

Abstract
To examine the mechanism of resistance to 7‐ethyl‐10‐hydroxycamptothecin (SN‐38) in lung cancer, we continuously exposed the non‐small‐cell lung cancer (NSCLC) cell line NCI‐H23 to SN‐38 and selected the SN‐38‐resistant clone H23/SN‐38. After 2 months of culturing in SN‐38‐free conditions, H23/SN‐38 cells recovered their sensitivity to SN‐38 and were subsequently established as the revertant H23/SN‐38REV cell line. Because H23/SN‐38 cells show cross resistance to certain anticancer drugs, such as topotecan, etoposide, doxorubicin and mitoxantrone, we examined the gene and protein expression levels of drug efflux transporters of the ATP‐binding cassette (ABC) family. We found that both gene and protein expression of ABCG2/BCRP (ABCG2) in H23/SN‐38 cells was increased compared with that in NCI‐H23 cells and H23/SN‐38REV cells. The cellular accumulation of topotecan in H23/SN‐38 cells was decreased compared with that in NCI‐H23 and H23/SN‐38REV cells, and treatment with reserpine (an inhibitor of ABCG2) increased the cellular accumulation of topotecan in H23/SN‐38 cells. Furthermore, treatment with reserpine also altered the sensitivity of H23/SN‐38 cells to SN‐38. These results indicate that the upregulation of ABCG2 was functional, and related to the resistance of H23/SN‐38 cells to SN‐38. Moreover, we found that gene expression levels of ABCG2 were significantly correlated with the concentration of SN‐38 for 50% cell survival in 13 NSCLC cells (r = 0.592, P < 0.05). The present results indicate that the induction of ABCG2 by SN‐38 does confer acquired resistance to CPT‐11/SN‐38, but the induction of ABCG2 and subsequent drug resistance are reversible. However, the expression level of ABCG2 may be a useful indicator of CPT‐11/SN‐38 activity in lung cancer. (Cancer Sci 2006; 97: 192–198)
7‐Ethyl‐10‐hydroxycamptothecin (SN‐38) is the active metabolite of irinotecan hydrochloride (CPT‐11), one of the topoisomerase inhibitors. Several mechanisms of resistance to CPT‐11/SN‐38 have been reported. A point mutation of the DNA topoisomerase I gene has been described in vitro ( 1 ) but no alteration of DNA topoisomerase I gene was observed in vivo, and low DNA topoisomerase I expression is associated with resistance to CPT‐11/SN‐38.( 2 ) SN‐38 has been shown to undergo glucuronidation by uridine 5′diphosphoglucuronosyltransferase (UGT) to form inactive SN‐38‐glucuronide, and hence increased intracellular drug detoxification through the upregulation of glucuronidation activity catalyzed by UGT is reportedly associated with resistance to CPT‐11/SN‐38.( 3 , 4 , 5 ) Furthermore, it has also been suggested that reduced drug accumulation mediated by the ATP‐dependent efflux pump is involved in the mechanism of resistance to CPT‐11/SN‐38.( 6 , 7 , 8 )
Lung cancer is one of the most common malignancies worldwide, and several randomized clinical trials and meta‐analyses have demonstrated that chemotherapy can slightly but significantly prolong survival in patients with lung cancer.( 9 , 10 ) CPT‐11 is one of the promising agents against human lung cancers( 11 , 12 ) and resistance to CPT‐11 is a common problem in the survival of lung cancer patients. However, biomarkers for predicting resistance to CPT‐11 have not been fully identified. To elucidate the biomarkers of resistance to CPT‐11, we developed an in vitro model of resistance to SN‐38 by continuous and progressive exposure of the non‐small‐cell lung cancer (NSCLC) cell line NCI‐H23 to SN‐38, and selected the SN‐38‐resistant clone H23/SN‐38. We then cultured H23/SN‐38 cells under SN‐38‐free conditions for 2 months and subsequently established the revertant H23/SN‐38REV cell line that exhibited sensitivity to SN‐38. Here we describe the determinants of resistance to CPT‐11/SN‐38 as elucidated using our selected SN‐38‐resistant cells.
