

Abstract
Peroxiredoxins (Prdxs) are thiol‐specific antioxidant proteins that are highly expressed in human cancer cells. Prdxs have been shown to be involved in tumor cell proliferation under conditions of microenvironmental stress such as hypoxia. We hypothesized that Prdxs could be categorized into two groups, stress‐inducible and non‐inducible ones. In this study, we analyzed the promoter activity and expression levels of five Prdx family members in human cancer cells. We found that both Prdx1 and Prdx5 are inducible after treatment with hydrogen peroxide or hypoxia, but that Prdx2, Prdx3, and Prdx4 are not or are only marginally inducible. We also found that Ets transcription factors are the key activators for stress‐inducible expression. High‐mobility group protein HMGB1 was shown to function as a coactivator through direct interactions with Ets transcription factors. The DNA binding of Ets transcription factors was significantly enhanced by HMGB1. Silencing of Ets1, Ets2, Prdx1, and Prdx5 expression sensitized cells to oxidative stress. These data indicate that transcription of Prdx genes mediated by Ets/HMG proteins might protect cells from oxidative stress. (Cancer Sci 2008; 99: 1950–1959)
Redox regulation is important for various metabolic functions of cells.( 1 ) Glutathione and thioredoxin are major reducing systems that regulate redox balance and communicate with cellular molecules to execute specific responses. Peroxiredoxins (Prdxs) are small antioxidant proteins that scavenge reactive oxygen species (ROS). Prdxs are also involved in the cellular response to ROS, and function as a cellular defense system against ROS. Like thioredoxin, expression of Prdxs increases during a variety of oxidative stress conditions. There are five members of the 2‐Cys Prdx family, including three cytoplasmic forms, one mitochondrial form, and one secretary form.
Mice lacking Prdx1 have a shortened lifespan owing to the development of severe hemolytic anemia and several malignant cancers.( 2 ) Prdx1‐deficient cells showed increased sensitivity to oxidative stress and decreased cell proliferation. Thus, Prdx1 may function as a tumor suppressor. Elevated expression of Prdxs has been reported in human cancers, including lung cancer, breast cancer, and bladder cancer.( 3 , 4 , 5 ) Knockdown of Prdx1 expression resulted in significant growth inhibition, radiosensitization, and reduced metastasis of lung cancer cells.( 6 ) Elucidating the molecular mechanisms underlying the expression of Prdx family members will be critical to understanding Prdx function under physiological or stress conditions in tumor cells.
In this study, we demonstrate that Prdx1 and Prdx5 are significantly induced after H2O2 and hypoxia treatment, through the activation of Ets transcription factors. Further, the high‐mobility group (HMG) protein HMGB1 was shown to interact directly with Ets transcription factors, enhancing their DNA binding ability and the promoter activity of target genes. This novel molecular interaction may contribute to our understanding of the functions of Prdxs and tumor biology from the aspect of redox regulation in cancer cells.
Materials and Methods
Cell culture. Human prostate cancer PC3 cells and human epidermoid cancer KB cells were cultured in Eagle's minimal essential medium. This medium was purchased from Nissui Seiyaku (Tokyo, Japan) and contained 10% fetal bovine serum. Cell lines were maintained in a 5% CO2 atmosphere at 37°C.
Antibodies. Antibodies against Sp1 (sc‐59), GST (sc‐138), Ets1 (sc‐111), Ets2 (sc‐351), Prdx2 (sc‐23967), and Prdx4 (sc‐23974) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti‐Flag (M2) antibody and anti‐Flag (M2) affinity gel were purchased from Sigma (St Louis, MO, USA). Anti‐HA‐peroxidase (3F10) and anti‐Thio antibody were purchased from Roche Molecular Biochemicals (Mannheim, Germany) and Invitrogen (San Diego, CA, USA), respectively. To generate anti‐Prdx1, anti‐Prdx5, and anti‐BAF57 antibodies, the synthetic peptides PDVQKSKEYFSKQK, GLTCSLAPNIISQL, and EPPTDPIPEDEKKE were used as immunogens, respectively, and the antisera obtained from immunized rabbits were affinity purified using the synthetic peptide antigens. Anti‐YB‐1 and anti‐HMGB1 antibodies were prepared as described previously.( 7 , 8 ) The anti‐Prdx3 antibody was kindly gifted by Dr H. Nanri (Seinan Jogakuin University).( 9 )
Plasmid construction. The plasmid construction of pGEX‐HMGB1 expressing GST‐HMGB1 and its deletion mutants (ΔA, ΔB) has been described previously.( 8 , 10 ) For construction of pcDNA3‐HA‐HMGB1, the HA‐tagged HMGB1 cDNA was ligated into the pcDNA3 vector (Invitrogen). To obtain full‐length cDNAs for Ets1 and Ets2, polymerase chain reaction (PCR) was carried out on a SuperScript cDNA library (Invitrogen) using the following primer pairs: 5′‐ATGAAGGCGGCCGTCGATCTCAAGC‐3′ and 5′‐TCACTCGTCGGCATCTGGCTTGACG‐3′ for Ets1; and 5′‐ATGAATGATTTCGGAATCAAGAATATGG‐3′ and 5′‐TCAGTCCTCCGTGTCGGGC‐3′ for Ets2. PCR products were cloned into the pGEM‐T easy vector (Promega, Madison, WI, USA). To construct pGEX‐Ets1 and pGEX‐Ets2, expressing GST‐Ets1 and GST‐Ets2, respectively, the NotI fragment of Ets1 cDNA and EcoRI fragment of Ets2 cDNA were ligated into the pGEX‐4T vector (Pharmacia Biotech, Tokyo, Japan). For construction of TH‐HA‐Ets1 and TH‐HA‐Ets2, expressing N‐terminal tagged HA‐Ets1 and HA‐Ets2, respectively, in bacteria, full‐length Ets1 and Ets2 cDNAs were ligated into the TH‐HA vector.( 11 ) Their deletion mutants (TH‐HA‐Ets1 Δ1, TH‐HA‐Ets1 Δ2, TH‐HA‐Ets2 Δ1, TH‐HA‐Ets2 Δ2) were constructed from TH‐HA‐Ets1/2 full‐length plasmids by deletion of the XagI‐Mph1103I, PaeI, PdiI‐XbaI, and Bpu1102I‐XbaI fragments, respectively. To generate ThioHis‐Ets1 Δ3, ThioHis‐Ets1 Δ4, and ThioHis‐Ets2 Δ3, PscI‐PstI fragment of TH‐HA‐Ets1 Δ1, XagI‐PstI fragment of TH‐HA‐Ets1, and XapI‐PstI fragment of TH‐HA‐Ets2 Δ1 were ligated into the pThioHis vector (Invitrogen), respectively. For construction of pcDNA3‐Flag‐Ets1 and pcDNA3‐Flag‐Ets2, N‐terminal Flag‐tagged Ets1 and Ets2 cDNAs were ligated into the pcDNA3 vector (Invitrogen). The constructs Prdx1‐Luc (–1065 to +83), Prdx2‐Luc (–338 to +136), Prdx3‐Luc (–357 to +42), Prdx4‐Luc (–306 to +36), and Prdx5‐Luc (–314 to +113) were made using the following primer pairs: 5′‐CTCGAGGCCAAGGCGGGCGGATCACCTG‐3′ and 5′‐AAGCTTAACCACCGACACCAGGCAAGAAC‐3′ for Prdx1‐Luc; 5′‐AGATCTTAGATGCTGCAGCCTCAGC‐3′ and 5′‐AAGCTTGGCAAAGGCTAGACGCACGG‐3′ for Prdx2‐Luc; 5′‐AGATCTTAGCTTATTAACGGACTAAAAC‐3′ and 5′‐AAGCTTCAGTGCACTCGGGCGCCACGG‐3′ for Prdx3‐Luc; 5′‐AGATCTGTGAGGGGCTTGTGTGCAG‐3′ and 5′‐AAGCTTCACGCGAGCGCAGAAACACG‐3′ for Prdx4‐Luc; and 5′‐AGATCTAAGATGCAAATCATATGC‐3′ and 5′‐AAGCTTCCCACGGCCACTTCCACTCC3′ for Prdx5‐Luc. These PCR products were cloned and ligated into the BglII‐HindIII sites or the XhoI‐HindIII sites of the pGL3‐basic vector (Promega). PCR was also performed to introduce the mutations of Ets binding sites into the Prdx1 and Prdx5 promoter, using the following primer pairs: 5′‐CTCGAGTGGTCCTGGAAGTAAAGAGAATCC‐3′ and 5′‐AAGCTTAACCACCGACACCAGGCAAGAAC‐3′ for Prdx1‐Luc WT; 5‐CTCGAGTGGTCCTAAAAGTAAAGAGAATCC‐3′ and 5′‐AAGCTTAACCACCGACACCAGGCAAGAAC‐3′ for Prdx1‐Luc MT; 5′‐AGATCTGGGTCCCGGAAGCTCTGTTCTGC‐3′ and 5′‐AAGCTTCCCACGGCCACTTCCACTCC‐3′ for Prdx5‐Luc WT; and 5′‐AGATCTGGGTCCCAAAAGCTCTGTTCTGC‐3′ and 5′‐AAGCTTCCCACGGCCACTTCCACTCC‐3′ for Prdx5‐Luc MT. Italicized nucleotides indicate wild‐type Ets binding sites and mutated ones. These PCR products were cloned and ligated into the BglII‐HindIII sites or the XhoI‐HindIII sites of the pGL3‐basic vector (Promega).
