymphoid neoplasms among Japanese people.( 1 , 2 ) It is a primary neoplasm that morphologically resembles extranodal marginal zone B‐cell lymphoma of mucosa‐associated lymphoid tissue (MALT) type but without evidence of extranodal or splenic disease.( 2 ) It is closely related to forms of B‐cell neoplasm thought to be derived from postgerminal center memory B‐cells. Most of the tumor cells closely resemble centrocytes or show features of monocytoid B‐cells, with abundant pale clear cytoplasm and well‐defined cell borders.( 1 , 3 ) However, NMZBL appears to be a heterogeneous tumor, showing marked cytological and histological diversity.( 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 ) Campo et al. described two variants of NMZBL, splenic type and MALT type, which are identified based on phenotypic and morphological findings reminiscent of splenic marginal zone lymphoma and MALT lymphoma, respectively.( 4 ) Moreover, in 2005, Karube et al. reported a new variant of NMZBL exhibiting ‘floral’ lymphoid follicles.( 14 )
In extranodal sites, transformed centroblast‐ or immunoblast‐like cells may be present in variable numbers in MALT lymphoma.( 3 ) According to the World Health Organization classification, when solid or sheet‐like proliferation of transformed cells are present, the tumor should be diagnosed as diffuse large B‐cell lymphoma (DLBCL) with the presence of accompanying MALT lymphoma (DLBCL + MALT). A variable number of transformed cells are also present in NMZBL.( 1 , 2 , 15 ) However, the number of transformed cells can present difficulties in grading MALT‐type NMZBL.( 4 , 5 , 6 , 15 ) Among the various forms of MALT‐type NMZBL, Berger et al. proposed the category large cell‐rich variant for cases with more than 50% of transformed cells or sheets of transformed cells.( 6 ) These cases were considered to be DLBCL + MALT type, based on the World Health Organization classification.( 1 , 3 )
The clinical characteristics and outcome of patients with MALT lymphoma have been very well described.( 3 , 4 , 5 , 15 , 16 ) However, little is known about the clinical findings of NMZBL.( 5 , 6 , 8 , 9 , 11 , 16 , 17 ) Thus, we here present the clinical findings of 65 Japanese patients with NMZBL. Furthermore, we classified these 65 cases into four different subtypes of patients according to the histological features: (1) MALT type; (2) splenic type; (3) floral type; and (4) DLBCL + MALT type. We then compared survival among the four different subtypes.( 4 , 6 , 14 )
Materials and Methods
A total of 65 cases of NMZBL were collected, 18 from a series by one of the authors (M. K.) and 47 from Aichi Cancer Center Hospital. All cases were treated between 1983 and 2003. Ten cases have been reported previously.( 10 , 13 ) Cases of small lymphocytic/chronic lymphocytic leukemia, follicular lymphoma and mantle cell lymphoma with marginal zone differentiation were excluded. None of the 65 cases had marginal zone B‐cell lymphoma arising from MALT organ or spleen.
The tissue specimens were fixed in formalin solution, processed routinely and embedded in paraffin. For light microscopic examination, the sections were stained with hematoxylin–eosin (HE).
A basic immunohistochemical panel including B‐cell markers (CD20, 79a), T‐cell markers (CD3, CD45RO) and bcl‐2 (Dako, Glostrup, Denmark) was used in all cases. When additional slides or paraffin blocks were available, immunohistochemical analysis was expanded to include antibodies against human immunoglobulin light chain (κ and λ Novocastra, Newcastle, UK), IgD (Dako or Novocastra), IgM (Dako or Novocastra), 4C7 (CD5; Novocastra), 56C6 (CD10; Novocastra), 1B12 (CD23; Novocastra) and DFT‐1 (CD43; Novocastra). Immunohistochemical studies were carried out using the antigen retrieval method with avidin–biotin–peroxidase or with Ventana automated (BenchMark, Tuscon, AZ, USA) stainer according to the manufacturer's instructions. If necessary, genomic DNA was extracted from formalin‐fixed tissues after dewaxing of paraffin sections, and analyzed for immunoglobulin heavy chain rearrangement by polymerase chain reaction as described previously.( 18 )
When the lymph node lesions were histologically suspected of being NMZL, cases in which atypical lymphoid cells lacked bcl‐2 or CD43 expression( 19 , 20 , 21 ) or those in which there was an absence of monoclonal nature by immunostaining, flow cytometry or Ig heavy chain rearrangement by polymerase chain reaction, were excluded from this study. In situ hybridization with Epstein–Barr virus (EBV)‐encoded small RNA oligonucleotides was carried out to test for the presence of EBV small RNA in formalin‐fixed paraffin‐embedded sections also using a Ventana automated (BenchMark) stainer. In 14 cases, the API2–MALT1 fusion transcript was examined using the formalin‐fixed and paraffin‐embedded tissue according to the method we described recently.( 22 )
Karyotype was examined according to the standard procedure. In brief, the prechemotherapy tissue was minced finely into cell suspension immediately after biopsy, and was cultured with non‐synchronized and non‐stimulating medium (RPMI‐1640 containing 10% fetal calf serum) for 2 days in a humidified atmosphere with 5% CO2 air. After exposure to colcemid, the cells were harvested and G‐banded using the Wright technique.
