

Abstract
Resistance of ovarian mucinous adenocarcinoma to standard chemotherapy with paclitaxel and carboplatin is associated with poor prognosis, and an effective treatment is needed. The present study aimed to identify an effective chemotherapy for ovarian mucinous adenocarcinoma. Five human ovarian mucinous adenocarcinoma cell lines (MN‐1, OMC‐1, RMUG‐L, RMUG‐S, TU‐OM‐1) were used in this study. Sensitivity of the cells to the anticancer agents was determined by 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide assay, and we assessed drug sensitivity by calculating the assay area under the curve for each agent. Protein expression was confirmed by Western blot analysis. We also examined the efficacy of combination chemotherapy on survival in a xenograft model of nude mice. The IC50 to anticancer agents ranged widely. The assay area under the curve indicated that two of five cell lines (MN‐1, TU‐OM‐1) were sensitive to oxaliplatin, 5‐fluorouracil and etoposide, and only one (TU‐OM‐1) was sensitive to 7‐ethyl‐10‐hydroxycamptothecin, which is an active metabolite of camptothecin. All cell lines were resistant to cisplatin and paclitaxel. The combination of oxaliplatin and 5‐fluorouracil resulted in additive or synergistic effects on all cell lines. The combination of oxaliplatin and 5‐fluorouracil significantly prolonged survival in a ovarian mucinous adenocarcinoma xenograft model of nude mice. Protein expression levels of the excision repair cross‐complementation group 1 were lower in oxaliplatin sensitive cell lines. Exposure to 5‐fluorouracil down‐regulated cross‐complementation group 1 expression in ovarian mucinous adenocarcinoma cells. We conclude that combination chemotherapy consisting of oxaliplatin and 5‐fluorouracil was an effective treatment for ovarian mucinous adenocarcinoma and may be a pivotal candidate for a novel treatment strategy. (Cancer Sci 2009; 100: 546–551)
Mucinous adenocarcinoma of the ovary (MAC) is the third most common type of epithelial ovarian cancers (EOC), comprising 10 to 12% of EOC. MAC appears to have a distinctly different clinical behavior from that of other EOC.( 1 , 2 , 3 ) Several studies show that MAC is often diagnosed at an early stage, and therefore, confers a relatively good prognosis.( 1 , 2 , 3 ) However, advanced MAC has a poorer prognosis than other histopathologic subgroups.( 4 , 5 , 6 ) MAC's low response (26–42%) to conventional platinum‐based chemotherapy is associated with poor prognosis because chemosensitivity is one of the main prognostic factors for patients with advanced EOC.( 4 , 5 , 6 , 7 , 8 , 9 ) Although MAC is known to be resistant to platinum‐ and taxane‐based chemotherapy, patients with EOC are usually treated with this first‐line chemotherapy regimen. A novel treatment strategy for advanced MAC is urgently needed.
Recently, new agents for EOC have been developed, including oxaliplatin (L‐OHP), etoposide (VP‐16) and camptothecin (CPT‐11).( 10 ) L‐OHP (1R,2R‐diaminocyclohexane oxalatoplatinum [II]) is a 1,2‐diaminocyclohexane (DACH) platinum compound with a partial lack of cross‐resistance to platinum analogs, cisplatin (CDDP) and carboplatin.( 11 , 12 , 13 ) VP‐16, which inhibits the activity of DNA topoisomerase II, is used as a second‐line treatment for patients with platinum‐resistant EOC.( 14 ) CPT‐11, which inhibits topoisomerase I by forming stable topoisomerase I–DNA cleavable complexes( 15 ) has been studied in relapsed EOC.( 16 ) The thymidylate synthase inhibitor, 5‐fluorouracil (5FU), was reported to be an active agent for recurrent EOC.( 17 , 18 )
We conducted the present study to identify an effective chemotherapy for MAC.
Materials and Methods
Cell lines and cell cultures. The five MAC cell lines used in the present study (MN‐1, OMC‐1, RMUG‐L, RMUG‐S, TU‐OM‐1) were obtained as follows: MN‐1 from Dr Yasuhiko Kiyozuka (Kansai Medical University, Osaka, Japan), OMC‐1 from Dr Tsuyoshi Saito (School of Medicine, Sapporo Medical University, Sapporo, Japan), and RMUG‐L and RMUG‐S from Dr Daisuke Aoki (Keio University, Tokyo, Japan). TU‐OM‐1 was established by our department. These cell lines were maintained in D‐MEM/Ham's F‐12 medium (Wako, Osaka, Japan) with 10% fetal bovine serum, 100 IU/mL penicillin and 50 µm/mL streptomycin in a humidified atmosphere containing 5% CO2 at 37°C.