Materials and Methods
Cell lines and chemicals
The following human lung cancer cell lines were used in this study:10 adenocarcinomas (NCI‐H23, PC‐9, ACC‐LC‐176, RERF‐LC‐MT, RERF‐LC‐AI, ACC‐LC‐94, ACC‐LC‐174, SK‐LC‐1, RERF‐LC‐OK and RERF‐LC‐MS), two squamous cell carcinomas (PC‐10 and QG56) and one large‐cell carcinoma (SK‐LC‐6). Cells from a human small‐cell lung cancer (SCLC) cell line, PC‐6, and the SN‐38‐resistant sub‐line PC‐6/SN2‐5 were kindly provided by Daiichi Pharmaceutical Co. (Tokyo, Japan).( 8 , 13 ) Cells were cultured in RPMI 1640 (or Dulbecco's modified Eagle's medium) supplemented with 10% heat‐inactivated fetal bovine serum and 1% (v/w) penicillin/streptomycin. SN‐38 was a gift from Yakult Honsha (Tokyo, Japan). Reserpine was obtained from Sigma‐Aldrich (St Louis, MO, USA), and gemcitabine was obtained from Eli Lilly Pharmaceuticals (Indianapolis, IN, USA). Cisplatin, etoposide and paclitaxel were gifts from Bristol Myers (Tokyo, Japan). Vinorelbine, 5‐fluorouracil and doxorubicin were obtained from Kyowa Hakko Kogyo (Tokyo, Japan), topotecan was obtained from Nihon Kayaku (Tokyo, Japan) and mitoxantrone was obtained from Wyeth Pharmaceuticals (Madison, NJ, USA).
SN‐38‐resistant cell lines were selected by culturing NCI‐H23 cells in the presence of SN‐38 at concentrations of 1 nM and higher for 3 months, with the drug‐containing medium changed every 4–7 days. They were made resistant by continuous exposure to stepwise‐increasing concentrations of the drug. Furthermore, variants of NCI‐H23 cells exhibiting intermediate resistance were cultured by limiting the dilution method for cloning. The resistant clone H23/SN‐38 was isolated and cultured again under SN‐38‐free conditions. After 2 months of culturing under SN‐38‐free conditions, H23/SN‐38 cells recovered their sensitivity to SN‐38 and were subsequently established as the revertant H23/SN‐38REV cell line.
Total RNA extraction and reverse transcription–polymerase chain reaction
Total RNA was extracted with TRIZOL reagent (Invitrogen, Carslbad, CA, USA) according to the manufacturer's instructions. cDNA was synthesized using a random hexamer (Amersham Biosciences, Buckinghamshire, UK) with Superscript RNase H‐reverse transcriptase (Gibco‐BRL, Bethesda, MD, USA). The reverse‐transcribed cDNA from each sample was subjected to polymerase chain reaction (PCR) amplification using Taq polymerase (Roche Applied Science, Indianapolis, IN, USA) and primers. The sequences of the ABCG2/BCRP (ABCG2) primers were 5′‐tggctgtcatggcttcagta‐3′ (forward) and 5′‐gccacgtgattcttccacaa‐3′ (reverse). The ABCB1/MDR1 (ABCB1), ABCC1/MRP1 (ABCC1) and ABCC2/MRP2 (ABCC2) primers and the PCR conditions have been described previously.( 14 ) Amplified products were separated by 2% agarose gel electrophoresis, and bands were visualized by staining with ethidium bromide. We also performed quantitative reverse transcription (RT)‐PCR with the LightCycler FastStart DNA SYBR Green kit (Roche Applied Science). We conducted a melting‐curve analysis to control for the specificity of the amplification products. The number of transcripts was calculated from a standard curve obtained by plotting the known input of six different concentrations against the number of PCR cycles at which the detected fluorescence intensity reached a fixed value. For each sample, results were normalized by comparison with the housekeeping gene glyceraldehyde‐3‐phosphate dehydrogenase.
Western blot analysis
All cancer cells were lysed with CelLyticTM‐M Mammalian Cell Lysis/Extraction Reagent (Sigma‐Aldrich) according to the manufacturer's instructions. Whole cell lysates (30 µg) were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to nitrocellulose membrane (Amersham Biosciences). After protein transfer, blots were incubated with the blocking solution and probed with anti‐ABCG2 antibody (BXP‐21; Chemicon, Temecula, CA, USA). Immunoreactive bands were visualized using the appropriate secondary horseradish peroxidase‐conjugated antibodies and enhanced chemiluminescence (Amersham Biosciences).
Chemosensitivity assay
Cells were cultured at 5000 cells/well in 96‐well tissue culture plates. To assess cell viability, stepwise 10‐fold dilutions of the anticancer drug were added 2 h after plating, and the cultures were incubated at 37°C for 96 h. At the end of the culture period, 20 µL of MTS [3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfophenyl)‐2H‐tetrazolium, inner salt] solution (CellTiter 96® AQueous One Solution Cell Proliferation Assay; Promega, Madison, WI, USA) was added and cells were incubated for a further 4 h, after which the absorbance was measured at 490 nm using an enzyme‐linked immunosorbent assay plate reader. Mean values were calculated from three independent experiments carried out in quadruplicate. The chemosensitivity is expressed here as the drug concentration for 50% cell survival (IC50), as determined from the concentration–effect relationship using Graph Pad Prism (version 4, GraphPad Software, San Diego, CA, USA).
