

Abstract
Nasal natural killer (NK)/T‐cell lymphoma (NKTCL) and chronic active Epstein–Barr virus infection (CAEBV) are relatively frequent, especially in Asia, and are poor in prognosis. Both diseases are proliferative diseases of NK/T cells that show highly complicated karyotypes, suggesting the involvement of chromosomal instability. ATR is an important gene for DNA damage response and chromosomal stability. To evaluate the role of ATR gene alterations in the pathogenesis of NKTCL and CAEBV, the whole coding region of the ATR gene was examined in cell lines derived from NKTCL and CAEBV, as well as tumor samples from patients. ATR alterations were detected in two of eight NKTCL and in one of three CAEBV lines. Most aberrant transcripts observed were deletions resulting from aberrant splicing. ATR alterations were also detected in four of 10 NKTCL clinical samples. Both NKTCL and CAEBV cell lines with ATR alterations showed a delay or abrogation in repair of ionizing radiation‐induced DNA double‐strand breaks and ultraviolet‐induced DNA single‐strand breaks, and both exhibited a defect in p53 accumulation. These findings show that alterations in the ATR gene result in an abnormal response to DNA double‐strand break and single‐strand break repair, suggesting a role for ATR gene alterations in NKTCL lymphomagenesis. (Cancer Sci 2006; 97: 605–610)
Generally, lymphoproliferative diseases of natural killer (NK)‐cell lineage are rare; however, nasal NK/T‐cell lymphoma (NKTCL) and chronic active EBV infection (CAEBV) are relatively frequent, especially in Asia. Both diseases are proliferative diseases of NK/T cells and are strongly associated with Epstein–Barr virus (EBV),( 1 , 2 , 3 , 4 ) and the clonal expansion of EBV‐positive NK/T cells constitutes an essential part of the diseases. A recent study demonstrated the common cytological and cytogenetic features of nasal NKTCL and CAEBV cell clones, including highly complicated karyotypes with common 6q abnormalities, suggesting chromosomal instability in these cells.( 5 , 6 ) Histologically, NKTCL shows a polymorphous pattern of proliferation consisting of large lymphoid cells and normal inflammatory cells. Free radicals and oxidative stress generated in the inflammatory lesions could cause DNA damage, thus provide a basis for lymphomagenesis.
Maintenance of genome stability partly depends on the proper regulation of cellular responses to DNA damage and the integrity of DNA repair systems.( 7 ) In mammalian cells, two members of the phosphatidylinositol 3‐kinase‐related kinase (PI3KK) family, ataxia‐telangiectasia mutated (ATM) kinase and Rad3‐related (ATR) kinase, play a central role in DNA damage recognition and the initial phosphorylation events.( 8 , 9 , 10 , 11 ) Previous studies suggested that ATR might participate in the signaling of ionizing radiation (IR)‐induced and ultraviolet (UV)‐induced DNA damage.( 8 , 9 , 12 ) Recently, O'Driscoll et al.( 13 ) reported that a aberrantly spliced transcript affecting ATR expression results in Seckel syndrome, an autosomal recessive disorder, and shares features with disorders showing impaired DNA‐damage responses, such as Nijmegen breakage syndrome. Recent studies have shown that the ATR gene plays a role in the maintenance of chromosomal stability.( 14 )
In the present study, ATR gene alterations were examined in eight NKTCL and three CAEBV cell lines, as well as tumor biopsy samples obtained from NKTCL patients. Two representative cell lines from NKTCL and CAEBV with aberrant transcripts in the ATR gene were further analyzed to evaluate the functional consequences in DNA repair.
Materials and Methods
Cell lines
Cell lines derived from five EBV‐positive NKTCL (HANK‐1,( 15 ) NK‐92,( 16 ) SNK‐6, SNT‐8( 17 ) and YT( 18 )), three EBV‐negative NKTCL KHYG‐1,( 19 ) MOTN‐1,( 20 ) MTA( 21 )), three CAEBV (AIK‐T4, SIS, SKN( 22 )) and one lymphoblasotoid cell line (IB4) were selected for this study. NK‐92 and YT were purchased from Deutsche Sammlung Von Mikroorganismen und Zellkulturen (Braunschweig, Germany). MTA was obtained from the Japanese Collection of Research Bioresources (Tokyo, Japan). Other cell lines were gifts from investigators listed in the Acknowledgments. All cell lines were incubated in RPMI‐1640 medium (Sigma, St Louis, MO, USA) supplemented with 10–20% heat‐inactivated fetal calf serum at 37°C in a humidified atmosphere of 5% CO2 and 95% air.