Western blotting analysis. Whole‐cell lysates were prepared as described previously.( 12 , 13 ) The indicated amounts of whole‐cell lysates were separated by SDS‐PAGE and transferred to polyvinylidine difluoride microporous membranes (Millipore, Bedford, MA, USA) using a semidry blotter. The blotted membranes were treated with 5% (w/v) skimmed milk in 10 mmol/L Tris, 150 mmol/L NaCl, and 0.2% (v/v) Tween 20, and incubated for 1 h at room temperature with primary antibody. The following antibodies and dilutions were used: a 1:5000 dilution of anti‐Sp1, a 1:5000 dilution of anti‐GST, a 1:500 dilution of anti‐Ets1, a 1:2000 dilution of anti‐Ets2, a 1:5000 dilution of anti‐Flag (M2), a 1:5000 dilution of anti‐Thio, a 1:10 000 dilution of anti‐Prdx1, a 1:5000 dilution of anti‐Prdx2, a 1:5000 dilution of anti‐Prdx3, a 1:1000 dilution of anti‐Prdx4, a 1:1000 dilution of anti‐Prdx5, a 1:10 000 dilution of anti‐YB‐1, a 1:2000 dilution of anti‐HMGB1, and a 1:10 000 dilution of anti‐BAF57. Membranes were then incubated for 40 min at room temperature with a peroxidase‐conjugated secondary antibody or a 1:5000 dilution of anti‐HA‐peroxidase. Bound antibody was visualized using an enhanced chemiluminescence kit (GE Healthcare Biosciences, Piscataway, NJ, USA) and membranes were exposed to Kodak X‐OMAT film (Kodak, Paris, France). For the correlation assay, the intensity of each signal was quantified by the NIH imaging program, version 1.62 (NIH, Bethesda, MD, USA).
Northern blotting analysis. Northern blotting analysis was performed as described previously.( 12 , 13 ) To obtain cDNAs for Prdx1, Prdx2, Prdx3, Prdx4, and Prdx5, PCR was carried out on a SuperScript cDNA library (Invitrogen) using the following primer pairs: 5′‐ATGTCTTCAGGAAATGCTAAAATTG‐3′ and 5′‐TCACTTCTGCTTGGAGAAATATTC‐3′ for Prdx1; 5′‐ATGGCCTCCGGTAACGCGCGC‐3′ and 5′‐CTAATTGTGTTTGGAGAAATATTCC‐3′ for Prdx2; 5′‐ATGGCGGCTGCTGTAGGACGG‐3′ and 5′‐CTACTGATTTACCTTCTGAAAGTAC‐3′ for Prdx3; 5′‐ATGGAGGCGCTGCCGCTGCTAGC‐3′ and 5′‐TCAATTCAGTTTATCGAAATACTTCAGC‐3′ for Prdx4; 5′‐ATGGGACTAGCTGGCGTGTGCG‐3′ and 5′‐TCAGAGCTGTGAGATGATATTGGG‐3′ for Prdx5. Ets1 and Ets2 cDNAs were obtained as described in “Plasmid construction”. RNA samples (20 µg/lane) were separated on a 1% formaldehyde–agarose gel and transferred to a Hybond N+ membrane (GE Healthcare Biosciences) with 10 × SSC. After prehybridization and hybridization with radiolabeled cDNA fragment of Ets1, Ets2, Prdx1, Prdx2, Prdx3, Prdx4, and Prdx5, signal intensities were analyzed using a bio‐imaging analyzer (FLA2000; Fujifilm, Tokyo, Japan).
Hypoxia conditions. Hypoxic conditions were generated as described previously.( 14 ) Briefly, plates seeded with PC3 cells or KB cells at 70% to 80% confluency, at a pH between 7.2 and 7.4, were incubated at 37°C in Anaerocult A mini (Merck, Darmstadt, Germany), creating oxygen‐free circumstances. After 4 h, plates were removed from the Anaerocult A mini and used in assays immediately or after further incubation under normal oxygen.
Knockdown analysis using siRNAs. The following double‐stranded RNA 25‐base‐pair oligonucleotides were commercially generated (Invitrogen): 5′‐GCAAAGAAAUGAUGUCUCAAGCAUU‐3′ for Ets1; 5′‐GCAGCAACUUGAAUUUGCUCACCAA‐3′ for Ets2; 5′‐UGACCAUCUGGCAUAACAGCUGUGG‐3′ for Prdx1; 5′‐AGAACCUCUUGAGACGUCGAUUCCC‐3′ for Prdx5. siRNA transfections were performed as described previously.( 12 , 13 ) Ten microliters of Lipofectamine 2000 (Invitrogen) was diluted in 250 µL of Opti‐MEM I medium (Invitrogen) and incubated for 5 min at room temperature. Next, 250 pmol of indicated siRNA or inverted control duplex Stealth RNAs (Invitrogen), diluted in 250 µL of Opti‐MEM I, was added gently and incubated for 20 min at room temperature. Oligomer‐lipofectamine complexes and aliquots of 1 × 106 cells in 500 µL of culture medium were combined and incubated for 10 min at room temperature. Aliquots of 1.5 × 105 PC3 cells were used in luciferase assays and aliquots of 2.5 × 103 PC3 cells were used in the WST‐8 assay, as described below. The remaining cells were seeded into 35‐mm dishes with 2 mL of culture medium and harvested after 72 h of culture for Western blotting analysis as described above.
Luciferase assay. Transient transfection and a luciferase assay were performed as described previously.( 12 , 13 ) PC3 cells (1 × 105) were seeded into 12‐well plates. The following day, cells were cotransfected with the indicated amounts of Prdx1, Prdx2, Prdx3, Prdx4, or Prdx5 reporter plasmid and expression plasmids using Superfect reagent (Qiagen, Hilden, Germany); cells were then transfected with siRNA as described above, followed by transfection with the indicated amounts of reporter plasmid at intervals of 12 h; finally cells were incubated under normal conditions or in the presence of 1 mmol/L H2O2, for 30 min, with or without recovery for 12 h, or incubated in hypoxic conditions for 4 h, with or without reoxygenation for 12 h. Forty‐eight hours post‐transfection, cells were lyzed with reporter lysis buffer (Promega) and luciferase activity was detected using a Picagene kit (Toyoinki, Tokyo, Japan); light intensity was measured using a luminometer (Luminescencer JNII RAB‐2300; ATTO, Tokyo, Japan). The results shown are normalized to protein concentrations measured using the Bradford method, and are representative of at least three independent experiments.
