

Abstract
The purpose of the present study was to investigate the mechanisms involved in the antiproliferative and apoptotic effects of MCS‐C2, a novel analog of the pyrrolo[2,3‐d]pyrimidine nucleoside toyocamycin and sangivamycin, in human prostate cancer LNCaP cells. MCS‐C2, a selective inhibitor of cyclin‐dependent kinase, was found to inhibit cell growth in a time‐ and dose‐dependent manner, and inhibit cell cycle progression by inducing the arrest of the G1 phase and apoptosis in LNCaP cells. When treated with 3 µM MCS‐C2, inhibited proliferation associated with apoptotic induction was found in the LNCaP cells in a concentration and time‐dependent manner, and nuclear DAPI staining revealed the typical nuclear features of apoptosis. Furthermore, MCS‐C2 induced cell cycle arrest in the G1 phase through the upregulated phosphorylation of the p53 protein at Ser‐15 and activation of its downstream target gene p21WAF1/CIP1. Accordingly, these results suggest that MCS‐C2 inhibits the proliferation of LNCaP cells by way of G1‐phase arrest and apoptosis in association with the regulation of multiple molecules in the cell cycle progression. (Cancer Sci 2006; 97: 430 –436)
Abbreviations:
- CDK
cyclin‐dependent kinases
- DAPI
4’,6‐diamidino‐2‐phenylindole
- MTT
3‐[4,5‐dimethylthiazol‐2‐yl]‐2,5‐diphenyltetrazolium bromide
- PBS
phosphate‐buffered saline
- PI
propidium iodide
- pRb
retinoblastoma protein
- RT‐PCR
reverse transcription–polymerase chain reaction
- SDS
sodium dodecylsulfate
- Ser
serine
- TUNEL
Tdt‐mediated dUTP nick‐end labeling.
The incidence of prostate cancer is extremely high, with an estimated 180 000 cases in the US in 1999 alone.( 1 ) The cancer predominantly occurs in the peripheral zone of the prostate,( 2 ) displays a short‐term responsiveness to androgen ablation therapy, and is characteristically resistant to chemotherapy.( 3 ) Despite various investigations of the roles of known oncogenes and tumor suppressor genes, the molecular events that initiate and drive the progression of this disease are still not well understood. As for cancer in general, the long‐term goal for the treatment of prostate cancer remains the targeting of tumors specific therapeutic manipulation of molecules or pathways that could regulate tumor cell growth or activate cell death.
Several recent studies have indicated that normal prostatic epithelial cells and many prostatic adenocarcinomas contain a wild‐type form of the tumor suppressor p53.( 4 ) p53 normally responds to different forms of cellular stress by targeting the activation of checkpoint genes that inhibit cell cycle progression (such as the cyclin‐dependent kinase inhibitor p21WAF1/CIP1,( 5 , 6 ) and/or trigger apoptotic cell death.( 7 ) Stimuli that activate p53 include ionizing and non‐ionizing radiation, ribonucleotide depletion, microtubule disruption, hypoxia, oncogenes, and chemotherapeutic drugs. Modulation of the p53 molecule is in part achieved by post‐translational modifications, such as phosphorylation and acetylation, which promote the formation of specific interactions with other proteins and target gene regulatory elements.( 8 ) The loss of p53 activity through mutation, deletion, or inactivation by endogenous or viral oncogenes leads to the propagation of DNA damage, resulting in genetic instability.