Patients
Tumor biopsy samples from 10 patients with nasal NKTCL who were admitted to the Osaka University Hospital and affiliated hospitals during the period 1996–2000 were analyzed with informed consent in accordance with the Declaration of Helsinki. The age of the patients at admission ranged from 36 to 74 (median 62.5) years, with a male‐to‐female ratio of 8:2. Histological specimens obtained by biopsy were fixed in 10% formalin and were processed routinely for paraffin embedding or snap frozen for extraction of total RNA. Histological sections cut at 4 µm were stained with hematoxylin and eosin and immunoperoxidase procedures. Histological slides were reviewed by two of the authors (TT, KA) for diagnosis. All tumors were classified histologically as extranodal NK/T‐cell lymphoma, nasal type, according to the World Health Organization classification.
Isolation of DNA and total RNA, reverse transcription–polymerase chain reaction and detection of alterations
DNA and total RNA were extracted from the cell lines with the TRIzol reagent (Invitrogen, Rockville, MD, USA) according to the manufacturer's instructions. Total RNA (5 µg) was reverse transcribed by random hexamer priming using the Superscript first strand synthesis system (Invitrogen). Semi‐nested reverse transcription–polymerase chain reaction was carried out to amplify the ATR transcripts using four sets of primers spanning the whole open reading frame, as described previously.( 23 ) Detection of aberrant ATR transcripts was carried out as described previously.( 23 )
Western blotting
Western blotting was carried out as described previously.( 23 ) Briefly, whole cells were lysed in 1 × sample buffer, separated using 10% sodium dodecylsulfate–polyacrylamide gel electrophoresis, and then blotted to polyvinylidene difluoride (PVDF) membrane using a wet‐blotting apparatus. The antibodies used to detect p53 were DO‐7 (epitope; 19–26 amino acids of p53; DakoCytomation, Glostrup, Denmark) and Pab240 (epitope; 213–217 amino acids of p53; Calbiochem, San Diego, CA, USA). Anti‐actin (Sigma‐Aldrich, Steinheim, Germany) was used as a control. Signals were visualized with ECLplus chemiluminiscent reagents (GE Healthcare Bio‐Sciences Corp., NJ, USA).
DNA double‐strand break repair assays
More than 1 × 105 cells from each cell line were embedded in agarose plugs as described previously.( 23 , 24 ) The plugs covered with RPMI‐1640 medium were exposed to 20 Gy of IR on ice, and kept at 37°C for up to 6 h to allow the cells to repair damaged DNA. The plugs were embedded into wells of a 0.8% agarose gel in 1 × TAE and subjected to pulsed‐field gel electrophoresis (PFGE) in a CHEF apparatus (Bio‐Rad, Hercules, CA, USA) at 3 V/cm, 14°C for 48 h with a pulse time of 45 s. Under these PFGE conditions, DNA fragments between 2 and 6 Mb migrate as a single band and form compression zones under each well, whereas most rejoined double‐strand breaks (DSB) are too large to enter the gel. DSB were quantified as the fraction of DNA in the compression zone relative to that in the wells using FMBIO Analysis V8.0 (TaKaRa, Kusatsu, Japan).
DNA single‐strand break repair assays
The alkaline single cell gel electrophoresis (Comet assay) was carried out as described previously with some modifications.( 23 ) Briefly, more than 5000 cells mixed with 50 µL of 0.5% low melting point agarose was layered onto a microscope slide coated with 80 µL of 0.65% normal agarose in phosphate‐buffered saline, and then exposed to 10 J/m2 of UV. Electrophoresis was carried out for 40 min at a constant current of 300 mA in electrophoretic buffer (1 mM ethylenediamine tetraacetic acid, 300 mM NaOH [pH 13]) at 4°C. Fifty cells were analyzed based on the methods for collecting comet data, in which repair of DNA single‐strand break (SSB) was quantitated and defined as the DNA migration length (DML), a ratio of the comet length to its width.
Statistical analysis
Comparisons were made using the Student's t‐test (unpaired): P‐values less than 0.05 were taken as statistically significant.