Dose–response studies. The sensitivity of the cell lines to the anticancer agents was determined by 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐dyphenyltetrazolium bromide (MTT) assay.( 19 ) Briefly, cells were diluted with culture medium to the seeding density of 1–3 × 104 cells/mL, plated on 96‐well tissue culture plates at 100 µL/well (Sumitomo Bakelite, Tokyo, Japan), and incubated at 37°C overnight. The next day, the cells were treated continuously with 20 µL of various concentrations of the anticancer agents to obtain a dose–response curve for each agent. Each drug concentration was as follows: 1–100 µM CDDP (Sigma, St. Louis, MO, USA), 0.1–50 µM 5FU (Wako), 1–100 µM L‐OHP (Wako), 0.01–10 µM paclitaxel (PTX) (Sigma), 0.01–10 µM 7‐ethyl‐10‐hydroxycamptothecin (SN38) (Yakult Honsha Co., Tokyo, Japan), which is an active metabolite of CPT‐11, and 0.1–100 µM VP‐16 (Sigma).
After incubation for 72 h, 10 µL MTT solution (5 mg/mL) (Sigma) was added to each well, and the plates were incubated for another 4 h. At the end of that incubation, 100 µL dimethylsulfoxide (Sigma) was added to each well to solubilize the MTT formazan product. Absorbance at 570 nm was measured with a microplate reader (model 550; Bio‐Rad, Richmond, CA, USA). Growth inhibition was calculated as the percentage of viable cells compared with untreated cultures.
L‐OHP or CDDP was combined with 5FU, PTX, SN38 and VP‐16 at a fixed ratio that spanned the individual (IC50) of each drug. The 50% inhibitory concentration of a substance (IC50) was determined on the basis of the dose–effect curves, using a standard MTT assay. Median‐effect plot analyses and calculation of the combination index (CI) were analyzed by the method of Chou and Talalay.( 20 ) CalcuSyn software (Biosoft, Cambridge, UK) was used to analyze data from the MTT assays in which cells were exposed to agents alone or in combination with anticancer drugs. The computer program provides a measure of the combined agents in an additive or synergistic manner. Chou and Talalay( 20 ) defined CI, which assesses synergism (CI < 0.9), additive behavior (CI = 0.9–1.1) or antagonism (CI > 1.1).
To assess drug sensitivity, the assay area under the curve (AUC) was calculated by means of the following formula: initial concentration × t 1/2 × 1.44{1 – exp(−0.693 × 72/t 1/2)}. Initial concentration was determined based on the IC50 of each agent in the present study and t 1/2 is the in vitro half‐life of the drug at 37°C.( 21 ) The half‐lives of CDDP and VP‐16 are 18.5 and 60 h, respectively.( 22 ) We confirmed that the half‐life of L‐OHP was 23 h. Those of 5FU, PTX and SN38 were reported as stable in serum‐containing medium at 37°C in the presence of 5% CO2; therefore, the assay AUC of those drugs were calculated as the drug concentration × 72 h.( 23 ) The assay AUC was then compared with the clinically achievable AUC with a standard dose of each drug. If the calculated assay AUC was less than the clinically achievable AUC, the drug was defined as sensitive.
Western blot analysis. Cells were washed twice with phosphate‐buffered saline (PBS) and then lyzed in lysis buffer (50 mM Tris‐HCl, 125 mM NaCl, 0.1% Nonidet P‐40, 5 mM ethylenediaminetetraacetic acid, 50 mM NaF, 0.1% phenylmethylsulfonyl fluoride, protease inhibitors (Protease Inhibitor Cocktail Set I; Calbiochem, Darmstadt, Germany). Protein concentrations were measured against a standardized control by using a protein assay kit (Bio‐Rad Laboratories, Hercules, CA, USA). A total of 50 µg protein was separated by electrophoresis on a 10% polyacrylamide gel and transferred to a polyvinylidene difluoride membrane (Millipore, Bedford, MA, USA). The specific antibodies used were rabbit anti‐Akt antibody (1:1000 dilution; Cell Signaling Technology, Beverly, MA, USA), rabbit antiphospho‐Akt (serine 473) antibody (1:1000 dilution; Cell Signaling Technology), rabbit anti‐MEK1 antibody (1:1000 dilution; Cell Signaling Technology), rabbit antiphospho‐MEK1 (threonine 286) antibody (1:500 dilution; Cell Signaling Technology), rabbit anti‐Bcl2 antibody (1:1000 dilution; Cell Signaling Technology), rabbit anti‐Bcl‐xL (1:1000 dilution; Cell Signaling Technology), mouse anti‐ERCC1 antibody (1:100 dilution; Abcam), mouse anti‐XPF antibody (1:100 dilution; Abcam), mouse anti‐XRCC1 antibody (1:500 dilution; Abcam) and mouse anti‐Actin antibody (1:1000 dilution; Sigma). These were visualized with secondary antirabbit or antimouse IgG antibody coupled with horseradish peroxidase, using enhanced chemiluminescence according to the manufacturer's recommendation.