Flow cytometry with topotecan
Topotecan was used as a fluorescent probe to detect the functional activity of ABCG2 as a transporter, because topotecan fluorescence is easily detectable using a conventional flow cytometer.( 15 ) Briefly, 2 × 106 cells were loaded for a minimum of 4 h at 37°C with 200 µM topotecan in RPMI 1640, and then washed twice in ice‐cold saline. The fluorescence of topotecan was analyzed with a flow cytometer (FACScan, Becton Dickinson, Franklin Lake, NJ, USA) equipped with an argon laser. The topotecan‐derived fluorescence of 2 × 104 events was measured through a 488‐nm bandpass filter at an excitation wavelength of 585 nm. In all flow cytometry assays, parallel samples were stored on ice to control for non‐specific drug binding to plasma membranes, and cells not exposed to topotecan were included as controls for autofluorescence.
Statistical analysis
Spearman's test was used to analyze the correlation between expression of the ABCG2 gene and the IC50 values for SN‐38. The differences between IC50 values for SN‐38 in ABCG2 protein expression were evaluated using the Mann–Whitney U‐test. The level of significance was set at 5%, using two‐sided analysis.
Results
Establishment of SN‐38‐resistant cell line
To obtain SN‐38‐resistant variants, we exposed the NSCLC cell line HCI‐H23 to increasing concentrations of SN‐38. Exposure of cultures to 1 nM SN‐38 for 6 weeks resulted in the isolation of several SN‐38‐resistant cell lines. One of these sub‐lines was further characterized, and the drug concentration was gradually increased every 2–4 weeks. Finally, cells (namely H23/SN‐38) growing vigorously in medium containing 10 nM SN‐38 were obtained 6 months later. The sensitivities of NCI‐H23 and H23/SN‐38 cells to a panel of chemotherapeutic drugs are summarized in Table 1. The IC50 for SN‐38 was 50 times higher for H23/SN‐38 cells (150.0 nM) than for the parent NCI‐H23 cells (3.03 nM). H23/SN‐38 cells showed cross‐resistance to topotecan, mitoxantrone, doxorubicin and etoposide, but not to the other four drugs tested: paclitaxel, vinorelbine, 5‐fluorouracil and cisplatin.
Table 1.
Drug concentrations for 50% cell survival (IC50) of various cytotoxic drugs in NCI‐H23 and H23/SN‐38 cells
| Drug | NCI‐H23 | H23/SN‐38 | RR | ||
|---|---|---|---|---|---|
| IC50 (nM) | 95% CI | IC50 (nM) | 95% CI | ||
| SN‐38 | 3.03 | 2.52–3.65 | 150 | 113–200 | 49.4 |
| Topotecan | 102.8 | 84.4–118.1 | 7870 | 5639–10 098 | 76.5 |
| Mitoxantrone | 2.48 | 1.76–3.50 | 214 | 147.5–312.1 | 86.2 |
| Etoposide | 761.2 | 537.2–1079 | 2626 | 1986–3472 | 3.4 |
| Doxorubicin | 13.9 | 11.6–16.7 | 97.7 | 79.5–111.4 | 7.0 |
| Cisplatin | 3750 | 2436–5728 | 4307 | 2163–8576 | 1.15 |
| Fluorouracil | 4817 | 3315–7001 | 7460 | 5724–9721 | 1.5 |
| Paclitaxel | 0.39 | 0.28–0.53 | 0.31 | 0.13–0.74 | 0.79 |
| Vinorelbine | 0.36 | 0.22–0.58 | 0.48 | 0.19–1.11 | 1.3 |
RR, resistant rate.
The stability of the acquired resistance of H23/SN‐38 cells was tested by growing them in drug‐free medium for 2 months. This resulted in their resistance to SN‐38, topotecan and mitoxantrone being reduced (Table 2). The cells were subsequently established as the revertant H23/SN‐38REV cell line.
Table 2.
Drug concentrations for 50% cell survival (IC50) of various cytotoxic drugs in H23/SN‐38REV cells
| Drug | NCI‐H23 | H23/SN‐38 | H23/SN‐38REV | |||
|---|---|---|---|---|---|---|
| IC50 (nm) | 95% CI | IC50 (nm) | 95% CI | IC50 (nm) | 95% CI | |
| SN‐38 | 3.03 | 2.52–3.65 | 150 | 113–200 | 69.1 | 51.5–93 |
| Topotecan | 102.8 | 84.4–118.1 | 7870 | 5639–10098 | 284 | 231–352 |
| Mitoxantrone | 2.48 | 1.76–3.50 | 214 | 147.5–312.1 | 128 | 105–157 |
Involvement of ABCG2 in resistance to SN‐38
Because H23/SN‐38 cells showed multidrug resistance, we examined the gene expression levels of drug efflux transporters of the ATP‐binding cassette (ABC) family. We found that gene expression of ABCG2 in H23/SN‐38 cells was increased compared with that in NCI‐H23 cells and H23/SN‐38REV cells (Fig. 1A), whereas the expression of MRP1/ABCC1 did not change (Fig. 1A) and we could not detect ABCB1 or ABCC2 in any of the three cell lines (data not shown). Further, we could not detect the UGT1A gene in any of the three cell lines (data not shown). We confirmed the differences in ABCG2 gene expression by real‐time PCR. We also confirmed that protein expression of ABCG2 in H23/SN‐38 cells was increased compared with that in NCI‐H23 cells and H23/SN‐38REV cells (Fig. 1B).