Chromatin immunoprecipitation assay (ChIP). The ChIP assay was performed as described previously.( 12 , 13 ) Briefly, PC3 cells were transiently transfected with Flag, Flag‐Ets1, or Flag‐Ets2 plasmid and cultured for 48 h as described above. Soluble chromatin from 1 × 106 cells was incubated with 2 µg of anti‐Flag (M2) affinity gel. Purified DNA was dissolved in 20 µL of dH2O and 2 µL of DNA was used for PCR analysis with the following primer pairs for Prdx1: 5′‐CTCGAGTCCTGATATTTTATTTTCCTTTAC‐3′ as a forward primer and 5′‐AAGCTTAACCACCGACACCAGGCAAGAAC‐3′ as a reverse primer; for Prdx2: 5′‐AGATCTTAGATGCTGCAGCCTCAGC‐3′ as a forward primer and 5′‐AAGCTTGGCAAAGGCTAGACGCACGG‐3′ as a reverse primer; for Prdx3: 5′‐AGATCTTAGCTTATTAACGGACTAAAAC‐3′ as a forward primer and 5′‐AAGCTTCAGTGCACTCGGGCGCCACGG‐3′ as a reverse primer; for Prdx4: 5′‐AGATCTGTGAGGGGCTTGTGTGCAG‐3′ as a forward primer and 5′‐AAGCTTCACGCGAGCGCAGAAACACG‐3′ as a reverse primer; and for Prdx5: 5′‐AGATCTGCAAAATAAACACATTTTACTCC‐3′ as a forward primer and 5′‐AAGCTTCCCACGGCCACTTCCACTCC‐3′ as a reverse primer. PCR products were separated by electrophoresis on 2% agarose gels and stained with ethidium bromide.
Coimmunoprecipitation assay. Transient transfection and immunoprecipitation assays were performed as described previously.( 12 , 13 ) Briefly, 1 × 105 PC3 cells were seeded into 6‐well plates. The following day, cells were transfected with 1 µg of each of HA‐ and Flag‐fused expression plasmids using Superfect reagent (Qiagen) according to the manufacturer's instructions. Three hours post‐transfection, the cells were washed with PBS, cultured at 37°C for 48 h in fresh medium, and then lyzed in buffer X containing 50 mmol/L Tris‐HCl (pH 8.0), 1 mmol/L ethylendiaminetetraacetic acid (EDTA), 120 mmol/L NaCl, 0.5% (v/v) Nonidet P‐40 (NP‐40), 10% (v/v) glycerol, 1 mmol/L phenylmethylsulfonyl fluoride (PMSF), and 1 mmol/L dithiothreitol (DTT). The lysates were centrifuged at 21 000 g for 10 min at 4°C and the supernatants (300 µg) were incubated for 2 h at 4°C with anti‐Flag (M2) affinity gel. Immunoprecipitated samples washed three times with buffer X and preimmunoprecipitated samples (30 µg) were subjected to Western blotting analysis with anti‐Flag antibody and anti‐HA antibodies, as described above. For immunoprecipitation assays without transient transfection, 70–80% confluent PC3 cells in 100‐mm tissue‐culture plates were harvested with PBS. After centrifugation at 500 g for 5 min, cells were resuspended in ice‐cold 10 mmol/L Hepes/KOH (pH 7.9), 10 mmol/L KCl, 0.1 mmol/L EDTA, 0.1 mmol/L EGTA, 1 mmol/L DTT, and 0.5 mmol/L PMSF and lyzed by adding NP40 to a final concentration of 0.5% (v/v), and lysates were centrifuged at 500 g for 5 min. The resulting nuclear pellet was resuspended in ice‐cold 20 mmol/L Hepes/KOH (pH 7.9), 0.4 mol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 1 mmol/L DTT, and 1 mmol/L PMSF, and incubated for 15 min on ice. The lysates were centrifuged at 21 000 g for 10 min at 4°C and the supernatants (500 µg) were incubated for 2 h at 4°C with 2 µg of rabbit immunoglobulin G (IgG) or anti‐Ets1 antibody. Immunoprecipitated samples washed three times with buffer X and preimmunoprecipitated samples (50 µg) were subjected to Western blotting analysis with indicated antibodies.
Expression of GST‐, ThioHis‐, and HA‐fusion proteins in E. coli. For expression of GST‐, ThioHis‐, and HA‐fusion proteins, bacteria transformed with expression plasmids were incubated with 1 mmol/L isopropyl‐β‐D‐thiogalactopyranoside (Boehringer Mannheim, Mannheim, Germany) for 2 h at room temperature, and collected by centrifugation. After sonication (TAITEC sonicator, Tokyo, Japan) in buffer X, the cell lysates were cleared by centrifugation at 21 000 g for 10 min at 4°C.
Purification of GST‐fusion proteins. Purification of GST‐fusion proteins has been described previously.( 12 , 13 ) GST‐fusion proteins prepared as described above were bound to glutathione‐sepharose 4B (GE Healthcare Biosciences) in a 50% slurry in buffer X for 4 h at 4°C, washed three times with buffer X, and eluted with 50 mmol/L Tris‐HCl (pH 8.0) and 20 mmol/L reduced glutathione, according to the manufacturer's protocol (GE Healthcare Biosciences). Purified GST‐fusion proteins were used in electrophoretic mobility shift assays (EMSAs).
GST pulldown assay. GST‐HMGB1 or its deletion mutants immobilized on glutathione‐sepharose 4B were incubated with soluble bacterial extracts containing the indicated fusion proteins for 2 h at 4°C in buffer X. Bound samples were washed three times with buffer X and subjected to Western blotting analysis with anti‐HA or anti‐Thio antibody.
EMSA. The sequences of the oligonucleotides used as probes in EMSAs were 5′‐GGATCTCGAGCAGGAAGTTCGA‐3′ and 5′‐GGTCGAACTTCCTGCTCGAGAT‐3′. Human Ets binding sites are italicized. Preparation of 32P‐labeled oligonucleotide probes and EMSAs with purified GST‐fusion proteins were performed as described previously.( 12 , 13 ) Briefly, oligonucleotides were annealed with their complementary strands. The double‐stranded products were end‐labeled with [α‐32P] dCTP (GE Healthcare Biosciences) using the Klenow fragment (Fermentas, Vilnius, Lithuania), and purified from gels. Then, GST‐fusion proteins were incubated for 5 min at room temperature in a final volume of 20 µL containing 10 mmol/L Tris‐HCl (pH 7.5), 50 mmol/L NaCl, 5 mmol/L MgCl2, 10 µmol/L ZnCl2, 1 mmol/L EDTA, 1 mmol/L DTT, 0.1 mg/mL bovine serum albumin (BSA), 10% glycerol, 0.05% NP‐40, and 4 ng of 32P‐oligonucleotide probe. The reaction mixtures were resolved by electrophoresis on 4% polyacrylamide gels (polyacrylamide/bisacrylamide, 80:1) at 10 V/cm for 90–120 min at room temperature in 0.5 × tris‐borate‐EDTA buffer (45 mmol/L Tris base, 45 mmol/L boric acid, and 1 mmol/L EDTA). Gels were dried and analyzed using a bio‐imaging analyzer (FLA2000).
Cytotoxicity analysis. The water‐soluble tetrazolium salt (WST‐8) assay was performed as described previously.( 12 , 13 ) Briefly, 2.5 × 103 PC3 cells transfected with the indicated amounts of siRNA were seeded into 96‐well plates. The following day, the indicated concentrations of H2O2 were applied. After 72 h, surviving cells were stained with TetraColor ONE (Seikagaku Corporation, Tokyo, Japan) for 90 min at 37°C. The absorbance was then measured at 450 nm.
Results
Prdx family gene expression under oxidative stress conditions. To examine whether oxidative stress can activate Prdx family gene expression, human prostate cancer PC3 cells were exposed either to 30 min of 1 mmol/L H2O2 or 4 h of hypoxia, followed by recovery/reoxygenation in a CO2 incubator. Western blot analysis showed a significant increase in the levels of Prdx1 and Prdx5 expression during recovery/reoxygenation (Fig. 1a). Similar results were also obtained by using human cancer KB cells (Suppl. Fig. 1a). Analysis of the nucleotide sequences of the promoters of Prdx family genes revealed multiple Ets transcription factor binding sites in the promoter regions of Prdx1 and Prdx5, suggesting that Ets transcription factors might mediate the stress induction of both Prdx1 and Prdx5.