Actuarial overall survival (OS) curves and failure‐free survival distributions were calculated using the Kaplan and Meier method,( 23 ) and differences were examined using the log‐rank test to detect significant prognostic factors.( 24 ) OS was defined as the time from diagnosis to death from any cause or from last follow‐up. Failure‐free survival (FFS) was the time from diagnosis to the occurrence of progression, relapse after complete remission or death from any cause. FFS had a value of 0 for patients who did not achieve complete remission.
Results
The neoplastic nature of the cases was demonstrated by Bcl‐2 or CD43 positivity (n = 61), or by light chain restriction of marginal zone B lymphocytes (n = 4) by immunostaining,( 19 , 20 , 21 ) light chain restriction by flow cytometric studies or immunoglobulin heavy chain rearrangement by polymerase chain reaction analysis.( 18 )
Clinical presentation. The main clinical findings are shown in Table 1. Among these 65 cases, the male‐to‐female ratio was 1:1.3 and the median patient age was 64 years. Most (90%) patients presented with asymptomatic peripheral lymphadenopathy, and 56 cases (92%) exhibited cervical lymphadenopathy. Extranodal involvement was recorded in six cases (9%). The extranodal sites consisted of tonsilla (n = 4), pharynx (n = 6) and psoas muscle (n = 1). However, none of these represented more than one extranodal site involvement. Fifty‐one cases (77%) demonstrated localized disease, whereas only 14 (23%) had advanced disease. Among the 51 patients with localized disease, 39 had lesions in the head and neck area. Only five patients (8%) had B symptoms, four (6%) showed a poor performance status and eight (12%) had a high lactate dehydrogenase (LDH) level. M‐protein was recorded in four cases. However, in three cases, M‐protein disappeared following treatment. Positivity of autoantibodies was detected in four cases (6%). Four patients (6%) were associated with autoimmune diseases (chronic thyroiditis, 2; rheumatoid arthritis, 1; Sjögren's syndrome, 1). In eight (12%) cases, another malignant tumor (carcinoma, 7; acute myeloid leukemia, 1) was found in addition to lymphoma. According to the International Prognostic Index (IPI),( 25 ) 46 (70%) of our patients had low risk, 17 (26%) had low‐intermediate risk, one (2%) had high‐intermediate risk and one (2%) had high risk.
Table 1.