CDK play pivotal roles in the signal transduction pathways that control the proliferation and differentiation of eukaryotic cells. The eukaryotic cell cycle progression is regulated by the sequential activation and subsequent inactivation of a series of CDK at different phases. The activities of CDK are positively regulated by cyclins and negatively regulated by CDK inhibitors. In the course of screening for a novel inhibitor of CDK2 and CDK1, the current authors isolated toyocamycin and sangivamycin from a culture both of Streptomyces sp. LPL931.( 9 ) Toyocamycin was first reported as an antibiotic in 1966,( 10 ) and later shown to inhibit RNA synthesis in mammalian cells.( 11 ) Yet, when toyocamycin was tested as a possible cancer treatment, a high toxicity was reported.( 12 ) Accordingly, many toyocamycin analogs have since been synthesized and evaluated for their antitumor and antiviral activities.( 13 , 14 , 15 , 16 ) However, although some analogs are potent inhibitors of many cancer cells, they are also invariably toxic to normal human cells. Therefore, in an attempt to identify a specific inhibitor that can inhibit CDK with minimal side‐effects on other serine/threonine protein kinase activity, the current authors previously synthesized an analog of toyocamycin, MCS‐C2 (Fig. 1), and evaluated its CDK inhibitory activity.( 17 , 18 ) The present study investigated the antineoplastic potential and mode of action of apoptotic induction by MCS‐C2 in LNCaP prostate cancer cells.
Figure 1.
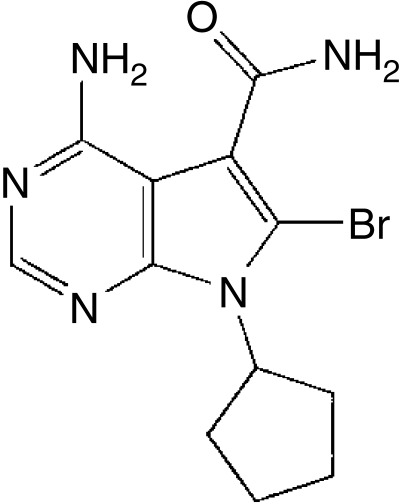
Chemical structure of MCS‐C2, a novel analog of the pyrrolo[2,3‐d]pyrimidine nucleoside toyocamycin and sangivamycin.
Materials and Methods
Chemicals
The PBS and RPMI‐1640 medium were purchased from Gibco (Grand Island, NY). The antibodies were all purchased from Santa Cruz Biotechnology (Santa Cruz, CA), except for p53‐phospho‐Ser‐392, −15 and caspase‐8 and‐9 (Cell Signaling Technology, Beverly, MA). The enhanced chemiluminescence kit was purchased from Amersham Pharmacia Biotech (Uppsala, Sweden) and the protein assay kit from Bio‐Rad Laboratories (Hercules, CA). All other materials were obtained from Sigma (St Louis, MO).
Cell line and cell culture
The human prostate LNCaP and PC‐3 cells were purchased from the American Type Culture Collection (Rockville, MD) and cultured at 37°C in a humidified atmosphere of 5% CO2‐air using RPMI‐1640 medium supplemented with 10% fetal bovine serum, 100 units/mL penicillin‐streptomycin. The cell density in the culture did not exceed 1 × 106 cells/mL.
Cell viability test
The LNCaP cells were seeded at a density of 1 × 104 cells/mL in a 96‐well culture dish and treated with various concentrations (0–1000 µM) of MCS‐C2 for 24 and 48 h. The viability test was assessed by the conventional colorimetric dye reduction method based on the reduction of MTT (Promega, Madison, WI), and the viable cell number was measured spectrophotometrically at 570 nm using an enzyme‐linked immunosorbent assay reader (Molecular Devices, Sunnyvale, CA).
Kinetic assay of CDKs
Cellular lysates (500–1000 µg) isolated from the MCS‐C2 treatment cells were incubated at 37°C with 20 µL of each antibody for 2 h, then with 30 µL of protein A/G agarose beads (Amersham Phamacia Biotech) at 4°C for 12 h. Immunocomplexes were washed four times with lysis buffer and followed by kinase buffer. Histone H1 kinase assays were then performed in the presence of 10 µg of histone H1 (Sigma), 20 mM HEPES, 20 mM β‐glycerophosphate, 5 mM NaF, 10 mM MgCl2, 1 mM dithiothreitol, 0.5 mM sodium orthovanadate and 10 µCi of γ‐32P ATP in 30 µL volume. After 30 min at 30°C, the reaction was mixed with an equal amount of standard 2 × SDS protein denature loading buffer and boiled. Then the supernatant was electrophoretically separated on a 12% SDS–polyacrylamide gel. The kinase activity was measured by exposure on X‐ray film.