Figure 1.

H2O2 or hypoxia followed by recovery/reoxygenation increases Ets1, Ets2, peroxiredoxin1 (Prdx1), and Prdx5 expression. (a,b) PC3 cells were treated with or without 1 mmol/L H2O2 for 30 min after which time the media were replaced with fresh media and cells were recovered for the indicated time periods, or hypoxia for 4 h followed by reoxygenation for the indicated time periods. Whole‐cell extracts (100 µg) were subjected to SDS‐PAGE, and Western blotting analysis was performed using the indicated antibodies. Immunoblotting of BAF57 is shown as a loading control. Relative intensity is shown at the bottom of the panel. (c) PC3 cells were treated as described in (a,b). Total RNA (20 µg) was separated on a 1% formaldehyde‐agarose gel and transferred to a Hybond N+ membrane. Northern blotting analysis was performed with the indicated cDNA probes. Gel staining with ethidium bromide is also shown.
Ets transcription factor expression under oxidative stress conditions. As expected, the expression levels of both Ets1 and Ets2 were also increased during recovery/reoxygenation after treatment with H2O2 and hypoxia in PC3 cells. More than two‐fold elevated expression of both Ets1 and Ets2 was observed after 12 h of H2O2 treatment (Fig. 1b), suggesting that stress induction of Prdx genes might be regulated by the transcription factors Ets1 and Ets2. In KB cells, similar results were also obtained (Suppl. Fig. 1b).
mRNA levels of Ets transcription factors and Prdx family genes under oxidative stress. To analyze the molecular mechanisms that up‐regulate both Ets and Prdx in response to oxidative stress, we examined the mRNA levels of these genes. Northern blotting analysis confirmed that the amounts of Ets1, Ets2, Prdx1, and Prdx5 mRNA were significantly increased in PC3 cells as a result of oxidative stress (Fig. 1c).
Ets proteins regulate Prdx1 and Prdx5 genes. To confirm these results, we first cloned the core promoter regions of all Prdx genes, including CpG islands, for reporter assays (Fig. 2a). Silencing of Ets expression significantly reduced the expression of Prdx1 and Prdx5, but not that of other Prdx genes (Fig. 2b). Reporter assays also showed that silencing of Ets expression significantly suppressed the promoter activities of both Prdx1 and Prdx5 genes to ~10–25% of the promoter activities of the other Prdx genes (Fig. 2c). Next, we performed cotransfection experiments using Ets1 and Ets2 expression plasmids. Both Ets1 and Ets2 could transactivate the Prdx1 and Prdx5 promoters, with ~three–four‐fold activation (Fig. 2d). Furthermore, Prdx1 and Prdx5 promoter activities were significantly induced during recovery/reoxygenation after oxidative stress (Fig. 2e).
Figure 2.
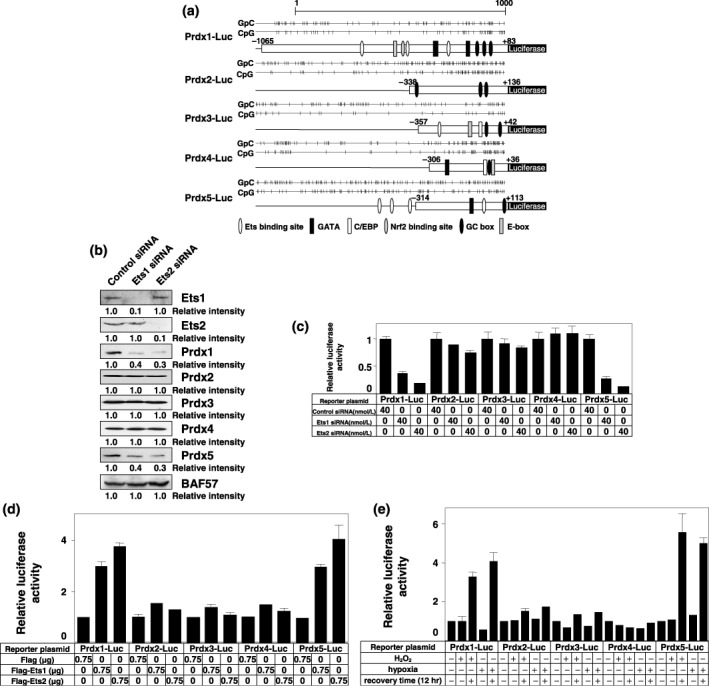
Ets transcription factors and oxidative stress regulate peroxiredoxin1 (Prdx1) and Prdx5 gene expression. (a) Schematic representations of Prdx1‐Luc, Prdx2‐Luc, Prdx3‐Luc, Prdx4‐Luc, and Prdx5‐Luc are shown with their CpG islands. (b) PC3 cells were transiently transfected with the indicated amounts of control, Ets1, or Ets2 siRNA. After 48 h, whole‐cell extracts (100 µg) were subjected to SDS‐PAGE, and Western blotting analysis was performed using the indicated antibodies. Immunoblotting of BAF57 is shown as a loading control. Relative intensity is shown at the bottom of the panel. (c) PC3 cells were transiently transfected with the indicated amounts of control, Ets1, or Ets2 siRNA followed by transfection with 1 µg of the indicated reporter plasmid at intervals of 12 h. The results were normalized to protein concentration measured using the Bradford method. All values are representations of at least three independent experiments. The luciferase activity of each Prdx‐Luc alone corresponds to 1. Bars indicate ± SD (d) PC3 cells were transiently cotransfected with 0.75 µg of Flag, Flag‐Ets1, or Flag‐Ets2 expression plasmid in addition to 0.75 µg of the indicated reporter plasmid. A luciferase assay was performed as described in (c). The luciferase activity of each Prdx‐Luc alone corresponds to 1. Bars indicate ± SD. (e) PC3 cells were transiently transfected with 1 µg of the indicated reporter plasmid. Thirty‐six hours post‐transfection, cells were incubated under normal conditions, treated with 1 mmol/L H2O2 for 30 min or incubated in hypoxic conditions for 4 h; cells were either lyzed immediately or after recovery/reoxygenation for 12 h. A luciferase assay was performed as described in (c). The luciferase activity of each Prdx‐Luc construct without oxidative stress corresponds to 1. Bars indicate ± SD.
To prove that Ets transcription factors were involved in the expression of Prdx1 and Prdx5 genes, we introduced the mutation in the Ets binding sites of both Prdx1 and Prdx5 promoters and examined the transactivation of reporter genes. The transfection of either Ets1 or Ets2 transactivated the wild‐type promoters of both Prdx1 and Prdx5 genes again. However, this transactivation was completely abolished by the introduction of mutations into Ets binding sites (Fig. 3a,b).
Figure 3.