Summary of the clinical findings of 65 cases
| Variable | Total cases | Splenic | Floral variant | MALT | Large cell‐rich |
|---|---|---|---|---|---|
| Number of patients | 65 | 7 | 9 | 29 | 20 |
| Age distribution (years) | 29–83 | 29–75 | 29–66 | 28–83 | 37–77 |
| Mean:median age (years) | 60:64 | 61:64 | 48:50 | 63:67 | 61:64 |
| Male‐to‐female ratio | 29:36 | 2:5 | 2:7 | 14:15 | 11:9 |
| Age >60 years | 38 | 5 | 2 | 19 | 12 |
| Cervical lymphadenopathy | 56 | 6 | 8 | 24 | 18 |
| Performance status = 2 | 4 | 0 | 0 | 2 | 2 |
| B symptoms | 5 | 0 | 1 | 2 | 2 |
| Clinical stage (I/II vs III/IV) | 51:14 | 7:0 | 5:4 | 26:3 | 13:7 |
| Extranodal involvement | 6 | 0 | 0 | 4 | 2 |
| Extranodal site = 2 | 0 | – | – | 0 | 0 |
| LDH > normal values | 8 | 0 | 1 | 4 | 3 |
| M‐protein | 4 | 1 | 0 | 2 | 1 |
| Associated with carcinoma | 7 | 1 | 1 | 3 | 2 |
| Associated with myeloid malignancy | 1 | 0 | 0 | 1 | 0 |
| Associated with autoimmune disease | 4 | 0 | 0 | 1 | 3 |
| Positive autoantibodies | 4 | 0 | 1 | 1 | 2 |
| Therapy | |||||
| Surgery alone | 17 | 1 | 5 | 7 | 4 |
| Radiation | 12 | 1 | 0 | 7 | 4 |
| Single agent | 2 | 0 | 0 | 2 | 0 |
| Single agent + radiation | 2 | 1 | 0 | 0 | 1 |
| CHOP‐like regimen | 18 | 2 | 3 | 6 | 7 |
| CHOP‐like regimen + radiation | 11 | 2 | 1 | 5 | 3 |
| High‐dose therapy | 3 | 0 | 0 | 2 | 1 |
| International prognostic factor | |||||
| Low | 46 | 7 | 7 | 23 | 9 |
| Low‐intermediate | 17 | 0 | 2 | 5 | 10 |
| High‐intermediate | 1 | 0 | 0 | 0 | 1 |
| High | 1 | 0 | 0 | 1 | 0 |
| Follow‐up period (months) | 2–256 | 6–216 | 3–132 | 2–206 | 7–256 |
| Mean:median follow‐up (months) | 35:51 | 47:18 | 37:18 | 54:56 | 52:39 |
CHOP, cyclophosphamide, doxarubicin, vincristine, and prednisolone; LDH, lactate dehydrogenase.
Therapy and follow up. Twenty‐one patients were treated with combination chemotherapy regimens, for the most part consisting of CHOP (cyclophosphamide, doxarubicin, vincristine, and prednisolone), 12 with radiotherapy alone, 11 with radiation and chemotherapy, two with radiation and single agent and two with single agent. The other 17 patients with local lymphadenopathy underwent total excision of the enlarged lymph node and were without chemotherapy.
Follow‐up information was available in all 65 cases and duration ranged from 2 to 256 months (mean, 35 months; median, 51 months). The OS rate of the 65 cases was 85% at 5 years and 69% at 10 years (Fig. 1). Complete remission was achieved in 66% of cases. However, as shown in Fig. 2, the FFS rate was 60% at 5 years, and 23% at 10 years.
Figure 1.
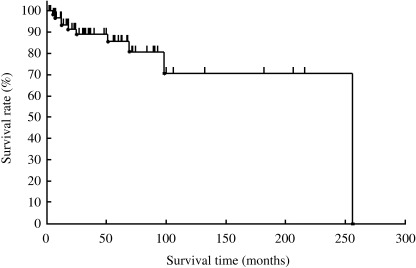
Overall survival of 65 patients with nodal marginal zone B‐cell lymphoma.
Figure 2.
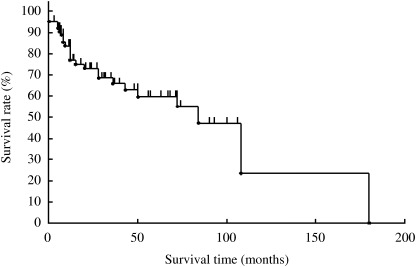
Failure‐free survival of 65 patients with marginal zone B‐cell lymphoma.
Clinical parameters associated with a good outcome. Patient characteristics that possibly influenced survival included age, sex, the presence of B symptoms, performance status (0–1 vs 2), serum LDH level above the normal level, stage (I/II vs III/IV), IPI (0–1 vs 2)( 25 ) and initial therapy (resection alone vs chemotherapy+ radiotherapy). Age less than 60 years was associated with a good OS. However, none of the studied clinical parameters was associated with a good FFS.
Cytogenetic abnormalities. Among the 65 cases in our study, 23 underwent cytogenetic analysis and 13 showed a normal karyotype. Ten patients had one or several cytogenetic abnormalities. In one case, a single cytogenetic abnormality existed, and in nine cases, multiple abnormalities were present. An abnormality of chromosome 1 was present in four cases, chromosome 3 in three cases (trisomy 3 in two cases), chromosome 7 in two cases, chromosome 8 in two cases, chromosome 10 in two cases and chromosome 18 in two cases. No API2–MALT1 fusion transcript was detected in any of the 14 cases examined.