Cell cycle analysis
After treatment with MCS‐C2, the cells (1 × 106 cells/mL) were washed twice with cold PBS and fixed in 70% ethanol. Immediately before the analysis, the cells were washed with PBS and stained with a solution containing 0.2 mg/mL PI for 1 h at 4°C and 0.1 mg/mL RNase A for 30 min at 37°C, then analyzed with a fluorescent‐activated cell sorter FACScan flow cytometer (Becton Dickinson, San Diego, CA) using CellQuest software (version 5.1.1; Becton Dickinson).
Nuclear staining with DAPI
The LNCaP cells (1 × 106 cells/mL) were cultured in RPMI‐1640 medium containing 10% fetal bovine serum in the absence or presence of MCS‐C2 (3 µM). After 12 h incubation, the cells were harvested and washed with PBS, then 4% neutral buffered formalin (100 µL) was added to the cell pellet. Thereafter, an aliquot (50 µL) of the cell suspension was smeared on slides and dried at room temperature, then the fixed cells were washed in PBS, air dried, and stained with DNA‐specific fluorochrome DAPI for 30 min at 37°C. The slides were then observed under a fluorescence microscope.
TUNEL assay for apoptosis‐induced DNA fragmentation
The Apoptosis Detection System developed by Promega was used according to the supplier's recommended protocol. Briefly, after the MCS‐C2 treatment, the cells were centrifuged, washed twice with PBS, and gently resuspended in 0.5 mL of PBS before adding 5 mL of 1% ice‐cold p‐formaldehyde for 20 min. The fixed cells were then washed with 5 mL of cold 70% ethanol, and the dehydrated cells incubated for 4 h/at 20°C. Thereafter, the cells were washed again with 5 mL of PBS, transferred to a 1.5 mL tube and centrifuged for 10 min at 20°C and 5000 g. The supernatant was discarded, and the pellet resuspended in 80 µL of an equilibration buffer for 5 min at room temperature. After reaction, the nuclei were incubated for 1 h at 37°C in the dark in 50 µL of an equilibration buffer containing fluorescein‐12‐dUTP in the presence of terminal deoxynucleotidyl transferase to label the 3′‐OH ends of the fragmented DNA. The reaction was stopped by the addition of 1 mL of 20 mM EDTA with gentle stirring. After being washed, the material was resuspended in 0.5 mL of PBS containing 5 µg/mL of PI and 250 µg of RNase A. The mixture was then incubated at room temperature in the dark for 30 min before being analyzed by a FACScan flow cytometer using CellQuest software.
Western blot analysis
The LNCaP cells were plated onto 60 mm dishes at a density of 2 × 105 cells/mL with or without MCS‐C2 (3 µM, 0–24 h) then harvested. To prepare the whole‐cell extract, the cells were washed with PBS and suspended in a protein lysis buffer (50 mM Tris, 5 mM EDTA, 150 mM NaCl, 1% NP‐40, 0.5% deoxycholic acid, 1 mM sodium orthovanadate, 100 µg/mL phenylmethylsulphonyl fluoride, and protease inhibitors). The protein content was determined using a Bio‐Rad protein assay reagent with bovine serum albumin as the standard. The protein extracts (30–50 µg) were analyzed based on 8–14% SDS–polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane (Millipore, Billerica, MA). The membranes were blocked with 5% w/v non‐fat dry milk, then incubated with the indicated antibodies in Tris‐buffered saline (10 mM Tris‐HCl, 150 mM NaCl/pH 7.6) containing 0.1% Tween‐20 with gentle shaking at 4°C for 2–12 h. The secondary antibody was a peroxidase‐conjugated goat antimouse, rabbit antibody. The signals were detected using an enhanced chemiluminescence Western blotting kit.