Annexin V staining. The Annexin V–PE Reagent (BioVision Research Products, Mountain View, CA, USA) was used to assess apoptosis in terms of the externalization of phosphatidylserine residues, according to the specifications of the manufacturer. Briefly, cells were washed twice with cold PBS and once with 1 × Annexin V binding buffer (10 mmol/L HEPES/NaOH [pH 7.4], 140 mmol/L NaCl, 2.5 mmol/L CaCl2). The cells were then stained with Annexin V–PE diluted 1:10 in 1 × Annexin V binding buffer for 15 min at room temperature. Finally, the cells were washed with 1 × Annexin V binding buffer, then 500 µL 1 × Annexin V binding buffer was added to each well and the cells analyzed with a flow cytometer (Olympus, Tokyo, Japan).
Ovarian cancer xenograft model. The present study was carried out at the Laboratory Animal Research Center under the control of the animal research committee, in accordance with the Guidelines for Animal Experimentation in the Faculty of Medicine, Tottori University, Yonago, Japan. For these experiments, TU‐OM‐1 cells in log‐phase growth were trypsinized, washed twice with PBS and centrifuged. Viable cells were counted, then 2 × 106 viable cells (in 0.5 mL PBS) were injected under aseptic conditions into the peritoneal cavities of female nude mice. Mice were then assigned randomly to one of four groups (five mice per group) and treatment was started five days later as follows. Group 1, intraperitoneal (i.p.) PBS weekly for 3 weeks; group 2, i.p. L‐OHP weekly (12.5 mg/kg per injection) for 3 weeks; group 3, i.p. 5FU weekly (25 mg/kg per injection) for 3 weeks; and group 4, i.p. L‐OHP and with 5FU weekly for 3 weeks.
Statistical analysis. Statistical analyses were carried out using the GraphPad Prism Version 5 program (GraphPad Software, San Diego, CA, USA). Data are presented as mean ± SD. Survival distributions were calculated using the Kaplan–Meier method, and the significance of apparent differences in survival distribution between groups was tested with log‐rank tests. P < 0.05 was considered statistically significant.
Results
Sensitivity to anticancer agents. The IC50 values of each cell line for anticancer agents are shown in Table 1. The IC50s ranged from 3.78 to 49.78 µM for CDDP, from 0.68 to 29.27 µM for 5FU, from 2.21 to 39.58 µM for L‐OHP, from 0.29 to 0.83 µM for PTX, from 0.02 to 4.18 µM for SN38 and from 0.31 to 38.72 µM for VP‐16, indicating that these cell lines showed various sensitivities to anticancer agents. When the assay AUC was compared with clinically achievable AUC using a standard dose of each drug, two of five cell lines (MN‐1, TU‐OM‐1) were defined as sensitive to L‐OHP, 5FU or VP‐16 and only one (TU‐OM‐1) was sensitive to SN38. All cell lines were resistant to CDDP and PTX (Table 2).
Table 1.
IC50 values to anticancer agents
| Cell lines | IC50 (µM) | |||||
|---|---|---|---|---|---|---|
| CDDP | 5FU | L‐OHP | PTX | SN38 | VP‐16 | |
| MN‐1 | 12.22 | 0.68 | 2.21 | 0.29 | 0.18 | 0.31 |
| OMC‐1 | 25.66 | 10.31 | 12.11 | 0.83 | 0.04 | 6.45 |
| RMUG‐L | 24.78 | 29.27 | 24.24 | 0.50 | 2.14 | 38.72 |
| RMUG‐S | 49.78 | 15.91 | 39.58 | 0.32 | 4.18 | 21.92 |
| TU‐OM‐1 | 3.78 | 4.50 | 2.91 | 0.67 | 0.02 | 0.65 |
5FU, 5‐fluorouracil; CDDP, cisplatin; L‐OHP, oxaliplatin; PTX, paclitaxel; SN38, 7‐ethyl‐10‐hydroxycamptothecin; VP‐16, etoposide.