Histological findings. Patients were subdivided into four subtypes based on the morphological features, as follows: (1) splenic type (n = 7); (2) floral type (n = 9); (3) MALT type (n = 29); and (4) DLBCL + MALT type (n = 20). The histological and immunohistochemical findings of each variant have been well described.( 4 , 5 , 6 , 8 , 9 , 10 , 11 , 12 , 13 , 14 )
In brief, the splenic type is characterized by a follicular and diffuse growth pattern with a marginal zone component in the low‐power field. In medium‐ and high‐power fields, the lymphoid follicles were occupied by medium‐sized or large neoplastic cells with various numbers of residual follicular center cells and mantle zone lymphocytes (Fig. 3a). Numerous regressive germinal centers representing hyaline‐vascular Castleman's disease were observed in one case (Fig. 3b).( 8 )
Figure 3.
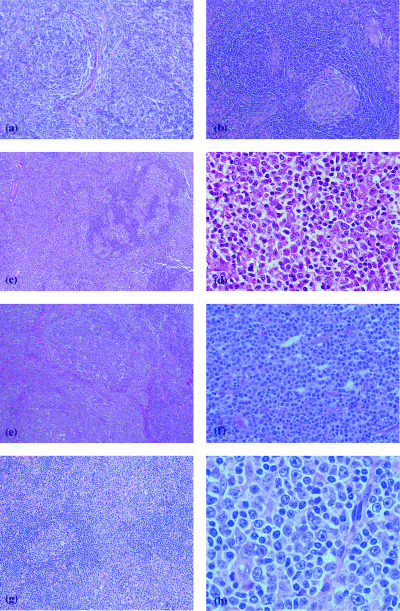
Marginal zone B‐cell lymphoma of the splenic type. (a) A nodular expansion of tumor cells surrounded a small residual germinal center. Note the follicular colonization. (b) A hyaline vascular lymphoid follicle (HE ×50) ‘floral’ type of marginal zone B‐cell lymphoma. (c) In a low‐power field, the lymph node showed many enlarged follicles surrounded by clear zones (HE ×10). (d) High‐power field of the clear zone. Medium‐sized cells with abundant pale cytoplasm and indented nuclei were observed. Scattered large transformed cells, plasma cells, histiocytes and eosinophiles (HE ×250). Marginal zone B‐cell lymphoma of mucosa‐associated lymphoid tissue type. (e) Parafollicular infiltration of the tumor cells. Note the ill‐defined mantle zone (HE ×10). (f) In a high‐power field, the interfollicular area contained numerous centrocyte‐like cells with plasma cell differentiation (HE ×100). Marginal zone B‐cell lymphoma of large cell rich type. (g) Parafollicular infiltration of the tumor cells. The mantle zone was well delineated (HE ×50). (h) In a high‐power field, the interfollicular area contained centrocyte‐like cells and numerous large transformed lymphocytes with plasma cell differentiation (HE ×250).
The floral type is characterized by hyperplastic lymphoid follicles with follicular fragmentation and the presence of atypical medium‐sized to large lymphoid cells forming a vague clear zone surrounding the mantle zone (Fig. 3c,d).
Morphologically, MALT type resembles extranodal MALT‐type lymphoma. The histological pattern on infiltration in lymph nodes was peri‐ or interfollicular with occasional follicular colonization (Fig. 3e). The tumor cells were of intermediate size and usually had round or indented nuclei and moderate to abundant cytoplasm (Fig. 3f). Sometimes the tumor cells exhibited a monocytoid appearance with abundant clear cytoplasm. Occasionally large cells resembling centroblasts or immunoblasts were also identified. Plasmacytoid differentiation of tumor cells was relatively frequent (Fig. 3f), and in four cases, it was the predominant feature of the neoplastic cells. Isolated or clustered polymorphonuclear leukocytes and histiocytes with or without epithelioid features were seen frequently intermingled with the tumor cells.
As described by Berger et al.,( 6 ) DLBCL + MALT type contained more than 50% transformed cells or sheets of transformed cells in addition to the characteristic histological findings of MALT type (Fig. 3g,h).