RNA extraction and real‐time RT‐PCR analysis
The total RNA extraction from the LNCaP cells was carried out with an Rneasy Mini Kit (Qiagen, Valencia, CA) as suggested by the manufacturer. The RT‐PCR for the mRNA quantification was performed with an iCycler iQ Multi‐Color Real‐Time PCR Detection System (Bio‐Rad). Specific primers were used for the human p53 (5′‐GTTCCGAGAGCTGAATGAGG‐3′ and 5′‐TGAGTCAGGCCCTTCTGTCT‐3′) and human p21 (5′‐CGCGACTGTGAYGCGCTA‐3′ and 5′‐AAGTCGAAGTTCCATCGCTCA‐3′). The total 50 µL reaction mixture contained a Hot‐star Taq DNA polymerase, 1 × PCR reaction buffer, 0.4 µM of each primer, 166 µM dNTP, and 1 × SYBR Green dye I (Molecular Probes, Eugene, OR). The reverse transcriptase reaction was performed at 42°C for 60 min. Each cycle of the subsequent PCR included denaturation (95°C, 1 min), primer annealing (55°C, 1 min), and an extension/synthesis step (72°C, 1 min). The relative quantization was calculated using the comparative threshold cycle method, where CT indicates the fractional cycle number at which the amplified gene amounted to a fixed threshold within the linear phase of amplification. The median CT of triplicate measurements was used to calculate ΔCT as the CT for the p53 and p21 genes, and the ΔCT for each sample was compared with the ΔCT for the control, expressed as ΔΔCT. The relative quantification was then expressed as the fold‐change in p53 and p21 expression compared with the control condition using the formula 2−ΔΔCT. Denaturing curves and agarose gel electrophoresis of the PCR product for p53, p21, and the housekeeping gene were used to confirm the homogeneity of the DNA products.( 19 )
Statistical analysis
The data are reported as the mean ± standard deviation of three independent experiments and were evaluated by Student's t‐test. Values of P < 0.05 were considered to be statistically significant. Results were obtained from the average of three experiments.
Results
MCS‐C2‐induced cell growth inhibition in LNCaP cells
To determine whether MCS‐C2 would induce cell growth inhibition in a human cancer cell line, the effect of MCS‐C2 on the viability of human prostate LNCaP cell line was assessed using an MTT assay. As shown in Figure 2, the growth of the LNCaP cells treated with 0.1–1000 µM of MCS‐C2 for 24 and 48 h was inhibited in a dose‐dependent manner. The IC50 value for growth inhibition during 24 h and 48 h was 1.9 µM and 0.3 µM, respectively.
Figure 2.

Effect of MCS‐C2 treatment on viability of LNCaP human prostate cancer cells. The LNCaP cells were exposed to the indicated amounts of MCS‐C2 for 24 and 48 h, and the cell viability determined by an MTT dye assay. IC50: 1.9 µM for 24 h, 0.3 µM for 48 h. The results represent the mean ± standard deviation of three independent experiments and a significant difference was established at *P < 0.05 and **P < 0.01 compared with the control group (0.1% dimethylsulphoxide) for the indicated time.
MCS‐C2 reduced kinase activity of cell cycle regulatory protein kinases
As shown in Figure 3, MCS‐C2 consistently produced a dose‐dependent decrease in several activity of several CDKs. Among the CDKs, MCS‐C2 strongly inhibits CDK2 activity. These results showed that MCS‐C2 was a very effective kinase inhibitor. Inhibition of CDK activities is the reason for cell cycle arrest occurring in LNCaP cells treated with MCS‐C2.
Figure 3.

Inhibition effects of MCS‐C2 on CDK activity. Cells were treated with MCS‐C2 at each concentration for 24 h and immunoprecipitated with target kinase (CDK1, CDK2, CDK4 and cyclin B) antibodies. Their kinetic activity was measured using histone H1 as substrate. Results were obtained from the average of three experiments.