Results
ATR alterations expressed in NKTCL and CAEBV cell lines
Aberrant transcripts of the ATR gene were observed in two of five cell lines derived from EBV‐positive NKTCL, zero of three EBV‐negative NKTCL, and one of three CAEBV cell lines (Table 1). All alterations were heterozygous deletions, which might have resulted from aberrant splicing. Deletion of exon 18 (131 bp) was observed in AIK‐T4, exons 29–34 (867 bp) in NK‐92, and exons 29–34 (867 bp) and a 65 bp deletion (nucleotides 7082–7146) in exon 41 in SNK‐6. Deletion of exons 29–34 might generate an in‐frame alteration. Other deletion alterations might generate a frameshift mutation, resulting in a premature stop codon. In addition to the wild‐type transcript, two aberrant transcripts of different types were detected in SNK‐6. In these lines, sequences around the skipping exon were also analyzed, but no alterations were detected in the acceptor/donor splice site, or around the insertion sequence (data not shown).
Table 1.
ATR alterations in nasal natural killer (NK)/T‐cell lymphoma (NKTCL) and chronic active Epstein–Barr virus infection (CAEBV) cell lines
| Cell line | Patient/ sample | Original disease | EBV | Nucleotide change | Predicted effect | Mutation‐positive clones (%) | Reference no. |
|---|---|---|---|---|---|---|---|
| NKTCL | |||||||
| HANK‐1 | 46F LN | Nasal‐type (at diagnosis) | + | WT | 15 | ||
| NK‐92 | 50 M PB | NHL with LGL cells (at diagnosis) | + | del exon 29–34 (867 bp) | In‐frame | 50 | 16 |
| SNK‐6 | 62 M TU | Nasal‐type | + | del exon 29–34 (867 bp) del 65 bp (in exon 41) | In‐frame Frameshift | 50 30 | 17 |
| SNT‐8 | 48F TU | Nasal‐type | + | WT | 17 | ||
| YT | 15 M PF | ALL + thymoma (at relapse) | + | WT | 18 | ||
| KHYG‐1 | 45F PB | Aggressive NK‐cell leukemia (at diagnosis) | – | WT | 19 | ||
| MOTN‐1 | 63F PB | (T‐LGL/T‐CLL) in chronic phase | – | WT | 20 | ||
| MTA | 58F PB | Aggressive NK‐like T‐cell leukemia/ lymphoma | – | WT | 21 | ||
| CAEBV | |||||||
| AIK‐T4 | 9F PB | Severe chronic active EBV infection | + | del exon 18 (131 bp) | Frameshift | 50% | 22 |
| SIS | 1 M PB | Severe chronic active EBV infection | + | WT | 22 | ||
| SKN | 6 M PB | Severe chronic active EBV infection | + | WT | 22 | ||
ALL, acute lymphoblastic leukemia; F, female; LN, lymph node; M, Male; NHL, non‐Hodgkin lymphoma; PB, peripheral blood; PF, pericardial fluid; T‐CLL, T‐cell chronic lymphocytic leukemia; T‐LGL, T‐cell large granular lymphocyte leukemia; TU, tumor tissue; WT, wild type.
Alteration analysis of clinical samples
Alterations in ATR were observed in four of 10 nasal NKTCL clinical samples (Fig. 1). Four alterations resulted from aberrant splicing. Deletion of exons 29 and 30 (257 bp) was found in case 1, exons 29–34 (867 bp) in case 2, exons 36 and 37 (241 bp) plus exons 39–41 (489 bp) in case 3, and exon 41 (144 bp) in case 4. Deletion of exons 29–34, 39–41 and 41 might generate an in‐frame alteration. Other deletions might generate a frameshift mutation, resulting in a premature stop codon. In addition to the wild‐type transcripts, two different aberrant transcripts were detected in case 3. The clinical samples contained 20–40% neoplastic cells, therefore it was not possible to determine whether the mutations were homozygous or heterozygous. In these cases, sequences around the skipping exon were analyzed, but no alterations were detected in the acceptor/donor splice site, or around the insertion sequence (data not shown).
Figure 1.