Table 2.
Calculated assay AUC at IC50 and clinically achievable AUC for each drug
| Cell lines | Calculated assay AUC (µM/h) | |||||
|---|---|---|---|---|---|---|
| CDDP | 5FU | L‐OHP | PTX | SN38 | VP‐16 | |
| MN‐1 | 303.2 | 49.1 † | 62.7 † | 21.0 | 13.1 | 14.9 † |
| OMC‐1 | 637.2 | 742.3 | 343.9 | 60.1 | 3.0 | 314.9 |
| RMUG‐L | 615.4 | 2107.4 | 688.4 | 36.6 | 154.1 | 1888.9 |
| RMUG‐S | 1236.4 | 1144.8 | 1124.0 | 23.3 | 301.1 | 1069.3 |
| TU‐OM‐1 | 94.1 | 324.3 † | 82.6 † | 48.6 | 1.2 † | 31.7 † |
| CA‐AUC | 85.8 | 418.2 | 88.7 | 19.3 | 1.5 | 55.6 |
Sensitive.
5FU, 5‐fluorouracil; AUC, area under the curve; CA, clinically achievable; CDDP, cisplatin; L‐OHP, oxaliplatin; PTX, paclitaxel; SN38, 7‐ethyl‐10‐hydroxycamptothecin; VP‐16, etoposide.
Combination effects of oxaliplatin and 5‐fluorouracil or etoposide. We then examined cell proliferation after treating with both L‐OHP and 5FU or VP‐16, because these drugs were effective for two of five MAC cell lines. When L‐OHP was combined with 5FU, the CI values at an effective dose of 50 (effective dose means the percentage of inhibition of cell growth using the drug combinations in the experiment) were less than 1.1 for all of the five MAC cell lines. The CI values were less than 1.1 for only one cell line when L‐OHP was combined with VP‐16 (Fig. 1). Thus, combination treatment of L‐OHP with 5FU might have more cytotoxic effect on MAC cells than that with VP‐16.
Figure 1.
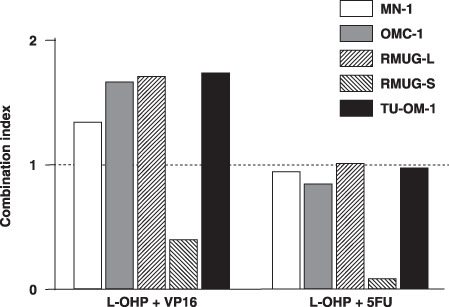
Effects of 5‐fluorouracil (5FU) are synergistic or additive with those of oxaliplatin (L‐OHP). L‐OHP was combined with 5FU or etoposide (VP‐16) at a fixed ratio that spanned the individual IC50 of each drug. The combination effects of L‐OHP and 5FU were synergistic or additive in all MAC cell lines tested. Data were analyzed by the method of Chou and Talalay( 20 ) to determine the combination index values. The results shown are an average of at least two independent experiments.
Next, to test whether combining L‐OHP with 5FU would suppress cell growth at the concentrations calculated by clinically achievable AUC, we treated MAC cell lines with 3.1 µM L‐OHP and 5.8 µM 5FU. Seventy‐two hours after exposure, growth inhibition was measured using the MTT assay. When L‐OHP was combined with 5FU, more than 50% of cell growth suppression was observed in four of five MAC cell lines (Fig. 2a). The number of Annexin V‐positive cells also increased additively 24 h after treating with L‐OHP and 5FU in all cell lines (Fig. 2b,c).
Figure 2.

The effects of oxaliplatin (L‐OHP) combined with 5‐fluorouracil (5FU) on mucinous adenocarcinoma (MAC) cell proliferation and apoptosis.(a) Five MAC cell lines were treated with 3.1 µM (L‐OHP) and/or 5.8 µM 5FU for 72 h compared with the control (phosphate‐buffered saline). Cell‐growth inhibition was determined using the 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐dyphenyltetrazolium bromide assay. Cell growth was suppressed more than 50% in four of five cell lines when L‐OHP was combined with 5FU at the same concentrations calculated by clinically achievable AUC. Points represent mean ± SD from six dishes. Treatment with L‐OHP combined with 5FU increased apoptotic cells. Cells were treated with 3.1 µM L‐OHP in the presence or absence of 5.8 µM 5FU for 24 h, then stained with Annexin V–PE. The number of apoptotic cells increased additively after treating with L‐OHP combined with 5FU in (b) MN‐1 cells and (c) TU‐OM‐1 cells. Similar results were obtained in the other three cell lines (data not shown). The results shown represent duplicate experiments.