Figure 1.
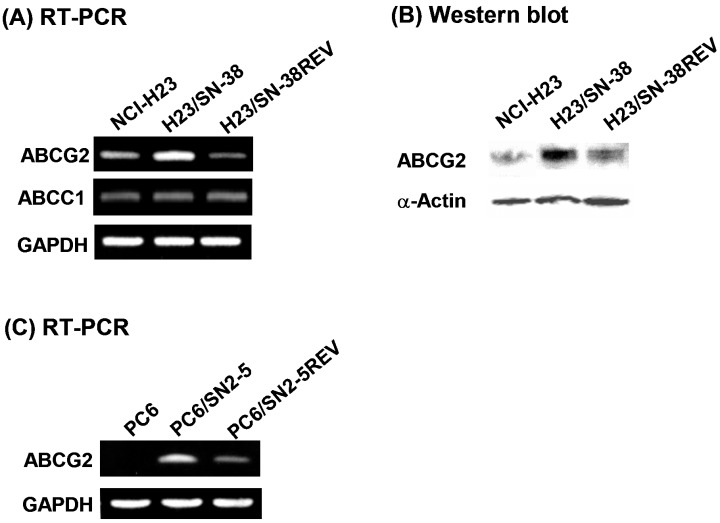
(A) Gene expression levels of ABCG2 and ABCC1 in NCI‐H23, H23/SN‐38 and H23/SN‐38REV cells. (B) Protein expression levels of ABCG2 in NCI‐H23, H23/SN‐38 and H23/SN‐38REV cells. (C) Gene expression levels of ABCG2 in PC‐6, PC‐6/SN2‐5 and PC‐6/SN2–5REV cells.
In order to elucidate whether the induction of ABCG2 expression by SN‐38 was reversible, we cultured another SN‐38‐resistant SCLC cell line (PC‐6/SN2‐5) in SN‐38‐free medium. The gene expression of ABCG2 has been shown to be higher in PC‐6/SN2‐5 cells than in parental PC‐6 cells, whereas the expression levels of ABCB1, ABCC1 and ABCC2 are the same in these two cell lines.( 13 ) After 2 months, the ABCG2 expression in PC‐6/SN2‐5 cells was partially decreased concomitant with a decreased in resistance to SN‐38 (namely, revertant cell line PC‐6/SN2–5REV, Fig. 1C; Table 3). We also confirmed the differences in ABCG2 gene expression by real‐time PCR.
Table 3.
Drug concentrations for 50% cell survival (IC50) of SN‐38 in PC‐6, PC‐6/SN2‐5 and PC‐6/SN2‐5REV cells
| Drug | PC‐6 | PC‐6/SN2‐5 | SN2‐5REV | |||
|---|---|---|---|---|---|---|
| IC50 (nm) | 95% CI | IC50 (nm) | 95% CI | IC50 (nm) | 95% CI | |
| SN‐38 | 0.003 | 0.0007–0.006 | 11.2 | 7.6–16.5 | 1.1 | 0.5–3.6 |
Functional activity of ABCG2
For the detection of functional activity of ABCG2 as a transporter, topotecan was used as a fluorescent probe because topotecan fluorescence is easily detectable by conventional flow cytometory and topotecan is a good substrate for ABCG2. The flow cytometric assay was carried out just after a 4 h‐exposure to 200 µM topotecan. After 4 h of incubation, the intracellular accumulation of topotecan was markedly lower in ABCG2‐overexpressing resistant clone H23/SN‐38 cells than in NCI‐H23 cells. We then treated the H23/SN‐38 cells with reserpine, an ABCG2 inhibitor,( 16 ) to investigate the restoration of topotecan accumulation. We found that a 30‐min pretreatment with 20 µM reserpine (a non‐cytotoxic concentration) increased the accumulation of topotecan in H23/SN‐38 cells. Furthermore, the H23/SN‐38REV cells also showed partial recovery of cellular accumulation of topotecan compared to H23/SN‐38 cells (Fig. 2).
Figure 2.