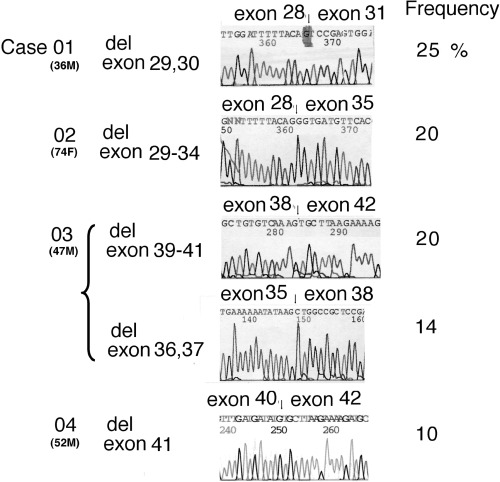
ATR gene alterations detected in biopsy samples from nasal natural killer (NK)/T‐cell lymphoma (NKTCL) patients. Four cases had alterations due to aberrant splicing. Deletion of exons 29 and 30 (257 bp) was found in case 1, exons 29–34 (867 bp) in case 2, exons 36 and 37 (241 bp) plus exons 39–41 (489 bp) in case 3, and exon 41 (144 bp) in case 4. Deletion of exons 29–34, 39–41 and 41 might generate an in‐frame alteration. Other deletions might generate a frameshift mutation, resulting in a premature stop codon.
Functional consequences of ATR alterations
To assess the functional consequence of ATR alterations for DNA‐damage response and p53 accumulation, functional studies on two cell lines (NK‐92, AIK‐T4) with heterozygous ATR alterations were carried out. IB4 and SIS, which carry no alterations within ATR, were used as negative controls.
p53 protein
Compared to unexposed cells, increased expression of p53 was found in IB4 and SIS cells after IR and UV exposure (Fig. 2). In contrast, p53 expression levels in NK‐92 and AIK‐T4 cells did not change after IR and UV exposure. These results indicate that IR and UV exposure did not induce p53 accumulation in these cell lines.
Figure 2.
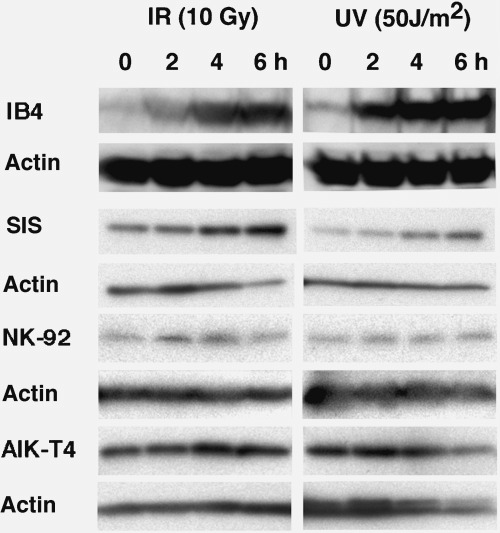
Accumulation of p53 protein. Equal numbers of cells were exposed to 10 Gy of irradiation (IR) and 50 J/m2 of ultraviolet (UV) light and incubated for up to 6 h. Whole cells were lysed and equal volumes of protein extracts were subjected to western blot analysis using anti‐p53 antibody. Accumulation of p53 protein in NK‐92 and AIK‐T4 cells was defective after IR and UV exposure. In control cell lines IB4 and SIS, p53 protein accumulated after IR and UV exposure as expected. Equivalence of loading cell lysates is shown by anti‐actin labeling.
Repair of DNA double‐strand breaks induced by irradiation
To evaluate whether ATR gene alterations result in impaired DNA DSB repair, the kinetics of DNA DSB repair was examined using PFGE (Fig. 3a). More than 87.0% of the DSB in IB4 and SIS lines were repaired within 1 h of IR exposure (Fig. 3b). In contrast, approximately 69.9% and 81.8% of DSB in NK‐92 and AIK‐T4 lines, respectively, were repaired after 1 h, reaching 92.5% and 91.5% after 6 h incubation. The difference in the rate of DSB repair 1 h after IR exposure between NK‐92, AIK‐T4 and IB‐4 was significant (P < 0.05). These results show that repair of DNA DSB after IR exposure is delayed in NK‐92 and AIK‐T4 lines.
Figure 3.