Oxaliplatin combined with 5‐fluorouracil on survival in a mouse xenograft model. After confirming that L‐OHP combined with 5FU reduced cell viability and enhanced apoptosis in vitro, we examined the effect of combination treatment of L‐OHP and 5FU on survival in a xenograft model of MAC. Mice treated with both L‐OHP and 5FU survived significantly longer than those treated with PBS, 5FU or L‐OHP (P < 0.005) (Fig. 3). The median survival times were 188 days for L‐OHP with 5FU treatment, 83 days for PBS treatment, 96 days for 5FU treatment and 124 days for L‐OHP treatment. This finding indicates that L‐OHP combined with 5FU resulted in prolonged survival in nude mice bearing TU‐OM‐1 cells.
Figure 3.

Treatment with oxaliplatin (L‐OHP) combined with 5‐fluorouracil (5FU) prolongs survival in mice with implanted TU‐OM‐1 cells. Female nude mice (five per group) were given an intraperitoneal (i.p.) injection of 2 × 106 TU‐OM‐1 cells followed by weekly i.p. injections of 250 µL PBS, 12.5 mg/kg L‐OHP, and/or 25 mg/kg 5FU for 3 weeks (days 5, 12, 19). Treatment with L‐OHP and 5FU prolonged survival relative to treatment with PBS, L‐OHP or 5FU (P < 0.005).
Protein expression of cell‐survival and DNA repair pathways. The protein expression levels of Akt, phosphorylated (p) Akt, MEK, pMEK, Bcl2 and Bcl‐XL were not related to the sensitivity of anticancer agents tested (Fig. 4a). Among DNA repair proteins, higher ERCC1 expression was observed in L‐OHP resistant cells (OMC‐1, RMUG‐L, RMUG‐S) (Fig. 4b).
Figure 4.
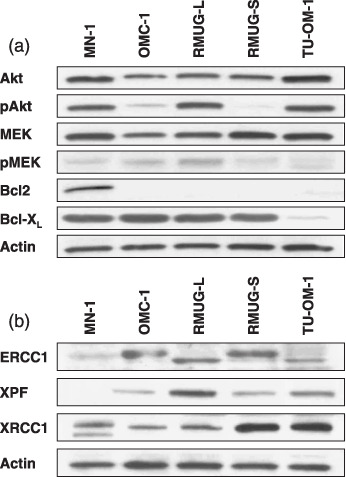
The high protein expression levels of ERCC1 in oxaliplatin‐resistant mucinous adenocarcinoma (MAC) cells. (a) Protein expression of the cell‐survival signaling pathways (Akt, MEK, Bcl2, and Bcl‐XL) and (b) DNA repair (ERCC1, XPF, and XRCC1) in MAC cells was determined by Western blot analysis. The results shown represent duplicate experiments.
Next, we examined the protein expression level of ERCC1 at 24 h after treatment with L‐OHP and/or 5FU. ERCC1 expression was suppressed after treating with 5FU with or without L‐OHP in RMUG‐L and TU‐OM‐1 cells (Fig. 5a). Similar results were obtained in the other three cell lines. Furthermore, Western blot analysis of tumor tissues verified that 5FU down‐regulated ERCC1 protein expression in tumor cells (Fig. 5b).
Figure 5.

5‐fluolouracil (5FU) down‐regulates ERCC1 expression. (a) Each of the cell lines was treated by 5FU with or without oxaliplatin (L‐OHP) for 24 h. The cells were then collected and protein expressions of ERCC1 were determined by Western blot analysis. After treatment with 5FU, the protein expression levels of ERCC1 were down‐regulated. The results shown represent duplicate experiments. (b) Nude mice bearing TU‐OM‐1 were treated with phosphate‐buffered saline (PBS) or 5FU intrapenitoneally for 24 h. Then, the expression levels of ERCC1 protein were determined by Western blot analysis. ERCC1 proteins were down‐regulated only in tumors from mice treated with 5FU. The results shown represent duplicate experiments.