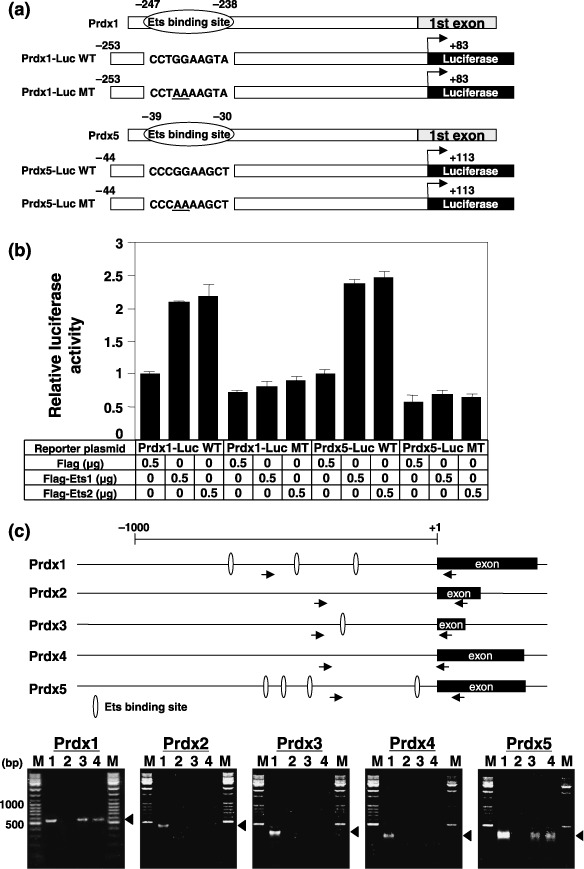
Ets transcription factors regulate peroxiredoxin1 (Prdx1) and Prdx5 gene expression through Ets binding sites located in the promoter region. (a) Schematic representation of the promoter regions of the Prdx1 and Prdx5 genes. Prdx1‐Luc WT, Prdx1‐Luc MT, Prdx5‐Luc WT, and Prdx5‐Luc MT used in (b) are also shown. (b) PC3 cells were transiently cotransfected with the indicated amounts of Ets1 or Ets2 expression plasmids and 0.5 µg of the indicated reporter plasmids shown in (a). The results were normalized to protein concentration measured using the Bradford method. All values are representations of at least three independent experiments. The luciferase activity of Prdx1‐Luc WT or Prdx5‐Luc WT alone corresponds to 1. Bars indicate ± SD (c) Schematic representation of the promoter region and 5′ end of Prdxs genes. White circles and arrows indicate Ets binding sites and polymerase chain reaction primer regions, respectively. Chromatin immunoprecipitation assays using PC3 cells transiently transfected with Flag vector (lane 2), Flag‐Ets1 plasmid (lane 3), or Flag‐Ets2 plasmid (lane 4) was performed using an anti‐Flag (M2) antibody. Soluble chromatin (lane 1) and immunoprecipitated DNAs (lane 2–4) were amplified by polymerase chain reaction using specific primer pairs for the Prdxs promoters (Prdx1, –491 to +83 bp; Prdx2, –338 to +136 bp; Prdx3, –357 to +42 bp; Prdx4, –306 to +36 bp; Prdx5; –264 to +113 bp). Lane M shows a DNA size marker and the arrowhead indicates PCR products (574 bp for Prdx1, 474 bp for Prdx2, 399 bp for Prdx3, 342 bp for Prdx4, 377 bp for Prdx5).
To determine whether the promoters of both Prdx1 and Prdx5 are direct target of Ets transcription factors, we performed ChIP assays covering the proximal promoter segments of five Prdx genes. We initially tried these experiments using commercial antibodies against Ets1 and Ets2, but they did not work well. Then, we employed transient transfection into PC3 cells with either Flag‐tagged Ets1 or Ets2 expression plasmid. After 48 h, ChIP assays were performed with anti‐Flag antibody. As shown in Fig. 3c, we found that Ets1 and Ets2 were observed in the promoters of both Prdx1 and Prdx5 genes. However, no binding of Ets was observed in Prdx2, Prdx3, and Prdx4 gene promoters. These results suggest that Ets binding sites in the proximal region of both Prdx1 and Prdx5 are the target of Ets binding in vivo.
Ets proteins interact with HMG protein. To investigate the mechanism of Ets‐mediated transcriptional regulation, we previously used a membrane pulldown assay to identify proteins that interact with Ets.( 15 ) One of the candidate proteins we found to preferentially interact with Ets was HMGB1. Coimmunoprecipitation experiments using expression plasmids clearly showed that both Ets1 and Ets2 interact with HMGB1 (Fig. 4a). Moreover, endogenous Ets1 protein was shown to interact with HMGB1 in human prostate cancer PC3 cells (Fig. 4b). We also observed that Ets1 interacts with Sp1 as a positive control.( 16 ) On the other hand, Ets1 could not interact with YB‐1 (Fig. 4b). Next, we mapped the domain in Ets proteins required for interactions with HMGB1 by a pulldown assay, using a GST‐fusion protein containing HMGB1 and HA‐ or ThioHis‐fusion proteins containing deletion mutants of Ets1 and Ets2 (Fig. 4c). A fusion protein lacking the D domain and part of the transcription‐activating domain was unable to bind HMGB1 (Fig. 4d). We next mapped the domain of HMGB1 responsible for the interaction with Ets1 and Ets2. A GST‐fusion protein containing HMGB1 with a deletion of the A box was unable to interact with Ets1 and Ets2 (Fig. 4e).
Figure 4.

Ets transcription factors interact with HMGB1. (a) Whole‐cell extracts (300 µg) prepared from PC3 cells cotransfected with 1 µg of each of the indicated expression plasmids were immunoprecipitated with anti‐Flag (M2) antibody. The resulting immunocomplexes and whole‐cell extracts (30 µg) were subjected to SDS‐PAGE, and Western blotting analysis was performed using anti‐HA and anti‐Flag (M2) antibodies. (b) Nuclear extracts (500 µg) prepared from PC3 cells were immunoprecipitated with 2 µg of rabbit IgG or anti‐Ets1 antibody. The resulting immunocomplexes and nuclear extracts (100 µg) were subjected to SDS‐PAGE, and Western blotting analysis was performed using indicated antibodies. (c) Schematic representations of GST‐, HA‐, or ThioHis‐fusion proteins containing deletion mutants of Ets1, Ets2, and HMGB1. TAD, transcription activating domain; DBD, DNA binding domain. (d) Equal amounts of GST and GST‐HMGB1 fusion proteins immobilized on glutathione‐sepharose 4B were incubated with HA‐ or ThioHis‐fusion proteins containing deletion mutants of Ets1 and Ets2 shown in (c). Bound protein samples and 10% of input were subjected to SDS‐PAGE, and Western blotting analysis was performed using anti‐HA or anti‐Thio antibody. Gel staining with Coomassie Brilliant Blue is also shown. (e) Equal amounts of GST, GST‐HMGB1, GST‐HMGB1‐ΔA, and GST‐HMGB1‐ΔB fusion proteins immobilized on glutathione‐sepharose 4B were incubated with HA‐Ets1 or HA‐Ets2 fusion proteins. Bound protein samples and 10% of input were subjected to SDS‐PAGE, and Western blotting analysis was performed using an anti‐HA antibody. Gel staining with Coomassie Brilliant Blue is also shown.
HMGB1 protein increases the DNA binding affinity of Ets proteins and enhances Prdx genes. It has been previously shown that HMGB1 can stimulate the DNA binding of p53.( 17 ) We examined the influence of HMGB1 on the sequence‐specific binding of Ets to DNA. GST‐fusion proteins for EMSAs were purified and stained with Coomassie Brilliant Blue (Fig. 5a). Fig. 5b shows the binding of Ets1 and Ets2 in the presence and absence of HMGB1. HMGB1 protein markedly enhanced the binding of Ets to DNA. HMGB1 protein increased the DNA binding affinity of Ets in a dose‐dependent manner (Fig. 5b). We next examined whether HMGB1 could transactivate the Ets‐dependent promoters of Prdx genes. As shown in Fig. 5c, the promoter activities of Prdx1 and Prdx5 were greatly enhanced by ~five–10‐fold by HMGB1 overexpression. Next, we tested whether HMGB1 as well as Ets transcription factors can increase expression of endogeneous Prdx genes. As shown in Fig. 5d, expression of HMGB1 could increase expression of both endogeneous Prdx1 and Prdx5 genes. Transfection of both Ets1 and Ets2 expression plasmids could also increase expression of these genes. These data also support the results shown in Fig. 2c.
Figure 5.