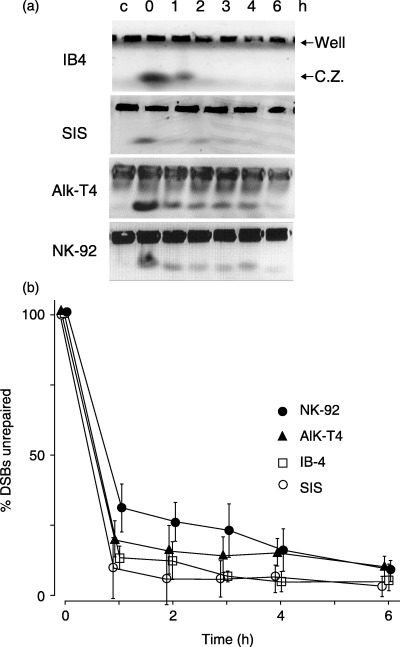
Induction and repair of DNA double‐strand breaks (DSB) after irradiation (IR) exposure determined by pulsed‐field gel electrophoresis (PFGE). (a) Ethidium bromide‐stained gel after PFGE. Equal numbers of IB4, SIS, NK‐92 and AIK‐T4 cells were embedded in 0.8% agarose plugs and exposed to 20 Gy IR. Cells were allowed to repair DSB for 0–6 h at 37°C, and were then lysed for DNA extraction and subjected to PFGE. Untreated control is shown in the first lane. The upper bands are derived from undamaged DNA or repaired DNA in wells. The lower bands show the compression zones (CZ) consisting of DSB DNA. (b) Quantification of DSB repair assays was performed. The percentage of unrepaired DSB was calculated using the ratio of signal from DSB DNA in the CZ‐to‐DNA in the corresponding well. The percentage of unrepaired DSB at 0 h incubation was set at 100%. Repair of DNA DSB after IR exposure was slightly delayed in NK‐92 and AIK‐T4 compared to control cell lines IB4 and SIS. The means (symbol) and standard deviation (error bar) from three experiments are depicted. Symbols represent cell lines: IB4 (□), SIS (○), NK‐92 (•), and AIK‐T4 (▴).
Repair of DNA single‐strand breaks induced by UV light
To evaluate whether alterations in the ATR gene result in impaired DNA SSB repair, NK‐92 and AIK‐T4 cell lines with ATR alterations together with IB4 and SIS cells were exposed to 10 J/m2 of UV, and repair of DNA SSB was evaluated using the Comet assay. After UV exposure, the comet tails were observed in all cell lines, indicating that DNA SSB had occurred (Fig. 4a). The maximum DML of IB4 was observed after an 6‐h incubation and a significant decrease in DML was observed at 18 h after exposure (Fig. 4b). The DML of SIS decreased, whereas the DML of NK‐92 and AIK‐T4 cells did not show an obvious decrease in DML after 18 h incubation. These results indicate that the NK‐92 and AIK‐T4 lines failed to repair DNA SSB after UV exposure.
Figure 4.
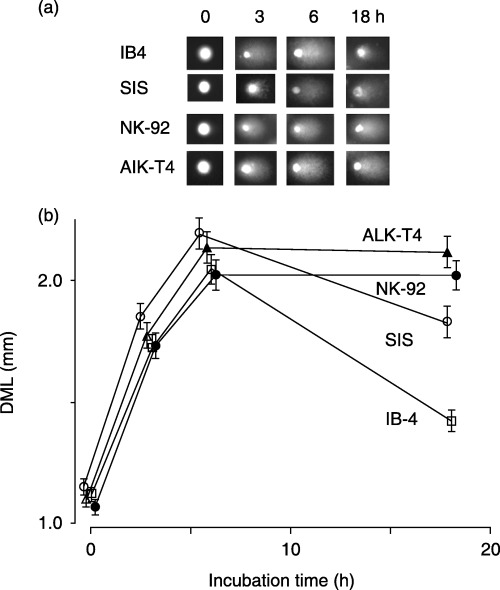
Induction and repair of DNA single‐strand breaks (SSB) after ultraviolet (UV) exposure determined using the Comet assay. IB4, SIS, NK‐92 and AIK‐T4 cells were exposed to 10 J/m2 of UV, incubated at 37°C for 0–18 h, then lysed and subjected to Comet assay. (a) The representative pattern of DNA migration (comet tails) from the Comet assay. (b) Quantification of SSB repair assays was carried out as described in Materials and Methods. Compared to control cell lines, IB4 and SIS, NK‐92 and AIK‐T4 cells showed failure and inability to repair DNA SSB after UV exposure. The means (symbol) and standard deviation (error bar) from three experiments is depicted. Symbols represent cell lines: IB4 (□), SIS (○), NK‐92 (•), and AIK‐T4 (▴).