Discussion
Determining cellular sensitivity is difficult due to the wide range of IC50 to anticancer agents. Additionally, protein binding of drug and drug stability in the culture medium are important pharmacodynamic factors. Assay AUC is a useful method to predict cellular sensitivity to anticancer agents, which correlates to clinical response.( 24 , 25 ) In the present study, we calculated assay AUC to assess drug sensitivity and found that two (MN‐1, TU‐OM‐1) of five cell lines were sensitive to VP‐16, 5FU and L‐OHP, and one cell line (TU‐OM‐1) to SN38, whereas all cell lines were resistant to CDDP and PTX, which have been used as a standard chemotherapy for EOC. Furthermore, combination treatment of L‐OHP with 5FU has more cytotoxic effect on MAC cells than that of L‐OHP with VP‐16 and suppressed cell proliferation more than 50% in four of five cell lines at the same concentrations calculated by clinically achievable AUC. We confirmed the combination effect of L‐OHP and 5FU in a MAC xenograft model. Combining L‐OHP and 5FU prolonged the survival of these mice compared with those treated with L‐OHP or 5FU alone. These data provide clear evidence that this combination therapy may be effective for MAC. To our knowledge, this is the first study to show that the combination of L‐OHP and 5FU is effective for MAC both in vitro and in vivo.
L‐OHP is a third‐generation platinum compound with a spectrum of activity and toxicity that differs from CDDP by the presence of a DACH ligand, although L‐OHP and CDDP act theoretically by the same mechanism of action.( 26 , 27 ) Like other DACH platinum compounds, L‐OHP has different sensitivity profiles in a broad range of cancer cell lines, and has shown activity in a number of cell lines, including ovarian cancer, that exhibit intrinsic or acquired resistance to CDDP.( 11 , 12 , 13 ) We also found two of five MAC cell lines to be sensitive to L‐OHP, although all cell lines were resistant to CDDP. Several explanations as to the mechanisms for this lack of cross‐resistance have been proposed. In contrast to CDDP, cellular accumulation of L‐OHP seems to be less dependent on copper transporter 1 (CTR1), which is the major copper influx transporter;( 28 ) a lower intracellular drug concentration and fewer platinum (Pt)–DNA adducts are sufficient for L‐OHP to exert its cytotoxicity;( 29 ) and mismatch repair protein does not bind to DACH–Pt–DNA adducts.( 30 )
Recently, several reports revealed the importance of the nucleotide excision repair (NER) process in the sensitivity to L‐OHP and CDDP.( 31 , 32 ) Furthermore, excision repair cross‐complementing rodent repair deficiency, complementation group 1 (ERCC1) is essential for NER process, and its mRNA or protein expression levels have been involved in L‐OHP sensitivity.( 31 , 32 , 33 ) Indeed, we observed high protein expression levels of ERCC1 in L‐OHP‐resistant cell lines. These results suggest that ERCC1 protein expression level may be related to L‐OHP sensitivity in MAC cells.
In this study, the combination of L‐OHP and 5FU revealed synergistic or additive effects in all MAC cell lines.( 20 ) We next attempted to elucidate the reason why this combination of L‐OHP and 5FU contributes to the increasingly cytotoxic effect for MAC cells, and found that the protein expression levels of ERCC1 were down‐regulated after treatment of 5FU both in vitro and in vivo. Ojima et al.( 34 ) reported that 5FU post‐treatment gradually inhibited mRNA expression of ERCC1 in colon cancer cell lines. Suppression of the protein expression of ERCC1 by 5FU may affect recombination DNA repair efficiency. Therefore, these results suggest that the down‐regulation of ERCC1 protein expression by treating with 5FU may enhance the cytotoxic effects of L‐OHP.
We found that the combination treatment of L‐OHP and 5FU had marked cytotoxic effects on MAC cells, even on cell types known to be resistant to conventional platinum‐ and taxane‐based chemotherapy. We also found that ERCC1 expression levels probably affected the cytotoxic effects of L‐OHP, and this effectiveness may be related to down‐regulation of ERCC1 protein expression by 5FU exposure. Therefore, we concluded that this combination treatment is worth exploring as a treatment modality for MAC. Our next step is to evaluate the ability of ERCC1 expression levels to predict response to chemotherapy in clinical studies. Currently, a phase III clinical trial of L‐OHP and capecitabine, which is a prodrug of 5FU, versus carboplatin and PTX in MAC, is underway at the Gynecologic Oncology Group (GOG). We hope that this combination therapy will prolong the survival of patients with advanced MAC.
Acknowledgments
This work was supported by a Grant‐in‐Aid for Scientific Research (C) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (17591737, H Itamochi).