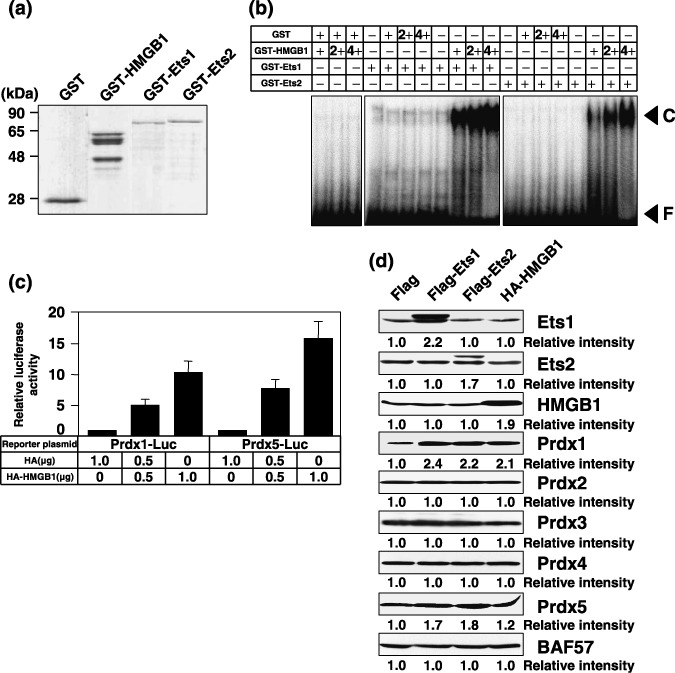
HMGB1 increases the DNA binding affinity of Ets and enhances the promoter activity of peroxiredoxin (Prdx) genes. (a) Purified GST, GST‐HMGB1, GST‐Ets1, and GST‐Ets2 fusion proteins used for EMSAs were stained with Coomassie Brilliant Blue. (b) GST or GST‐HMGB1 fusion proteins (+, 50 ng; 2+, 100 ng; 4+, 200 ng) were mixed with GST‐Ets1 or GST‐Ets2 fusion proteins (+, 50 ng), and incubated with 4 ng of 32P‐labeled oligonucleotides containing the Ets binding site. Reaction mixtures were resolved by electrophoresis on 4% polyacrylamide gels in 0.5 × tris‐borate‐EDTA buffer. Gels were dried and analyzed using a bio‐imaging analyzer (FLA2000). F and C indicate free probe and DNA‐protein complex, respectively. (c) PC3 cells were transiently cotransfected with the indicated amounts of HA or HA‐HMGB1 expression plasmid in addition to 0.5 µg of Prdx1‐Luc or Prdx5‐Luc. The results were normalized to protein concentration, which was measured using the Bradford method. All values are representations of at least three independent experiments. The luciferase activity of Prdx1‐Luc alone or Prdx5‐Luc alone corresponds to 1. Bars indicate ± SD (d) PC3 cells were transiently transfected with 2 µg of the indicated expression plasmid. After 48 h, whole‐cell extracts (100 µg) were subjected to SDS‐PAGE, and Western blotting analysis was performed using the indicated antibodies. Immunoblotting of BAF57 is shown as a loading control. Relative intensity is shown at the bottom of the panel.
Down‐regulation of Ets1, Ets2, Prdx1, or Prdx5 sensitizes cells to oxidative stress. Initially, we analyzed whether the expression of Prdx genes correlates with sensitivity to H2O2. Knockdown of each Prdx gene by siRNA rendered cells more sensitive to H2O2 (Fig. 6a). We next examined whether Ets1 and Ets2 could protect cells against oxidative stress. As shown in Fig. 6b, silencing of either Ets1 or Ets2 sensitized cells to oxidative stress, suggesting that Ets activity influences the sensitivity to ROS through the expression of Prdx1 and Prdx5.
Figure 6.
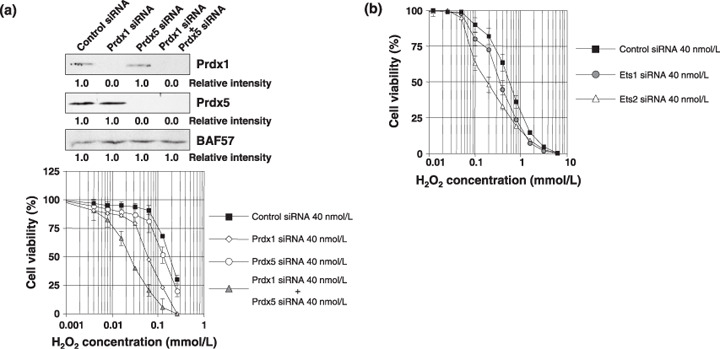
Down‐regulation of Ets1, Ets2, peroxiredoxin1 (Prdx1), or Prdx5 sensitizes cells to oxidative stress. (a, upper panel) PC3 cells were transiently transfected with 40 nmol/L of control, Prdx1, and/or Prdx5 siRNAs. After 48 h, whole‐cell extracts (100 µg) were subjected to SDS‐PAGE, and Western blotting analysis was performed using the indicated antibodies. Immunoblotting of BAF57 is shown as a loading control. Relative intensity is shown at the bottom of the panel. (lower panel) 2.5 × 103 PC cells were transfected with 40 nmol/L of Control siRNA (black square), Prdx1 siRNA (white triangle), Prdx5 siRNA (white circle), or Prdx1 siRNA and Prdx5 siRNA (gray triangle). The following day, various concentrations of H2O2 were applied. After 72 h, cell survival was analyzed by a WST‐8 assay. Cell survival in the absence of H2O2 corresponds to 100%. All values are the means of at least three independent experiments. Bars indicate ± SD (b) 2.5 × 103 PC cells were transfected with 40 nmol/L of control siRNA (black square), Ets1 siRNA (gray circle), or Ets2 siRNA (white triangle). WST‐8 assay was performed as described in (a).
Discussion
ROS are highly reactive oxygen metabolites that include the superoxide radical, hydrogen peroxide (H2O2) and the hydroxyl radical.( 1 , 18 ) ROS can be produced under aerobic conditions or when an organism is exposed to a variety of stress conditions.( 1 , 18 ) Human tumor cells produce significant amounts of ROS. To protect cells from oxidative stress, all organisms are equipped with antioxidant proteins, including superoxide dismutase/catalase, glutathione, and many kinds of peroxidase.( 1 , 18 ) Peroxiredoxins (Prdxs) are a new type of antioxidant protein that reduces ROS. Prdx1 and Prdx2 are mainly found in the cytoplasm, Prdx3 is present in mitochondria, and Prdx4 is secreted into the extracellular space. Prdx5 is an atypical, cytosolic type, which possesses more effective antioxidant activity against ROS than other Prdxs. It has been shown that ROS are involved in signaling in biological processes, including cell proliferation and apoptosis.( 1 ) The expression of Prdxs is closely associated with human cancers, such as thyroid cancer,( 19 ) breast cancer,( 4 ) and lung cancer.( 3 ) However, little is known about the regulation of Prdx gene expression in cancer cells. We speculated that Prdxs may be categorized into stress‐inducible and non‐inducible types.
Identification of the target genes regulated by specific transcription factors and cofactors interacting with these transcription factors is critical for understanding gene expression. Our observations allow us to construct a model that shows a novel relation between Ets transcription factors, HMG proteins, and Prdx gene expression (Fig. 7). The experimental data clearly demonstrate that Prdxs can be categorized into two groups from the viewpoint of transcriptional regulation. Prdx1 and Prdx5 are stress inducible, whereas the other Prdxs are constitutively expressed (Fig. 1a and Suppl. Fig. 1a). In addition, we showed that Ets1 and Ets2 are key transcription factors for the stress induction of Prdx1 and Prdx5 expression (1, 2, 3), and that HMGB1 functions as a coactivator through direct interactions with Ets1 and Ets2 (4, 5). Although we found one Ets binding site in the promoter region of Prdx3 (Fig. 2a), the expression of Prdx3 was not induced by oxidative stress or by the transfection of Ets expression plasmids (1, 2). This site is also a high‐affinity binding site of the other transcription factor NRF2, indicating that this site of the Prdx3 promoter may not function as an Ets binding site. Furthermore, it has been shown that Prdx3 expression is regulated by c‐Myc, suggesting that E‐box located in the core promoter region may function mainly through c‐Myc dependent transcription.( 20 )
Figure 7.