MCS‐C‐induced upregulation of p53 through phosphorylation at Ser‐15, −392
As shown in Figure 4a, the protein levels of p53 and p21 were significantly increased in the LNCaP cells treated with MCS‐C2. Interestingly, the RT‐PCR analysis demonstrated that the mRNA levels of p21WAF1/CIP1 were clearly increased after 12 h (68‐fold compared to the control), whereas the mRNA levels of p53 showed no increase (Fig. 4b). Furthermore, the amount of phosphorylated p53 at position Ser‐392 gradually increased after 6 h of treatment with 3 µM MCS‐C2 (Fig. 4a), followed by an increase at position Ser‐15 after 12 h. As such, the increased protein level of p53 was due to phosphorylation at Ser‐15, −392, attributable to a post‐translational mechanism causing a prolonged half‐life of p53. Therefore, these results indicate that MCS‐C2 induced the activation of p53 by post‐translational modification (phosphorylation) and subsequent transcriptional activation of p21WAF1/CIP1. Furthermore, the level of the p53‐regulated pro‐apoptotic Bax protein was also increased similar to that of p21WAF1/CIP1.
Figure 4.

MCS‐C2 upregulated protein and mRNA levels of p53 and p1WAF1/CIP1. The LNCaP cells were treated with 3 µM MCS‐C2 and the total proteins and RNAs extracted. To detect the level of mRNA, a real‐time quantitative RT‐PCR was performed. (a) Protein level in p53 and its downstream genes (p21, Bax, Bcl‐2) based on western blotting. (b) Relative Ct value for p53 and p21 genes when compared to the reference gene (β‐actin). The relative protein and mRNA levels under each treatment were the average of three independent experiments.
Inhibition of cell cycle regulation by MCS‐C2
Considering the induction of p53 and p21WAF1/CIP1 in the LNCaP cells after MCS‐C2 treatment, the occurrence of a cell cycle arrest was also investigated. As shown in Figure 5a, a flow cytometric analysis revealed an appreciable arrest of cells in the G1 phase of the cell cycle and apoptosis after treatment with MCS‐C2 in LNCaP cells. The LNCaP cell population gradually increased from 52% at 0 h to 60% after 6 h in the G1 phase with exposure to 3 µM of MCS‐C2. In PC‐3 (–/– p53) cells, MCS‐C2 induced G1 phase arrest but there was no apoptotic induction after treatment of 10 µM MCS‐C2 (Fig. 5b). The p53 transcriptional transactivation of the endogenous p21WAF1/CIP1 promoter may have contributed to the induction of the G1 cell cycle arrest in the LNCaP cells. In a Western blot analysis, the increased dephosphorylated pRb indicated a G1‐phase arrest after 12 and 24 h. Yet there were no significant changes in the G1‐related proteins, such as CDK/cyclin proteins (Fig. 7b).
Figure 5.

Cell cycle regulation of LNCaP and PC‐3 cells induced by MCS‐C2. The LNCaP cells were treated with 3 µM MCS‐C2 for the indicated time (a), PC‐3 cells were treated with 10 µM MCS‐C2 for the indicated time (b), then stained with PI. The nuclei were analyzed for their DNA content by flow cytometry using CellQuest software. A total of 10 000 nuclei were analyzed from each sample. Results were obtained from the average of three experiments.
Figure 7.

Effect of MCS‐C2 on expression of cell cycle and apoptosis‐related proteins. The cells were pretreated with 3 µM MCS‐C2 for different lengths of time, then washed with phosphate‐buffered saline, lyzed, and a Western blot analysis performed. MCS‐C2 induced the proteolytic cleavage of caspase‐8, ‐9, and ‐3, while inactivating PARP (a) and the dephosphorylation of pRb. However, there was no significant change in the G1‐related protein (CDK and cyclin) levels (b). β‐actin was used as the internal control.