References
- 1. McGuire V, Jesser CA, Whittemore AS. Survival among U.S. women with invasive epithelial ovarian cancer. Gynecol Oncol 2002; 84: 399–403. [DOI] [PubMed] [Google Scholar]
- 2. Seidman JD, Kurman RJ, Ronnett BM. Primary and metastatic mucinous adenocarcinomas in the ovaries: incidence in routine practice with a new approach to improve intraoperative diagnosis. Am J Surg Pathol 2003; 27: 985–93. [DOI] [PubMed] [Google Scholar]
- 3. Chan JK, Teoh D, Hu JM, Shin JY, Osann K, Kapp DS. Do clear cell ovarian carcinomas have poorer prognosis compared to other epithelial cell types? A study of 1411 clear cell ovarian cancers. Gynecol Oncol 2008; 109: 370–6. [DOI] [PubMed] [Google Scholar]
- 4. Hess V, A’Hern R, Nasiri N et al . Mucinous epithelial ovarian cancer: a separate entity requiring specific treatment. J Clin Oncol 2004; 22: 1040–4. [DOI] [PubMed] [Google Scholar]
- 5. Cannistra SA. Cancer of the ovary. N Engl J Med 2004; 351: 2519–29. [DOI] [PubMed] [Google Scholar]
- 6. Winter WE 3rd, Maxwell GL, Tian C et al . Prognostic factors for stage III epithelial ovarian cancer: a Gynecologic Oncology Group Study. J Clin Oncol 2007; 25: 3621–7. [DOI] [PubMed] [Google Scholar]
- 7. Pectasides D, Fountzilas G, Aravantinos G et al . Advanced stage mucinous epithelial ovarian cancer: the Hellenic Cooperative Oncology Group experience. Gynecol Oncol 2005; 97: 436–41. [DOI] [PubMed] [Google Scholar]
- 8. Pisano C, Greggi S, Tambaro R et al . Activity of chemotherapy in mucinous epithelial ovarian cancer: a retrospective study. Anticancer Res 2005; 25: 3501–5. [PubMed] [Google Scholar]
- 9. Nakayama K, Takebayashi Y, Nakayama S et al . Prognostic value of overexpression of p53 in human ovarian carcinoma patients receiving cisplatin. Cancer Lett 2003; 192: 227–35. [DOI] [PubMed] [Google Scholar]
- 10. Agarwal R, Linch M, Kaye SB. Novel therapeutic agents in ovarian cancer. Eur J Surg Oncol 2006; 32: 875–86. [DOI] [PubMed] [Google Scholar]
- 11. Pendyala L, Creaven PJ. In vitro cytotoxicity, protein binding, red blood cell partitioning, and biotransformation of oxaliplatin. Cancer Res 1993; 53: 5970–6. [PubMed] [Google Scholar]
- 12. Rixe O, Ortuzar W, Alvarez M et al . Oxaliplatin, tetraplatin, cisplatin, and carboplatin: spectrum of activity in drug‐resistant cell lines and in the cell lines of the National Cancer Institute's Anticancer Drug Screen panel. Biochem Pharmacol 1996; 52: 1855–65. [DOI] [PubMed] [Google Scholar]
- 13. Noordhuis P, Laan AC, Van De Born K, Losekoot N, Kathmann I, Peters GJ. Oxaliplatin activity in selected and unselected human ovarian and colorectal cancer cell lines. Biochem Pharmacol 2008; 76: 53–61. [DOI] [PubMed] [Google Scholar]
- 14. Rose PG, Blessing JA, Mayer AR, Homesley HD. Prolonged oral etoposide as second‐line therapy for platinum‐resistant and platinum‐sensitive ovarian carcinoma: a Gynecologic Oncology Group study. J Clin Oncol 1998; 16: 405–10. [DOI] [PubMed] [Google Scholar]
- 15. Tsuruo T, Matsuzaki T, Matsushita M, Saito H, Yokokura T. Antitumor effect of CPT‐11, a new derivative of camptothecin, against pleiotropic drug‐resistant tumors in vitro and in vivo . Cancer Chemother Pharmacol 1988; 21: 71–4. [DOI] [PubMed] [Google Scholar]
- 16. Sehouli J, Stengel D, Oskay‐Oezcelik G et al . Nonplatinum topotecan combinations versus topotecan alone for recurrent ovarian cancer: results of a phase III study of the North‐Eastern German Society of Gynecological Oncology Ovarian Cancer Study Group. J Clin Oncol 2008; 26: 3176–82. [DOI] [PubMed] [Google Scholar]