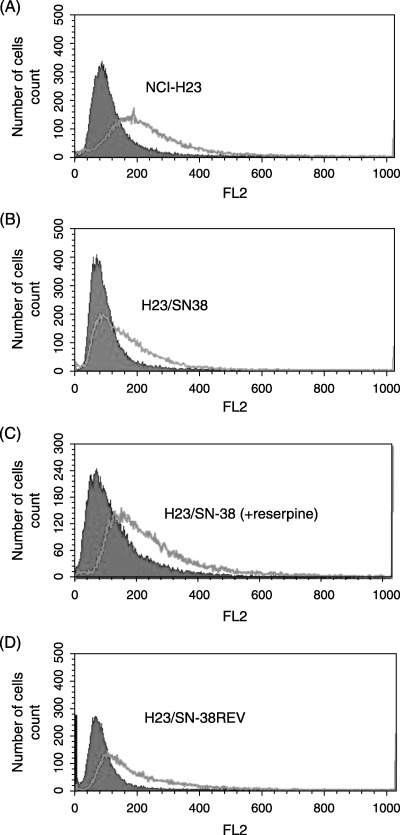
Flow cytometry analysis of the cellular accumulation of topotecan. The flow cytometric assay was carried out just after a 4 h‐exposure to 200 µM topotecan. Uptake of topotecan was measured using a fluorescence‐activated cell sorter. The presence of topotecan is shown by a single line, and absence is shown by a shaded line. A fluorescence peak shift to the right was observed in NCI‐H23 cells. In H23/SN‐38 cells, a fluorescence peak shift was not observed in the absence of reserpine.
Inhibition of SN‐38 cytotoxicity by reserpine
To elucidate whether ABCG2 actually affects the sensitivity to SN‐38, we pretreated H23/SN‐38 cells with reserpine and then measured the SN‐38 cytotoxicity. We found that the sensitivity of H23/SN‐38 cells to SN‐38 was increased by 30‐min pretreatment with 20 µM reserpine (IC50 for SN‐38 decreased from 49.4 nM to 20.3 nM), whereas that of the parental NCI‐H23 cells did not change (Fig. 3).
Figure 3.
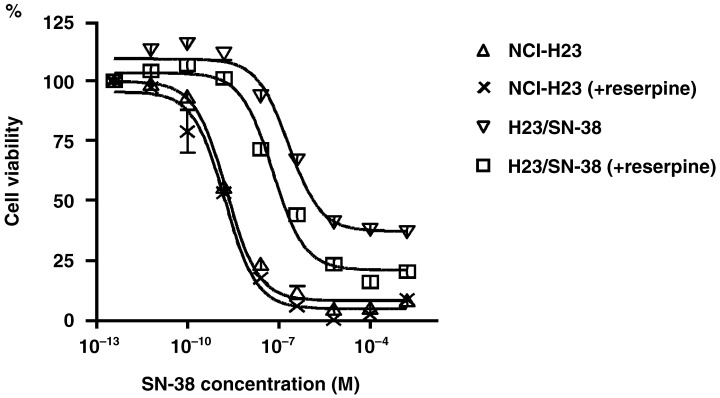
(A) Pretreatment with the ABCG2 inhibitor reserpine affects the SN‐38 cytotoxicity of H23 and H23/SN‐38 cells. The sensitivity of H23/SN‐38 cells to SN‐38 was increased by 30‐min pretreatment with 20 µM reserpine, whereas that of the parental NCI‐H23 cells did not change.
Relationship between cytotoxicity of SN‐38 and expression of ABCG2 genes
Using real‐time PCR, we found a clear correlation between ABCG2 gene expression and SN‐38 sensitivity in these 13 NSCLC cells (r = 0.592, P < 0.05; Fig. 4A). We detected the gene expression of ABCG2 in 9 of 13 NSCLC cells (Fig. 4A) by RT‐PCR, and we also found protein expression of ABCG2 in seven of the NSCLC cells in which moderate or high expression of ABCG2 was detected (Fig. 4B). Subsequently, we found that IC50 values for SN‐38 in NSCLC cells that express ABCG2 protein is significantly higher than that in NSCLC cells that does not express ABCG2 protein (P < 0.05; Fig. 4C).
Figure 4.
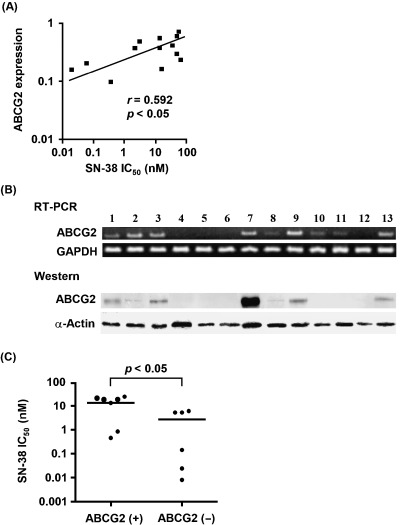
(A) Relationship between the basal expression levels of the ABCG2 gene by real‐time polymerase chain reaction and SN‐38 sensitivity in 13 NSCLC cell lines. (B) Gene and protein expression levels of ABCG2 in 13 NSCLC cell lines: 1, NCI‐H23; 2, RERF‐LC‐AI; 3, SK‐LC‐1; 4, ACC‐LC‐176; 5, RERF‐LC‐MT; 6, QG56; 7, RERF‐LC‐MS; 8, RERF‐LC‐OK; 9, PC‐9; 10, ACC‐LC‐94; 11, ACC‐LC‐174; 12, SK‐LC‐6; and 13, PC‐10. (C) Comparison of the drug concentration for 50% cell survival (IC50) values for SN‐38 between ABCG2‐expressed and ABCG2‐not expressed NSCLC cells by western blot.