Apoptotic induction by MCS‐C2
To verify whether the growth inhibitory effect of MCS‐C2 was due to apoptosis, nuclear DAPI staining was used to examine the morphologic changes in the nuclei of the LNCaP cells treated with MCS‐C2, and the changes found were typical of apoptosis, that is, fragmented apoptotic bodies with 3 µM MCS‐C2 after 24 h incubation (Fig. 6a). This apoptotic morphological change was also confirmed with flow cytometric analysis, where PI staining of the LNCaP cells revealed that the apoptotic cell population gradually increased from <1% at 0 h to 19% after 24 h with exposure to 3 µM MCS‐C2 (Fig. 5). Moreover, as shown in Figure 6b, a TUNEL assay equally exhibited the induction of apoptosis after treatment with MCS‐C2. A Western blot analysis of the procaspase and cleaved products was carried out to obtain direct evidence on the involvement of caspases in the process of the MCS‐C2‐induced apoptosis. As such, the activation of procaspase‐3, ‐9, and ‐8 and cleavage of PARP, which is the target protein of caspase‐3, are also shown in Figure 7a.
Figure 6.

Induced apoptosis of LNCaP cells by MCS‐C2, as determined by TUNEL and DAPI assay. The cells were treated with 3 µM MCS‐C2 for 24 h, then fluorescence microscopic examination followed by DAPI staining. Arrow indicates the apoptotic bodies of nuclear fragmentation (a). The nuclei stained with d‐UTP fluorescein‐isothiocyanate and propidium iodide of the untreated and treated cells with MCS‐C2 were analyzed for their DNA content by flow cytometry using CellQuest software. A total of 10 000 nuclei were analyzed from each sample (b).
Discussion
The pharmacological manipulation of growth inhibition and the antiproliferative effect of malignant cells through the induction of apoptosis or suicidal cell death have recently been recognized as novel strategies for the identification and screening of potential chemotherapeutic agents. Many chemotherapeutic agents have been found to induce apoptosis,( 20 , 21 ) yet the mechanism of apoptosis is remarkably conserved and arbitrated with a greater than expected complexity. Apoptosis can be executed by activating the caspase family, among which caspase‐3 is a key, being responsible either partially or wholly for the proteolytic cleavage of a large number of substrates, such as PARP.( 22 ) Conversely, the Bcl‐2 protein is recognized as a significant negative regulator of apoptosis and acts upstream of caspase‐3 to prevent apoptosis. Caspase‐3 has also been shown to play an important role in apoptosis induced by several conditions, and necessary in determining the nuclear alteration of apoptosis.( 23 )
Targeting the activation of p53 in tumors that contain the wild‐type gene is a promising strategy for cancer therapies. Commonly used chemotherapeutic agents, which in many cases act by inducing DNA damage, activate cellular pathways that inhibit proliferation and lead to growth arrest or apoptosis. Although many prostate cancers contain wild‐type p53 and proliferate slowly, radiation and chemotherapeutic strategies used to control this disease are relatively ineffective.