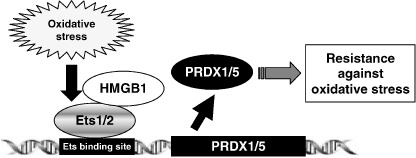
A model that shows a novel relation between peroxiredoxin (Prdx) gene expression and Ets/HMGB1 transcription system. Oxidative stress induces the expression of Ets transcription factors. Then, Ets transcription factors induce both Prdx1 and Prdx5 expression via binding to Ets binding site. HMG proteins enhance the DNA binding ability of Ets transcription factors and function as a coactivator. Increased Prdxs lead to resistance against oxidative stress.
The human Ets transcription factor family comprises more than 25 members that retain a conserved DNA binding domain called the Ets domain.( 21 , 22 ) The Ets transcription factor family is involved in various important biological processes.( 23 , 24 , 25 ) Protein–protein interactions often regulate DNA binding, subcellular localization, and transcriptional activity. So far, many partners that interact with Ets family members have been identified. There has been only one report on the interaction between an Ets family member and an HMG protein.( 26 ) In the present study, we showed that Ets1 and Ets2 interact with HMGB1, and that this molecular interaction is involved not only in the DNA binding ability of these transcription factors, but also in the transactivation of their target genes (Fig. 5). The region in Ets1 and Ets2 that is required for the interaction with HMGB1 is located between amino acid residues 250–330, and includes a phosphorylation site.( 27 ) Since the Ets domain is not necessary for this interaction, HMGB1 may not participate in target gene selection, but exclusively inhibit the Ets domain–dependent interaction with other molecules. As in vitro phosphorylation of Ets proteins has been shown to inhibit sequence‐specific DNA binding,( 28 ) the molecular interaction with HMGB1 might inhibit phosphorylation of Ets proteins and thereby stimulate sequence‐specific DNA binding.
Both Ets proteins and HMGB1 have been shown to be highly expressed in tumor cells.( 29 , 30 , 31 ) Thus, we speculate that mutual cooperation of these proteins is involved in various aspects of tumor biology, including transformation, antiapoptotic ability, and malignant progression.( 30 , 32 , 33 ) Ets transcription factors have been shown to be involved in tumor angiogenesis and metastasis.( 30 , 34 , 35 ) The expression levels of Prdx3, Prdx4, and Prdx5 were significantly higher in tumors that were poorly differentiated. A significant positive association between prognosis and Prdx expression has previously been shown in breast cancers.( 4 ) Taken together, these data do not contradict our results that Prdx1 and Prdx5 are Ets target genes. As shown in Fig. 6, our data clearly indicate that Ets activity influences sensitivity to ROS through the expression of both Prdx1 and Prdx5 genes. These results supports previous reports that overexpression of either Prdx1 or Prdx5 contributes to protect cells against apoptosis caused by oxidative stress.( 36 , 37 , 38 , 39 ) Thus, both Prdx1 and Prdx5 may be required for cancer cells to remain viable under conditions of oxidative stress. There are also data to show that the level of ROS is increased by the knockdown of either Prdx1 or Prdx5 expression.( 40 , 41 ) Therefore, the expression of Ets transcription factors might play a critical role in the early development of tumors in a hypoxic environment. Further investigations are required to show the correlation between Ets and Prdx expression in clinical tumors.
In summary, although Prdxs function as antioxidant proteins, Prdx1 and Prdx5 expression are stress inducible. The current work suggests the presence of functional differences among members of the Prdx family and reveals important information contributing to our understanding of tumor biology.
Supporting information
Fig. S1. H2O2 or hypoxia followed by recovery/reoxygenation increases Ets1, Ets2, peroxiredoxin1 (Prdx1), and Prdx5 expression in KB cells. (a,b) (left panel) KB cells were treated with or without 1 mmol/L H2O2 for 30 min, after which time the media were replaced with fresh media and cells were recovered for the indicated time periods. (right panel) KB cells were treated with or without hypoxia for 4 h followed by reoxygenation for the indicated time periods. Whole‐cell extracts (100 µg) were subjected to SDS‐PAGE, and Western blotting analysis was performed using the indicated antibodies. Immunoblotting of BAF57 is shown as a loading control. Relative intensity is shown at the bottom of the panel.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Acknowledgments
This work was supported in part by the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Mext), Kakenhi (13218132 and 17590257), Kobayashi Institute for Innovative Cancer Chemotherapy, and a Grant‐in‐Aid for Cancer Research from the Fukuoka Cancer Society, Japan. We greatly appreciate the gift of anti‐Prdx3 antibody from Dr H. Nanri (Seinan Jogakuin University, Kitakyushu, Japan). We would like to thank Satoko Takazaki for her technical assistance.
References
- 1. Finkel T. Oxygen radicals and signaling. Curr Opin Cell Biol 1998; 10: 248–53. [DOI] [PubMed] [Google Scholar]
- 2. Neumann CA, Krause DS, Carman CV et al . Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature 2003; 424: 561–5. [DOI] [PubMed] [Google Scholar]
- 3. Chang JW, Jeon HB, Lee JH et al . Augmented expression of peroxiredoxin I in lung cancer. Biochem Biophys Res Commun 2001; 289: 507–12. [DOI] [PubMed] [Google Scholar]
- 4. Karihtala P, Mäntyniemi A, Kang SW, Kinnula VL, Soini Y. Peroxiredoxins in breast carcinoma. Clin Cancer Res 2003; 9: 3418–24. [PubMed] [Google Scholar]
- 5. Quan C, Cha EJ, Lee HL, Han KH, Lee KM, Kim WJ. Enhanced expression of peroxiredoxin I and VI correlates with development, recurrence and progression of human bladder cancer. J Urol 2006; 175: 1512–16. [DOI] [PubMed] [Google Scholar]
- 6. Chen MF, Keng PC, Shau H et al . Inhibition of lung tumor growth and augmentation of radiosensitivity by decreasing peroxiredoxin I expression. Int J Radiat Oncol Biol Phys 2006; 64: 581–91. [DOI] [PubMed] [Google Scholar]
- 7. Ohga T, Koike K, Ono M et al . Role of the human Y box‐binding protein YB‐1 in cellular sensitivity to the DNA‐damaging agents cisplatin, mitomycin C, and ultraviolet light. Cancer Res 1996; 56: 4224–8. [PubMed] [Google Scholar]
- 8. Imamura T, Izumi H, Nagatani G et al . Interaction with p53 enhances binding of cisplatin‐modified DNA by high mobility group 1 protein. J Biol Chem 2001; 276: 7534–40. [DOI] [PubMed] [Google Scholar]
- 9. Araki M, Nanri H, Ejima K et al . Antioxidant function of the mitochondrial protein SP‐22 in the cardiovascular system. J Biol Chem 1999; 274: 2271–8. [DOI] [PubMed] [Google Scholar]
- 10. Ise T, Nagatani G, Imamura T et al . Transcription factor Y‐box binding protein 1 binds preferentially to cisplatin‐modified DNA and interacts with proliferating cell nuclear antigen. Cancer Res 1999; 59: 342–6. [PubMed] [Google Scholar]
- 11. Uramoto H, Izumi H, Ise T et al . p73 Interacts with c‐Myc to regulate Y‐box‐binding protein‐1 expression. J Biol Chem 2002; 277: 31694–702. [DOI] [PubMed] [Google Scholar]
- 12. Igarashi T, Izumi H, Uchiumi T et al . Clock and ATF4 transcription system regulates drug resistance in human cancer cell lines. Oncogene 2007; 26: 4749–60. [DOI] [PubMed] [Google Scholar]