Discussion
In the present study we successfully isolated an SN‐38‐resistant clone (H23/SN‐38) by continuously exposing NCI‐H23 cells to SN‐38. The H23/SN‐38 cells showed enhancement of functional ABCG2 expression, which demonstrates that overexpression of ABCG2 is related to the acquired resistance to SN‐38 in lung cancer.
Multidrug resistance is often attributed to ABC transporters in cancer cells. ABCB1 was the first of the ABC transporters to be identified and characterized, and the isolation of the second distantly related ABCC1 transporter facilitated the discovery of eight more genes. ABCG2 belongs to another ABC transporter family, and has its own characteristic substrate specificity and biology. ABCB1 confers high levels of resistance to bulky amphipathic drugs. The spectrum of drugs affected by ABCC transporters is similar to that for ABCB1; however, ABCC transporters efflux anticancer drugs by cotransporting drugs with glutathione, or glutathione–drug or glucuronide–drug conjugates. In contrast, ABCG2 affects a narrower range of anticancer agents compared to the ABCB1 and ABCC transporters, including anthracyclines, mitoxantrone and topoisomerase I inhibitors.( 17 ) Our finding that H23/SN‐38 cells that overexpress ABCG2 showed cross‐resistance to topotecan, mitoxantrone, doxorubicin and etoposide − but not to paclitaxel, vinorelbine, 5‐fluorouracil or cisplatin − is similar to the ABCG2 drug‐resistance profiles in previous reports.
It has been reported recently that overexpression of ABCG2 directly confers resistance to SN‐38 in lung cancer cells.( 8 ) SN‐38 undergoes glucuronidation by UGT to form inactive SN‐38‐glucuronide, and ABCC2 effluxes mainly SN‐38‐glucuronide products and those related with CPT‐11/SN‐38 resistance.( 6 , 18 ) In contrast, ABCG2 transports both SN‐38 and SN‐38‐glucuronide products with a higher affinity for SN‐38.( 19 ) We did not detect ABCC2 gene expression in H23/SN‐38 cells. Furthermore, we did not detect gene expression of UGT1A − which is mainly involved in SN‐38 glucuronidation − in H23/SN‐38 cells. These results indicate that overexpression of ABCG2 may be sufficient to confer resistance to SN‐38 through efflux of SN‐38 itself.
The mechanism underlying the upregulation of ABCG2 is still uncertain. ABCC family transporters confer resistance to methotrexate for only short‐term exposure to this drug. In contrast, ABCG2 transports the polyglutamylated forms of methotrexate and confers resistance to prolonged exposure to this agent.( 17 , 20 ) Candeil et al.( 21 ) also hypothesize − from their in vitro and in vivo experiments − that in colon cancer a longer exposure to CPT‐11 can lead to a larger proportion of ABCG2‐overexpressing cells in the tissue samples. These results indicate that short‐term exposure to anticancer drugs does not affect ABCG2 expression and that the mechanism underlying the upregulation of ABCG2 may be quite different from those of other ABC transporters.
After 2 months culturing under SN‐38‐free conditions, H23/SN‐38 cells recovered their sensitivity to SN‐38 concomitant with a decreased expression of ABCG2. To determine whether this phenomenon was also present in other established SN‐38‐resistant cells, we cultured the SN‐38‐resistant SCLC cell line PC‐6/SN2‐5 under SN‐38‐free conditions for 2 months, which resulted in ABCG2 expression decreasing concomitant with a decreased resistance to SN‐38. We believe this to be the first report that the induction of ABCG2 by anticancer drugs is reversible; the present results indicate that the effect of ABCG2 on the acquired resistance to CPT‐11/SN‐38 is reversible. However, we found that resistance to SN‐38 does not recover completely when ABCG2 expression decreased. A similar result was obtained following treatment with an ABCG2 inhibitor, which failed to restore the complete sensitivity of SN‐38‐resistant colon cancer cells to SN‐38.( 21 ) Taken together, the above results indicate that the mechanism of acquired resistance to SN‐38 is multifactorial. Indeed, CPT‐11 activates transcriptional factor nuclear factor (NF)‐κB, which plays an important role for underlying mechanisms of the antiapoptotic pathway and inducible drug resistance.( 22 ) Therefore, further studies are required to elucidate the determinants in relation to the persistent acquired resistance to SN‐38. Nonetheless, the recovery of ABCG2 expression may be an important concept for determining lung cancer chemotherapies, because we are able to use ABCG2‐mediated drugs when patients are pretreated with CPT‐11‐containing chemotherapy at least a few months prior.