Epstein–Barr virus was not detected in any of the 26 cases examined.
Histological subtypes and survival. DLBCL + MALT type exhibited a poorer 5‐year OS than splenic variant (P > 0.005) (Fig. 4). However, there was no difference observed between the four histological subtypes in the FFS.
Figure 4.
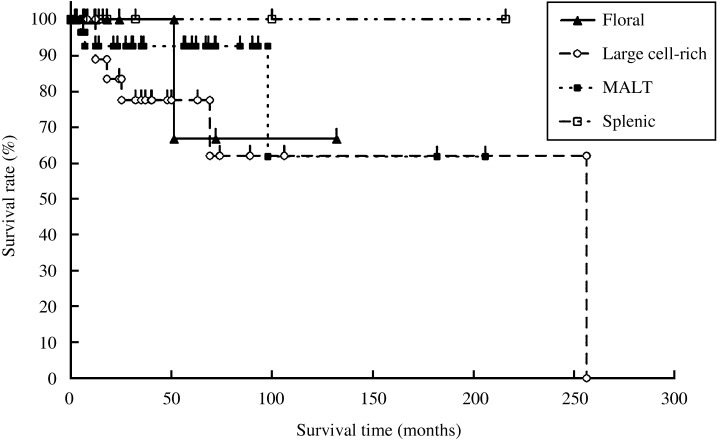
Overall survival of the 65 patients with nodal marginal zone B‐cell lymphoma, according to the histological subtypes.
Discussion
In comparison with other NMZBL series published in the western literature (Table 2), the clinical findings of our 65 cases were similar to those described in previous reports in terms of the following aspects: (1) 38 (58%) patients were >60 years old (median age, 64 years); (2) there was a slight female predominance; (3) the majority of the patients (92%) exhibited asymptomatic lymphadenopathy in the head and neck area; (4) only a minority of the patients had B symptoms (6%) and poor performance status (8%); and (5) only 5% of patients were positive for M‐protein.( 5 , 6 , 9 , 10 , 14 , 15 , 16 ) However, the 65 patients in the present series exhibited relatively longer 5‐year OS (85%) and FFS (60%) than the NMZBL series published in the western literature.( 5 , 6 , 9 , 11 , 17 ) This could be related to the high proportions of low clinical stage (stages I and II, 77%), good performance state (0–1, 92%) and low IPI score (0–1, 71%) seen in our series. As Berger et al. previously described,( 6 ) the present study indicates that NMZBL should be classified as an indolent lymphoma. Moreover, in Japan, the clinical picture of NMZBL, particularly the prognosis, appears to differ from that of western countries. However, further study is needed to clarify this issue. Except for patient age 60 years or over, none of the initial parameters studied was associated with a poorer survival. This may be related to the fact that OS and FFS were both good in our series.
Table 2.
Comparison of the clinical features with published series in western countries
| Feature | Reference | |||||
|---|---|---|---|---|---|---|
| ILSG( 17 ) | Nathwani et al.( 5 ) | Berger et al.( 6 ) | Camacho et al.( 9 ) | Traverse‐Glehen et al.( 11 ) | Present series | |
| Number of cases | 20 | 20 | 37 | 24 | 21 | 65 |
| Male‐to‐female ratio | 1:1.4 | 1:1.3 | 1:1.3 | 1:2.1 | 2:1 | 1:1.3 |
| Median age (years) | 58 | 59 | 35% > 60 years | 62 | 57 | 64 |
| Performance status = 2 (%) | NA | 100 | 13 | NA | 81 | 6 |
| B symptoms (%) | NA | 14 | 14 | 20 | 10 | 8 |
| Extranodal involvement = 2 | NA | NA | 25 | NA | NA | 0 |
| Stage I/II (%) | 18 | 29 | 32 | 59 | 24 | 77 |
| Bone marrow infiltration (%) | 41 | 28 | 43 | 29 | 62 | 0 |
| LDH > normal values | NA | 36 | 40 | 42 | 48 | 12 |
| M‐protein | NA | NA | 8 | NA | 33 | 6 |
| International prognostic index 0/1 | 36 | 57 | 31 | 48 | NA | 71 |
| International prognostic index 4/5 | 9 | 7 | 5 | 5 | NA | 3 |
| 5‐year overall survival (%) | 57 | 56 | 55 | 79 | 70 | 85 |
| 5‐year failure‐free survival (%) | 29 | 28 | 29 | 22 | 35 | 65 |
LDH, lactate dehydrogenase; NA, not available.