- 17. Look KY, Blessing JA, Muss HB, DeGeest K. 5‐fluorouracil and low‐dose leucovorin in the treatment of recurrent epithelial ovarian carcinoma. A phase II trial of the Gynecologic Oncology Group. Am J Clin Oncol 1992; 15: 494–6. [DOI] [PubMed] [Google Scholar]
- 18. Look KY, Muss HB, Blessing JA, Morris M. A phase II trial of 5‐fluorouracil and high‐dose leucovorin in recurrent epithelial ovarian carcinoma. A Gynecologic Oncology Group Study. Am J Clin Oncol 1995; 18: 19–22. [DOI] [PubMed] [Google Scholar]
- 19. Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Meth 1983; 65: 55–63. [DOI] [PubMed] [Google Scholar]
- 20. Chou TC, Talalay P. Quantitative analysis of dose–effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 1984; 22: 27–55. [DOI] [PubMed] [Google Scholar]
- 21. Park JG, Kramer BS, Steinberg SM et al . Chemosensitivity testing of human colorectal carcinoma cell lines using a tetrazolium‐based colorimetric assay. Cancer Res 1987; 47: 5875–9. [PubMed] [Google Scholar]
- 22. Gorai I, Nakazawa T, Miyagi E, Hirahara F, Nagashima Y, Minaguchi H. Establishment and characterization of two human ovarian clear cell adenocarcinoma lines from metastatic lesions with different properties. Gynecol Oncol 1995; 57: 33–46. [DOI] [PubMed] [Google Scholar]
- 23. Ohta I, Gorai I, Miyamoto Y et al . Cyclophosphamide and 5‐fluorouracil act synergistically in ovarian clear cell adenocarcinoma cells. Cancer Lett 2001; 162: 39–48. [DOI] [PubMed] [Google Scholar]
- 24. Ozawa S, Sugiyama Y, Mitsuhashi Y, Kobayashi T, Inaba M. Cell killing action of cell cycle phase‐non‐specific antitumor agents is dependent on concentration–time product. Cancer Chemother Pharmacol 1988; 21: 185–90. [DOI] [PubMed] [Google Scholar]
- 25. Mitsuhashi Y, Inaba M, Sugiyama Y, Kobayashi T. In vitro measurement of chemosensitivity of human small cell lung and gastric cancer cell lines toward cell cycle phase‐nonspecific agents under the clinically equivalent area under the curve. Cancer 1992; 70: 2540–6. [DOI] [PubMed] [Google Scholar]
- 26. Extra JM, Marty M, Brienza S, Misset JL. Pharmacokinetics and safety profile of oxaliplatin. Semin Oncol 1998; 25: 13–22. [PubMed] [Google Scholar]
- 27. Fracasso PM, Blessing JA, Morgan MA, Sood AK, Hoffman JS. Phase II study of oxaliplatin in platinum‐resistant and refractory ovarian cancer: a gynecologic group study. J Clin Oncol 2003; 21: 2856–9. [DOI] [PubMed] [Google Scholar]
- 28. Holzer AK, Manorek GH, Howell SB. Contribution of the major copper influx transporter CTR1 to the cellular accumulation of cisplatin, carboplatin, and oxaliplatin. Mol Pharmacol 2006; 70: 1390–4. [DOI] [PubMed] [Google Scholar]
- 29. Hector S, Bolanowska‐Higdon W, Zdanowicz J, Hitt S, Pendyala L. In vitro studies on the mechanisms of oxaliplatin resistance. Cancer Chemother Pharmacol 2001; 48: 398–406. [DOI] [PubMed] [Google Scholar]
- 30. Fink D, Nebel S, Aebi S et al . The role of DNA mismatch repair in platinum drug resistance. Cancer Res 1996; 56: 4881–6. [PubMed] [Google Scholar]
- 31. Arnould S, Hennebelle I, Canal P, Bugat R, Guichard S. Cellular determinants of oxaliplatin sensitivity in colon cancer cell lines. Eur J Cancer 2003; 39: 112–19. [DOI] [PubMed] [Google Scholar]
- 32. Youn CK, Kim MH, Cho HJ et al . Oncogenic H‐Ras up‐regulates expression of ERCC1 to protect cells from platinum‐based anticancer agents. Cancer Res 2004; 64: 4849–57. [DOI] [PubMed] [Google Scholar]
- 33. Reed E. Platinum‐DNA adduct, nucleotide excision repair and platinum based anti‐cancer chemotherapy. Cancer Treat Rev 1998; 24: 331–44. [DOI] [PubMed] [Google Scholar]
- 34. Ojima E, Inoue Y, Watanabe H et al . The optimal schedule for 5‐fluorouracil radiosensitization in colon cancer cell lines. Oncol Rep 2006; 16: 1085–91. [PubMed] [Google Scholar]