- 13. Wakasugi T, Izumi H, Uchiumi T et al . ZNF143 interacts with p73 and is involved in cisplatin resistance through the transcriptional regulation of DNA repair genes. Oncogene 2007; 26: 5194–203. [DOI] [PubMed] [Google Scholar]
- 14. Ryuto M, Ono M, Izumi H et al . Induction of vascular endothelial growth factor by tumor necrosis factor alpha in human glioma cells. Possible roles of SP‐1. J Biol Chem 1996; 271: 28220–8. [DOI] [PubMed] [Google Scholar]
- 15. Shiota M, Izumi H, Onitsuka T et al . Twist and p53 reciprocally regulate target genes via direct interaction. Oncogene Epub ahead of print. [Cited 26 May 2008.] [DOI] [PubMed] [Google Scholar]
- 16. Dittmer J, Pise‐Masison CA, Clemens KE, Choi KS, Brady JN. Interaction of human T‐cell lymphotropic virus type I Tax, Ets1, and Sp1 in transactivation of the PTHrP P2 promoter. J Biol Chem 1997; 272: 4953–8. [DOI] [PubMed] [Google Scholar]
- 17. Jayaraman L, Moorthy NC, Murthy KG, Manley JL, Bustin M, Prives C. High mobility group protein‐1 (HMG‐1) is a unique activator of p53. Genes Dev 1998; 12: 462–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature 2000; 408: 239–47. [DOI] [PubMed] [Google Scholar]
- 19. Yanagawa T, Ishikawa T, Ishii T et al . Peroxiredoxin I expression in human thyroid tumors. Cancer Lett 1999; 145: 127–32. [DOI] [PubMed] [Google Scholar]
- 20. Wonsey DR, Zeller KI, Dang CV. The c‐Myc target gene PRDX3 is required for mitochondrial homeostasis and neoplastic transformation. Proc Natl Acad Sci USA 2002; 99: 6649–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Oikawa T. ETS transcription factors: possible targets for cancer therapy. Cancer Sci 2004; 95: 626–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Seth A, Watson DK. ETS transcription factors and their emerging roles in human cancer. Eur J Cancer 2005; 41: 2462–78. [DOI] [PubMed] [Google Scholar]
- 23. Maroulakou IG, Bowe DB. Expression and function of Ets transcription factors in mammalian development: a regulatory network. Oncogene 2000; 19: 6432–42. [DOI] [PubMed] [Google Scholar]
- 24. Bartel FO, Higuchi T, Spyropoulos DD. Mouse models in the study of the Ets family of transcription factors. Oncogene 2000; 19: 6443–54. [DOI] [PubMed] [Google Scholar]
- 25. Sementchenko VI, Watson DK. Ets target genes: past, present and future. Oncogene 2000; 19: 6533–48. [DOI] [PubMed] [Google Scholar]
- 26. John S, Reeves RB, Lin JX et al . Regulation of cell‐type‐specific interleukin‐2 receptor alpha‐chain gene expression: potential role of physical interactions between Elf‐1, HMG‐I (Y), and NF‐kappa B family proteins. Mol Cell Biol 1995; 15: 1786–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Koizumi S, Fisher RJ, Fujiwara S et al . Isoforms of the human ets‐1 protein: generation by alternative splicing and differential phosphorylation. Oncogene 1990; 5: 675–81. [PubMed] [Google Scholar]
- 28. Fisher RJ, Fivash M, Casas‐Finet J et al . Real‐time DNA binding measurements of the ETS1 recombinant oncoproteins reveal significant kinetic differences between the p42 and p51 isoforms. Protein Sci 1994; 3: 257–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sapi E, Flick MB, Rodov S, Kacinski BM. Ets‐2 transdominant mutant abolishes anchorage‐independent growth and macrophage colony‐stimulating factor‐stimulated invasion by BT20 breast carcinoma cells. Cancer Res 1998; 58: 1027–33. [PubMed] [Google Scholar]
- 30. Sementchenko VI, Schweinfest CW, Papas TS, Watson DK. ETS2 function is required to maintain the transformed state of human prostate cancer cells. Oncogene 1998; 17: 2883–8. [DOI] [PubMed] [Google Scholar]
- 31. Nagatani G, Nomoto M, Takano H et al . Transcriptional activation of the human HMG1 gene in cisplatin‐resistant human cancer cells. Cancer Res 2001; 61: 1592–7. [PubMed] [Google Scholar]
- 32. Zhang C, Kavurma MM, Lai A, Khachigian LM. Ets‐1 protects vascular smooth muscle cells from undergoing apoptosis by activating p21WAF1/Cip1: ETS‐1 regulates basal and inducible p21WAF1/Cip1 transcription via distinct cis‐acting elements in the p21WAF/Cip1 promoter. J Biol Chem 2003; 278: 27903–9. [DOI] [PubMed] [Google Scholar]
- 33. Myers E, Hill AD, Kelly G et al . Associations and interactions between Ets‐1 and Ets‐2 and coregulatory proteins, SRC‐1, AIB1, and NCoR in breast cancer. Clin Cancer Res 2005; 11: 2111–22. [DOI] [PubMed] [Google Scholar]
- 34. Elvert G, Kappel A, Heidenreich R et al . Cooperative interaction of hypoxia‐inducible factor‐2alpha (HIF‐2alpha) and Ets‐1 in the transcriptional activation of vascular endothelial growth factor receptor‐2 (Flk‐1). J Biol Chem 2003; 278: 7520–30. [DOI] [PubMed] [Google Scholar]
- 35. Ito H, Duxbury M, Benoit E et al . Prostaglandin E2 enhances pancreatic cancer invasiveness through an Ets‐1‐dependent induction of matrix metalloproteinase‐2. Cancer Res 2004; 64: 7439–46. [DOI] [PubMed] [Google Scholar]
- 36. Bae JY, Ahn SJ, Han W, Noh DY. Peroxiredoxin I and II inhibit H2O2‐induced cell death in MCF‐7 cell lines. J Cell Biochem 2007; 101: 1038–45. [DOI] [PubMed] [Google Scholar]
- 37. Berggren MI, Husbeck B, Samulitis B, Baker AF, Gallegos A, Powis G. Thioredoxin peroxidase‐1 (peroxiredoxin‐1) is increased in thioredoxin‐1 transfected cells and results in enhanced protection against apoptosis caused by hydrogen peroxide but not by other agents including dexamethasone, etoposide, and doxorubicin. Arch Biochem Biophys 2001; 392: 103–9. [DOI] [PubMed] [Google Scholar]
- 38. Yuan J, Murrell GA, Trickett A, Landtmeters M, Knoops B, Wang MX. Overexpression of antioxidant enzyme peroxiredoxin 5 protects human tendon cells against apoptosis and loss cellular function during oxidative stress. Biochem Biophys Acta 2004; 1693: 37–45. [DOI] [PubMed] [Google Scholar]
- 39. Banmeyer I, Marchand C, Verhaeghe C, Vucic B, Rees JF, Knoops B. Overexpression of human peroxiredoxin 5 in subcellular compartments of Chinese hamster ovary cells: effects on Cytotoxicity and DNA damage caused by peroxides. Free Radic Biol Med 2004; 36: 65–77. [DOI] [PubMed] [Google Scholar]
- 40. Egler RA, Fernandes E, Rothermund K et al . Regulation of reactive oxygen species, DNA damage, and c‐Myc function by peroxiredoxin 1. Oncogene 2005; 24: 8038–50. [DOI] [PubMed] [Google Scholar]
- 41. Kropotov AV, Grudinkin PS, Pleskach NM, Gavrilov BA, Tomilin NV, Zhivotovsky B. Downregulation of peroxiredoxin V stimulates formation of etoposide‐induced double‐strand DNA breaks. FEBS Lett 2004; 572: 75–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. H2O2 or hypoxia followed by recovery/reoxygenation increases Ets1, Ets2, peroxiredoxin1 (Prdx1), and Prdx5 expression in KB cells. (a,b) (left panel) KB cells were treated with or without 1 mmol/L H2O2 for 30 min, after which time the media were replaced with fresh media and cells were recovered for the indicated time periods. (right panel) KB cells were treated with or without hypoxia for 4 h followed by reoxygenation for the indicated time periods. Whole‐cell extracts (100 µg) were subjected to SDS‐PAGE, and Western blotting analysis was performed using the indicated antibodies. Immunoblotting of BAF57 is shown as a loading control. Relative intensity is shown at the bottom of the panel.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item