Discussion
Maintenance of genome stability depends on the appropriate response to DNA damage.( 7 ) This response is based on complex networks of signaling pathways that activate numerous processes and lead ultimately to damage repair and cellular survival and apoptosis. The protein kinases ATM and ATR are master controllers of some of these networks.( 8 , 9 , 10 , 11 ) These two kinases act either in concert or separately to orchestrate the response to specific types of DNA damage or stalled replication. ATM responds mainly to DNA DSB, whereas ATR is activated by DNA SSB and stalled DNA replication forks.
The greatest frequency of genetic alteration encountered in any of the cell lines was 50%, indicating heterozygous mutations. ATR is an essential gene required for cell proliferation, therefore homozygous ATR mutation is lethal.( 25 , 26 ) O'Driscoll et al.( 13 ) reported that a heterozygous aberrantly spliced transcript affecting ATR expression results in Seckel syndrome, an autosomal recessive disorder. They demonstrated that the cells derived from patients with Seckel syndrome had an abnormal response to DNA breaks. Lewis et al.( 27 ) demonstrated that cells with heterozygous truncating ATR mutations in exon 10 show abnormalities in DNA repair as well as cell cycle checkpoints.
In the present study, heterozygous aberrant transcripts affecting the PI3K domain of ATR were detected in two of eight NKTCL cell lines and one of three CAEBV cell lines. In the clinical samples, alterations in ATR were also detected in four of 10 cases. The cell lines with ATR alterations showed a delay in the repair of DSB, an abrogation in the repair of SSB, and exhibited a defect in p53 accumulation after IR and UV exposure. These findings indicate that the alterations in ATR result in functional deficiencies in the cell lines.
In the present study, two cell lines with heterozygous ATR aberrant transcripts showed abnormal responses to DNA damage induced by UV and IR exposure. Because UV‐induced SSB mainly triggers ATR but not ATM, the abnormal response after UV exposure may result from abrogation of ATR, whereas IR‐induced DSB mainly trigger ATM. Tibbetts et al.( 8 ) suggested that ATM and ATR might be induced sequentially by IR. They proposed that, in response to DSB, ATM is activated first, phosphorylating specific substrates, and then ATR kicks in as a maintenance enzyme to prolong phosphorylation of the substrates targeted initially by ATM. Delay of DSB repair as observed in NK‐92 and AIK‐T4 lines may be consistent with their hypothesis.
Histologically, NKTCL usually shows a polymorphous pattern of proliferation of large lymphoid cells intermingling with small lymphocytes, plasma cells, macrophages and, less frequently, eosinophils.( 28 ) Free radicals secreted by the infiltrating inflammatory cells might cause DNA damage. Recent studies have demonstrated the common cytological and cytogenetic features of nasal NKTCL and CAEBV cell clones, including highly complicated karyotypes, indicative of chromosomal instability in these cells.( 5 , 6 ) Alterations in ATR, the gene responsive to DNA damage, might facilitate accumulation of structural abnormalities of chromosomes as well as mutations of the genes.
ATR alterations were observed in cancers of the stomach,( 29 ) endometrium,( 30 ) urothelium ( 31 ) and colorectum( 32 ) with mismatch repair phenotypes. In these cases, 10 repeats of mononucleotide A in exon 10 were changed, which resulted in frameshift mutations. The same kind of mutation was not observed in either the present study or our previous study on 50 cell lines derived from hematopoietic malignancies.( 33 ) Although the deletion mutations observed in the present study have not been reported in cancers, this might be due to differences in the cell lineages used, and in the methods employed for investigation of ATR alterations (i.e. mRNA was used in the present study, whereas previous studies on cancers used genomic DNA. Because detection of large deletions and exon skipping is difficult using genomic DNA, further study is necessary to clarify the extent of ATR alteration in cancers and hematopoietic malignancies.
The present study showed that alterations in ATR were relatively frequent in NKTCL, suggesting a role for ATR alterations in the initiation and progression of nasal NKTCL.
Acknowledgments
We thank Dr E. Kieff (Brigham and Women's Hospital, Boston, MA, USA) for providing IB‐4, Drs M. Daibata and S. Imai (Kochi University, Japan) for AIK‐T4, SIS and SKN, Dr N. Shimizu (Tokyo Medical and Dental University, Japan) for SNT‐8 and SNK‐6, Dr Y. Kagami (Aichi Cancer Center Hospital, Japan) for HANK‐1, Dr Y. Matsuo (Hayashibara Biochemical Laboratories, Japan) for MOTN‐1, and Dr M. Yagita (Tazuke‐Kofukai Medical Research Institute, Japan) for the KHYG‐1 cell line. Supported by grants from the Ministry of Education, Science, Culture, and Sports, Japan (16790206, 15406013, 17590343, 16390105) and Japan Society for the Promotion of Science (17.05219).