Previous reports have emphasized the histological diversity of NMZBL. In particular, the number of transformed cells can give rise to difficulties in grading MALT‐type lesions of NMZBL.( 4 , 6 , 12 , 15 ) However, to our knowledge the prognostic significance of histological variety has not been studied previously. Thus, based on the histomorphological features, we classified NMZBL into four subtypes and compared the prognosis among the four histological subtypes. Only DLBCL + MALT type had a significantly poorer 5‐year OS than splenic type. The recognition of DLBCL + MALT type appears important based on the following points:
-
1
Both MALT type and splenic type exhibited similar OS. However, DLBCL + MALT type showed a poorer 5‐year OS than splenic type.
-
2
In our series, the 5‐year OS of DLBCL + MALT type was 77% (data not shown). DLBCL + MALT type showed similar 5‐year OS to that of germinal center‐derived DLBCL and had a longer 5‐year OS than CD10‐negative non‐germinal center DLBCL.( 26 , 27 )
Among CD10‐negative non‐germinal center DLBCL, the large cell‐rich type of NMZBL is categorized as an indolent tumor. The present study indicated that DLBCL arising from MALT‐type NMZBL appears to be a high‐grade NMZBL and may be separated from conventional non‐germinal center DLBCL. Finally, the present study indicates that the API2–MALT1 fusion transcript dose not appear to be associated with NMZBL.
References
- 1. Isaacson PG, Nathwani BN, Piris MA et al. Nodal marginal zone B‐cell lymphoma. In: Jaffe ES, Harris NL, Stein H, Vardiman JW, eds. Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press, 2001; 1–6. [Google Scholar]
- 2. Lymphoma Study Group of Japanese Pathologists. The World Health Organization classification of malignant lymphoma in Japan: Incidence of recently recognized entities. Pathol Int 2000; 50: 696–702. [DOI] [PubMed] [Google Scholar]
- 3. Isaacson PG, Mûller‐Hermelink HK, Piris MA et al. Extranodal marginal zone B‐cell lymphoma of mucosa associated lymphoid tissue (MALT lymphoma). In: Jaffe ES, Harris NL, Stein H, Vardiman JW, eds. Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press, 2001; 157–60. [Google Scholar]
- 4. Campo E, Miquel R, Krenacs L, Sorbara L, Raffeld M, Jaffe ES. Primary modal marginal zone lymphomas of splenic and MALT type. Am J Surg Pathol 1999; 23: 59–68. [DOI] [PubMed] [Google Scholar]
- 5. Nathwani BN, Drachenberg MR, Hernanzez AM, Levine AM, Sheibani K. Nodal monocytoid B‐cell lymphoma (nodal marginal‐zone B‐cell lymphoma). Semin Hematol 1999; 36: 128–38. [PubMed] [Google Scholar]
- 6. Berger F, Felman P, Thieblemont C et al. Non‐MALT marginal zone B‐cell lymphomas: a description of clinical presentation and outcome in 124 patients. Blood 2000; 95: 1950–6. [PubMed] [Google Scholar]
- 7. Conconi A, Bertoni F, Pedrinis E et al. Nodal marginal zone B‐cell lymphomas may arise from different subsets of marginal zone B lymphocytes. Blood 2001; 98: 781–6. [DOI] [PubMed] [Google Scholar]
- 8. Taddesse‐Heath L, Pittaluga S, Sorbara L, Bussey M, Raffeld M, Jaffe ES. Nodal marginal zone B‐cell lymphoma in young adults. Am J Surg Pathol 2003; 27: 522–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Camacho FI, Algara P, Mollejo M et al. Nodal marginal zone B‐cell lymphoma: a heterogenous tumor. A comprehensive analysis of a series of 27 cases. Am J Surg Pathol 2003; 27: 762–71. [DOI] [PubMed] [Google Scholar]
- 10. Kojima M, Nakamura S, Motoori T et al. Primary marginal zone B‐cell lymphoma of the lymph node resembling plasmacytoma arising from a plasma cell variant of Castleman's disease. A clinicopathological and immunohistochemical study of seven patients. APMIS 2002; 110: 875–80. [DOI] [PubMed] [Google Scholar]