In the present study, MCS‐C2 was found to regulate the cell cycle through the activation of p53 by phosphorylation at Ser‐15, −392 and the upregulation of the p21WAF1/CIP1 expression level. Ser‐15 is a functionally important residue within the p53 amino‐terminal region,( 24 ) and the phosphorylation of Ser‐15 represents an early cellular response to a variety of genotoxic stresses.( 25 ) Furthermore, the p53 phosphorylated at Ser‐15 displays a reduced binding to the inhibitor protein MDM2.( 26 ) Thus, because the association of p53 with MDM2 means p53 is targeted for proteosome‐mediated degradation that inhibits its transactivating function,( 27 ) Ser‐15 phosphorylation promotes both the accumulation and functional activation of p53 in response to DNA damage. Also, p53 can be phosphorylated at Ser‐392 in vivo, and by CAK in vitro. Phosphorylation of p53 at Ser‐392 is altered in human tumors and has been reported to influence the growth suppressor function, DNA binding and transcriptional activation of p53.( 28 )
A cyclin–CDK complex hyperphosphorylates the pRb, leading to its release from E2F.( 29 ) The released transcription factor E2F then activates the genes responsible for cellular proliferation by progression through the G1 into the S phase. The impairment of a growth stimulatory signaling pathway has been shown to induce the expression of CDK inhibitors, which binds to and subsequently inhibits cyclin–CDK activity. This in turn interferes with the hyperphosphorylation of pRb by keeping it in the hypophosphorylated form and bound to E2F, thereby blocking cell proliferation and inducing cell‐growth arrest. Thereafter, the inhibitors of cell cycle progression can also be used as antiproliferation agents.( 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 )
The present study revealed that MCS‐C2 had a growth‐inhibiting and antiproliferative effect on LNCaP cells (Fig. 2), which appeared to be associated with the induction of apoptosis and cell cycle arrest through a p53 pathway (4, 5). In an immunoblot analysis, MCS‐C2 was found to activate p53 through phosphorylation at Ser‐15 and the subsequent transcriptional activation of p21WAF1/CIP1. The significant increase in the level of p21WAF1/CIP1 mRNA after treatment with MCS‐C2 revealed the reason for the G1 cell cycle arrest (Fig. 4b), as the p21WAF1/CIP1 inhibited CDK4,6/cyclin D and induced dephosphorylation of the pRb protein (Fig. 7b). To identify the apoptotic induction by MCS‐C2 through a p53 pathway in LNCaP cells, the apoptotic induction in PC‐3(–/–p53) prostate cancer cells was tested. In PC‐3 cells, MCS‐C2 induced G1 phase arrest of cell cycle progression. However, no apoptotic induction was detectable in PC‐3 cells treated with MCS‐C2 (Fig. 5b). Accordingly, the protein levels of Bax and p21 in PC‐3 cells were not changed (data not shown). These results demonstrated that the apoptosis induction activity of MCS‐C2 was not effective in p53 mutant cell lines, such as PC‐3 cells. Therefore, the induction of apoptosis in LNCaP cells by MCS‐C2 was mediated by p53‐dependent pathways.
The MCS‐C2‐induced apoptosis was confirmed based on the nuclear morphologic changes and DNA fragmentation (Fig. 6b), as well as flow cytometric analysis (5, 6). The apoptosis‐related proteins caspase‐8, caspase‐9, and caspase‐3 were all activated, whereas PARP was inactivated (Fig. 7a). It is already known that Bcl‐2 and Bax play a major role in determining whether cells experience apoptosis under experimental conditions that promote cell death. For example, Bcl‐2 protects cells from apoptosis, but an increased expression of Bax can induce apoptosis. The ratio of Bax to Bcl‐2 is also important for the survival of drug‐induced apoptosis in leukemia cell lines.( 33 ) In the current study, a decrease in Bcl‐2 expression was observed after treatment with MCS‐C2 for 24 h. However, the expression of Bax was upregulated. Hence, the ratio of Bax to Bcl‐2 was altered in favor of apoptosis (Fig. 4a). As such, these results indicate that the MCS‐C2‐induced apoptosis was mediated through the upregulation of Bax by a p53 pathway.
In conclusion, MCS‐C2 was found to induce cell cycle arrest and apoptosis in a p53‐dependent manner, whereby MCS‐C2 activated p53 and upregulated the expression level of p21WAF1/CIP1.
Acknowledgments
This work was supported by research funding from Hanyang University (HY‐2002‐I) and by grant no. R01‐2003‐000‐10594‐0 from the Basic Research Program of the Korea Science and Engineering Foundation.