References
- 1. Kikuta H, Taguchi Y, Tomizawa K et al. Epstein–Barr virus genome‐positive T lymphocytes in a boy with chronic active EBV infection associated with Kawasaki‐like disease. Nature 1988; 333: 455–7. [DOI] [PubMed] [Google Scholar]
- 2. Jones JF, Shurin S, Abramowsky C et al. T‐cell lymphomas containing Epstein–Barr viral DNA in patients with chronic Epstein–Barr virus infections. N Engl J Med 1988; 318: 733–41. [DOI] [PubMed] [Google Scholar]
- 3. Harabuchi Y, Yamanaka N, Kataura A et al. Epstein–Barr virus in nasal T‐cell lymphomas in patients with lethal midline granuloma. Lancet 1990; 335: 128–30. [DOI] [PubMed] [Google Scholar]
- 4. Ho FC, Srivastava G, Loke SL et al. Presence of Epstein–Barr virus DNA in nasal lymphomas of B‐ and T‐cell type. Hematol Oncol 1990; 8: 271–81. [DOI] [PubMed] [Google Scholar]
- 5. Zhang Y, Nagata H, Ikeuchi T et al. Common cytological and cytogenetic features of Epstein–Barr virus (EBV)‐positive natural killer (NK) cells and cell lines derived from patients with nasal T/NK‐cell lymphomas, chronic active EBV infection and hydroa vacciniforme‐like eruptions. Br J Haematol 2003; 121: 805–14. [DOI] [PubMed] [Google Scholar]
- 6. Wong KF, Zhang YM, Chan JK. Cytogenetic abnormalities in natural killer cell lymphoma/leukaemia − is there a consistent pattern? Leuk Lymphoma 1999; 34: 241–50. [DOI] [PubMed] [Google Scholar]
- 7. Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 2003; 3: 155–68. [DOI] [PubMed] [Google Scholar]
- 8. Tibbetts RS, Cortez D, Brumbaugh KM et al. Functional interactions between BRCA1 and the checkpoint kinase ATR during genotoxic stress. Genes Dev 2000; 14: 2989–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature 2000; 408: 433–9. [DOI] [PubMed] [Google Scholar]
- 10. Khanna KK, Jackson SP. DNA double‐strand breaks: signaling, repair and the cancer connection. Nat Genet 2001; 27: 247–54. [DOI] [PubMed] [Google Scholar]
- 11. Rouse J, Jackson SP. Interfaces between the detection, signaling, and repair of DNA damage. Science 2002; 297: 547–51. [DOI] [PubMed] [Google Scholar]
- 12. Cliby WA, Roberts CJ, Cimprich KA et al. Overexpression of a kinase‐inactive ATR protein causes sensitivity to DNA‐damaging agents and defects in cell cycle checkpoints. EMBO J 1998; 17: 159–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. O'Driscoll M, Ruiz PV, Woods CG, Jeggo PA, Goodship JA. A splicing mutation affecting expression of ataxia‐telangiectasia and Rad3‐related protein (ATR) results in Seckel syndrome. Nat Genet 2003; 33: 497–501. [DOI] [PubMed] [Google Scholar]
- 14. Casper AM, Nghiem P, Arlt MF, Glover TW. ATR regulates fragile site stability. Cell 2002; 111: 779–89. [DOI] [PubMed] [Google Scholar]
- 15. Kagami Y, Nakamura S, Suzuki R et al. Establishment of an IL‐2‐dependent cell line derived from ‘nasal‐type’ NK/T‐cell lymphoma of CD2+, sCD3–, CD3epsilon+, CD56+ phenotype and associated with the Epstein–Barr virus. Br J Haematol 1998; 103: 669–77. [DOI] [PubMed] [Google Scholar]
- 16. Gong JH, Maki G, Klingemann HG. Characterization of a human cell line (NK‐92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia 1994; 8: 652–8. [PubMed] [Google Scholar]
- 17. Nagata H, Konno A, Kimura N et al. Characterization of novel natural killer (NK)‐cell and gammadelta T‐cell lines established from primary lesions of nasal T/NK‐cell lymphomas associated with the Epstein–Barr virus. Blood 2001; 97: 708–13. [DOI] [PubMed] [Google Scholar]