We also found a correlation between the IC50 for SN‐38 and ABCG2 expression in 13 NSCLC cells. There is reportedly a strong correlation between mRNA and protein expression levels for ABCG2 in breast cancer.( 23 ) Moreover, the gene expression levels of ABCG2 reportedly reflect the functional activity of ABCG2 in lung cancer.( 15 ) These results indicate that the expression levels of ABCG2 may be a useful indicator of CPT‐11/SN‐38 activity in lung cancer.
Many substrates and inhibitors against ABCG2 have been reported.( 24 ) In the present study, we found that the ABCG2 inhibitor reserpine modified the resistance of H23/SN‐38 cells to SN‐38. The epidermal growth factor receptor kinase inhibitor gefitinib and some flavonoids have recently been shown to act as ABCG2‐substrate anticancer drugs, and coadministration of these agents with ABCG2‐inhibitors increases the toxicity for cancers.( 25 , 26 ) Because ABCG2 is expressed in many types of cancers( 27 ) treatment with ABCG2‐inhibitors may be useful to overcome ABCG2‐mediated drug resistance in cancer treatments. Whereas ABCG2 expression in normal tissues is also found in placental syncytiotrophoblasts, the epithelia of the small intestine and colon, the liver canalicular membrane, and in ducts and lobules of the breast,( 27 ) the physiological role of ABCG2 in normal tissues remains uncertain. It is therefore necessary to consider possible toxicity when substrates or inhibitors to ABCG2 are used in cancer chemotherapy.
In conclusion, the present study shows that the induction of ABCG2 by SN‐38 does not confer permanent acquired resistance to CPT‐11/SN‐38. However, the expression level of ABCG2 may be a useful determinant for CPT‐11/SN‐38 activity in lung cancer. Future studies should investigate the clinical and functional involvement of ABCG2 in resistance to CPT‐11/SN‐38 in vivo.
References
- 1. Kubota N, Kanzawa F, Nishio K et al. Detection of topoisomerase I gene mutation in CPT‐11 resistance lung cancer cell lines. Biochem Biophys Res Commun 1992; 188: 571–7. [DOI] [PubMed] [Google Scholar]
- 2. Ohashi N, Fujiwara Y, Yamaoka N, Katoh O, Satow Y, Yamakido M. No alteration in DNA topoisomerase I gene related to CPT‐11 resistance in human lung cancer. Jpn J Cancer Res 1996; 87: 1280–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Iyer L, King CD, Whiington PF et al. Genetic predisposition of the metabolism of irinotecan (CPT‐11). J Clin Invest 1998; 101: 847–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Takahashi T, Fujiwara Y, Yamakido M, Katoh O, Watanabe H, Mackenzie PI. The role of glucuronidation in 7‐ethyl‐10‐hydroxycamptothecin resistance in vitro . Jpn J Cancer Res 1997; 88: 1211–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Oguri T, Takahashi T, Miyazaki M et al. UGT1A10 is responsible for SN‐38 glucuronidation and its expression in human lung cancers. Anticancer Res 2004; 24: 2893–6. [PubMed] [Google Scholar]
- 6. Koike K, Kawabe T, Tanaka T et al. A canalicular multispecific organ anion transporter (cMOAT) antisense cDNA enhances drug sensitivity in human hepatic cancer cells. Cancer Res 1997; 57: 5475–9. [PubMed] [Google Scholar]
- 7. Brabgi M, Litman T, Ciotti M et al. Camptothecin resistance: role of the ATP‐binding cassette (ABC), mitoxantrone‐resistance half‐transporter (MXR), and potential for glucronidation in MXR‐expression cells. Cancer Res 1999; 59: 5983–46. [PubMed] [Google Scholar]
- 8. Kawabata S, Oka M, Shiozawa K et al. Breast cancer resistance protein directly confers SN‐38 resistance of lung cancer cells. Biochem Biophys ResCommun 2001; 280: 1216–23. [DOI] [PubMed] [Google Scholar]
- 9. Non‐Small Cell Lung Cancer Collaborative Group. Chemotherapy in non‐small cell lung cancer. A meta‐analysis using updated data on individual patients from 52 randomized clinical trials. BMJ 1995; 311: 899–909. [PMC free article] [PubMed] [Google Scholar]