- 11. Traverse‐Glehen A, Felman P, Gallet‐Bauchu E et al. A clinicopathological study of nodal marginal zone B‐cell lymphoma: a report of 21 cases. Histopathology 2006; 48: 162–73. [DOI] [PubMed] [Google Scholar]
- 12. Feller AC, Diebold J. Histopathology of Nodal and Extranodal Non‐Hodgkin's Lymphomas. Berlin: Springer, 2004. [Google Scholar]
- 13. Kojima M, Nakamura S, Murase T et al. Follicular colonization of nodal marginal zone B‐cell lymphoma resembling follicular lymphoma: report of six cases. Int J Surg Pathol 2005; 13: 73–8. [DOI] [PubMed] [Google Scholar]
- 14. Karube K, Ohshima K, Tsuchiya T et al. A ‘floral’ variant of nodal marginal zone lymphoma. Hum Pathol 2005; 36: 202–6. [DOI] [PubMed] [Google Scholar]
- 15. Diebold J, Tourneau AL, Comperat E, Molina T, Audouín J. Primary splenic and nodal marginal zone lymphoma. J Clin Exp Hematopathol 2005; 45: 1–14. [Google Scholar]
- 16. Nathwani BN, Anderson JR, Armitage JO et al. Marginal zone B‐cell lymphoma: a clinical comparison of nodal and mucosa‐associated lymphoid tissue types. J Clin Oncol 1999; 17: 2486–92. [DOI] [PubMed] [Google Scholar]
- 17. The Non‐Hodgkin's Lymphoma Classification Project. A clinical evaluation of the International Lymphoma Study Group classification of non‐Hodgkin's lymphoma. Blood 1997; 89: 3909–18. [PubMed] [Google Scholar]
- 18. Wan JH, Trainor KJ, Brisco MJ, Morley AA. Monoclonality in B cell lymphoma detected in paraffin wax embedded sections using the polymerase chain reaction. J Clin Pathol 1990; 43: 888–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang T, Lasota J, Hanau CA, Miettinen M. Bcl‐2 oncoprotein is widespread in lymphoid tissue and lymphoma but its different expression in benign versus malignant follicles and monocytoid B‐cell proliferation is of diagnostic value. APMIS 1995; 103: 655–62. [DOI] [PubMed] [Google Scholar]
- 20. Lai R, Arber DA, Chang KL, Wilson CS, Weiss LM. Frequency of bcl‐2 expression in non‐Hodgkin's lymphoma: a study of 778 cases with comparison of marginal zone lymphoma and monocytoid B‐cell hyperplasia. Mod Pathol 1998; 11: 864–9. [PubMed] [Google Scholar]
- 21. Lai R, Weiss LM, Chang KL, Arber DA. Frequency of CD43 expression in non‐Hodgkin lymphoma: A survey of 742 cases and further characterization of rare CD43+ follicular lymphomas. Am J Clin Pathol 1999; 111: 488–94. [DOI] [PubMed] [Google Scholar]
- 22. Inagaki H, Chan JK, Ng JWM et al. Primary thymic ectranodal marginal zone B‐cell lymphoma of mucosa‐associated lymphoid tissue type exhibits distinctive clinicopathological and molecular features. Am J Pathol 2002; 160: 1435–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kaplan EL, Meier P. Non‐parametric estimation from incomplete observation. Am J Stat Assoc 1958; 53: 457–81. [Google Scholar]
- 24. Peto PE, Pike MC, Armitage P et al. Design and analysis of randomized clinical trails requiring prolonged observation of each patients. Br J Cancer 1977; 35: 1–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. The International Non‐Hodgkin's Lymphoma Prognostic Factors Project. A predictive model for aggressive non‐Hodgkin's lymphoma. N Engl J Med 1993; 329: 987–94. [DOI] [PubMed] [Google Scholar]
- 26. Allzadeh AA, Elsen MB, Davis RE et al. Distinct type of diffuse large B‐cell lymphoma identified by gene expression profiling. Nature 2000; 40: 3503–11. [DOI] [PubMed] [Google Scholar]
- 27. Hans CP, Weisenburger DD, Greiner TC et al. Confirmation of the molecular classification of diffuse large B‐cell lymphoma by immunohistochemistry using a tissue microarray. Blood 2004; 103: 275–82. [DOI] [PubMed] [Google Scholar]