- 18. Yodoi J, Teshigawara K, Nikaido T et al. TCGF (IL 2)‐receptor inducing factor(s). I. Regulation of IL 2 receptor on a natural killer‐like cell line (YT cells). J Immunol 1985; 134: 1623–30. [PubMed] [Google Scholar]
- 19. Yagita M, Huang CL, Umehara H et al. Novel natural killer cell line (KHYG‐1) from a patient with aggressive natural killer cell leukemia carrying a p53 point mutation. Leukemia 2000; 14: 922–30. [DOI] [PubMed] [Google Scholar]
- 20. Matsuo Y, Drexler HG, Takeuchi M, Tanaka M, Orita K. Establishment of the T‐cell large granular lymphocyte leukemia cell line MOTN‐1 carrying natural killer‐cell antigens. Leuk Res 2002; 26: 873–9. [DOI] [PubMed] [Google Scholar]
- 21. Emi N, Abe A, Kasai M et al. CD4‐ and CD56‐positive T‐cell line, MTA, established from natural killer‐like T‐cell leukemia/lymphoma. Int J Hematol 1999; 69: 180–5. [PubMed] [Google Scholar]
- 22. Imai S, Sugiura M, Oikawa O et al. Epstein–Barr virus (EBV)‐carrying and ‐expressing T‐cell lines established from severe chronic active EBV infection. Blood 1996; 87: 1446–57. [PubMed] [Google Scholar]
- 23. Liu A, Takakuwa T, Fujita S et al. Alterations of DNA damage‐response genes ATM and ATR in pyothorax‐associated lymphoma. Lab Invest 2005; 85: 436–46. [DOI] [PubMed] [Google Scholar]
- 24. Abbott DW, Freeman ML, Holt JT. Double‐strand break repair deficiency and radiation sensitivity in BRCA2 mutant cancer cells. J Natl Cancer Inst 1998; 90: 978–85. [DOI] [PubMed] [Google Scholar]
- 25. Brown EJ, Baltimore D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev 2000; 14: 397–402. [PMC free article] [PubMed] [Google Scholar]
- 26. De Klein A, Muijtjens M, Van Os R et al. Targeted disruption of the cell‐cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr Biol 2000; 10: 479–82. [DOI] [PubMed] [Google Scholar]
- 27. Lewis KA, Mullany S, Thomas B et al. Heterozygous ATR mutations in mismatch repair‐deficient cancer cells have functional significance. Cancer Res 2005; 65: 7091–5. [DOI] [PubMed] [Google Scholar]
- 28. Teruya‐Feldstein J, Jaffe ES, Burd PR et al. The role of Mig, the monokine induced by interferon‐gamma, and IP‐10, the interferon‐gamma‐inducible protein‐10, in tissue necrosis and vascular damage associated with Epstein–Barr virus‐positive lymphoproliferative disease. Blood 1997; 90: 4099–105. [PubMed] [Google Scholar]
- 29. Menoyo A, Alazzouzi H, Espin E, Armengol M, Yamamoto H, Schwartz S Jr. Somatic mutations in the DNA damage‐response genes ATR and CHK1 in sporadic stomach tumors with microsatellite instability. Cancer Res 2001; 61: 7727–30. [PubMed] [Google Scholar]
- 30. Vassileva V, Millar A, Briollais L, Chapman W, Bapat B. Genes involved in DNA repair are mutational targets in endometrial cancers with microsatellite instability. Cancer Res 2002; 62: 4095–9. [PubMed] [Google Scholar]
- 31. Mongiat‐Artus P, Miquel C, Van Der Aa M et al. Microsatellite instability and mutation analysis of candidate genes in urothelial cell carcinomas of upper urinary tract. Oncogene 2006; 25: 2133–8. [DOI] [PubMed] [Google Scholar]
- 32. Li LS, Kim NG, Kim SH et al. Chromosomal imbalances in the colorectal carcinomas with microsatellite instability. Am J Pathol 2003; 163: 1429–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ham MF, Takakuwa T, Luo WJ et al. Impairment of double‐strand breaks repair and aberrant splicing of ATM and MRE11 in leukemia–lymphoma cell lines with microsatellite instability. Cancer Sci 2006; 97: 226–34. [DOI] [PMC free article] [PubMed] [Google Scholar]