- 10. Schiller JH, Harrington D, Belani CP et al. Comparison of four chemotherapy regimens for advanced non‐small‐cell lung cancer. N Engl J Med 2002; 346: 92–8. [DOI] [PubMed] [Google Scholar]
- 11. Noda K, Nishiwaki Y, Kawahara M et al. Irinotecan plus cisplatin compared with etoposide plus cisplatin for extensive small‐cell lung cancer. N Engl J Med 2002; 346: 85–91. [DOI] [PubMed] [Google Scholar]
- 12. Negoro S, Masuda N, Takada Y et al. Randomised phase III trial of irinotecan combined with cisplatin for advanced non‐small‐cell lung cancer. Br J Cancer 2003; 88: 335–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ishii M, Iwahana M, Mitsui I et al. Growth inhibitory effect of a new camptothecin analog, DX‐8951f, on various drug‐resistant sublines including BCRP‐mediated camptothecin derivative‐resistant variants derived from the human lung cancer cell line PC‐6. Anticancer Drugs 2002; 11: 353–62. [DOI] [PubMed] [Google Scholar]
- 14. Hirata S, Katoh O, Oguri T, Watanabe H, Yajin K. Expression of drug resistance‐related genes in head and neck squamous cell carcinomas and normal mucosa. Jpn J Cancer Res 2000; 91: 84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kawabata S, Oka M, Soda H et al. Expression and functional analyses of breast cancer resistance protein in lung cancer. Clin Cancer Res 2003; 9: 3052–7. [PubMed] [Google Scholar]
- 16. Wierdl M, Wall A, Morton CL et al. Carboxylesterase‐mediated sensitization of human tumor cells to CPT‐11 cannot override ABCG2‐mediated drug resistance. Mol Pharmacol 2003; 64: 279–88. [DOI] [PubMed] [Google Scholar]
- 17. Haimeur A, Conseil G, Deeley RG, Cole SP. The MRP‐related and BCRP/ABCG2 multidrug resistance proteins: biology, substrate specificity and regulation. Curr Drug Metab 2004; 5: 21–53. [DOI] [PubMed] [Google Scholar]
- 18. Chu XY, Kato Y, Ueda K et al. Biliary excretion mechanism of CPT‐11 and its metabolites in humans: involvement of primary active transporters. Cancer Res 1998; 58: 5137–43. [PubMed] [Google Scholar]
- 19. Nakatomi K, Yoshikawa M, Oka M et al. Transport of 7‐ethyl‐10‐hydroxycamptothecin (SN‐38) by breast cancer resistance protein ABCG2 in human lung cancer cells. Biochem Biophys Res Commun 2001; 288: 827–32. [DOI] [PubMed] [Google Scholar]
- 20. Volk EL, Rohde K, Rhee M et al. Methotrexate cross‐resistance in a mitoxantrone‐selected multidrug‐resistant MCF7 breast cancer cell line is attributable to enhanced energy‐dependent drug efflux. Cancer Res 2000; 60: 3514–21. [PubMed] [Google Scholar]
- 21. Candeil L, Gourdier I, Peyron D et al. ABCG2 overexpression in colon cancer cells resistant to SN38 and in irinotecan‐treated metastases. Int J Cancer 2004; 109: 848–85. [DOI] [PubMed] [Google Scholar]
- 22. Wang CY, Cusack JC Jr, Liu R, Baldwin AS Jr. Control of inducible chemoresistance: enhanced anti‐tumor therapy through increased apoptosis by inhibition of NF‐κB. Nat Med 1999; 5: 412–17. [DOI] [PubMed] [Google Scholar]
- 23. Faneyte IF, Kristel PM, Maliepaard M et al. Expression of the breast cancer resistance protein in breast cancer. Clin Cancer Res 2002; 8: 1068–74. [PubMed] [Google Scholar]
- 24. Doyle LA, Ross DD. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2). Oncogene 2003; 22: 7340–58. [DOI] [PubMed] [Google Scholar]
- 25. Imai Y, Tsukahara S, Asada S, Sugimoto Y. Phytoestrogens/flavonoids reverse breast cancer resistance protein/ABCG2‐mediated multidrug resistance. Cancer Res 2004; 64: 4346–52. [DOI] [PubMed] [Google Scholar]
- 26. Nakamura Y, Oka M, Soda H et al. Gefitinib (“Iressa”, ZD1839), an epidermal growth factor receptor tyrosine kinase inhibitor, reverses breast cancer resistance protein/ABCG2‐mediated drug resistance. Cancer Res 2005; 65: 1541–54. [DOI] [PubMed] [Google Scholar]
- 27. Maliepaard M, Scheffer GL, Faneyte IF et al. Subcellular localization and distribution of the breast cancer resistance protein transporter in normal human tissues. Cancer Res 2001; 61: 3458–64. [PubMed] [Google Scholar]