References
- 1. Landis SH, Murray T, Bolden S, Wingo PA. Cancer statistics, 1999. CA Cancer J Clin 1999; 49: 8–31. [DOI] [PubMed] [Google Scholar]
- 2. McNeal JE. Origin and development of carcinoma in the prostate. Cancer 1969; 23: 24–34. [DOI] [PubMed] [Google Scholar]
- 3. Abbas F, Scardino PT. The natural history of clinical prostate carcinoma. Cancer 1997; 80: 827–33. [DOI] [PubMed] [Google Scholar]
- 4. Effert PJ, McCoy RH, Walther PJ, Liu ET. P53 gene alterations in human prostate carcinoma. J Urol 1993; 150: 257–61. [DOI] [PubMed] [Google Scholar]
- 5. El‐Deiry WS, Tokino T, Velculescu VE et al. WAF1, a potential mediator of p53 tumor suppression. Cell 1993; 75: 817–25. [DOI] [PubMed] [Google Scholar]
- 6. Lim H, Kim MK, Cho YH, Kim JM, Lim Y, Lee CH. Inhibition of cell cycle progression and induction of apoptosis in HeLa cells by HY558‐1, a novel CDK inhibitor isolated from Penicillium minioluteum F558. J Microbiol Biotechnol 2004; 14: 978–84. [Google Scholar]
- 7. Giaccia AJ, Kastan MB. The complexity of p53 modulation: emerging patterns from divergent signals. Genes Dev 1998; 12: 2973–83. [DOI] [PubMed] [Google Scholar]
- 8. Colman MS, Afshari CA, Barrett JC. Regulation of p53 stability and activity in response to genotoxic stress. Mutat Res 2000; 462: 179–88. [DOI] [PubMed] [Google Scholar]
- 9. Park SG, Cheon JY, Lee YH et al. A specific inhibitor of cyclin‐dependent protein kinase, CDC2 and CDK2. Mol Cells 1996; 6: 679–83. [Google Scholar]
- 10. Aszalos A, Lemanski P, Robison R, Davis S, Berk B. Identification of antibiotic 1037 as toyocamycin. J Antibiot (Tokyo) 1966; 19: 285. [PubMed] [Google Scholar]
- 11. Sverak L, Bonar RA, Langlois AJ, Beard JW. Inhibition by toyocamycin of RNA synthesis in mammalian cells and in normal and avian tumor virus‐infected chick embryo cells. Biochim Biophys Acta 1970; 224: 441–50. [DOI] [PubMed] [Google Scholar]
- 12. Wilson WL. Phase I study with toyocamycin (NSC‐63701). Cancer Chemother Report 1968; 52: 301–3. [PubMed] [Google Scholar]
- 13. Clercq EJ, Balzarini D, Madej F, Hansske Robins MJ. Nucleic acid related compounds. 51. Synthesis and biological properties of sugar‐modified analogues of the nucleoside antibiotics tubercidin, toyocamycin, sangivamycin, and formycin. J Med Chem 1987; 30: 481–6. [DOI] [PubMed] [Google Scholar]
- 14. Finch RA, Revankar GR, Chan PK. Structural and functional relationships of toyocamycin on NPM‐translocation. Anticancer Drug Des 1997; 12: 205–15. [PubMed] [Google Scholar]
- 15. Gupta PK, Nassiri MR, Coleman LA, Wotring LL, Drach JC, Townsend LB. Synthesis, cytotoxicity, and antiviral activity of some acyclic analogues of the pyrrolo[2,3‐d]pyrimidine nucleoside antibiotics tubercidin, toyocamycin, and sangivamycin. J Med Chem 1989; 32: 402–8. [DOI] [PubMed] [Google Scholar]
- 16. Krawczyk SH, Nassiri MR, Kucera LS et al. Synthesis and antiproliferative and antiviral activity of 2′‐deoxy‐2′‐fluoroarabinofuranosyl analogs of the nucleoside antibiotics toyocamycin and sangivamycin. J Med Chem 1995; 38: 4106–14. [DOI] [PubMed] [Google Scholar]
- 17. Kim MK, Cho YH, Kim JM et al. Inhibition of cell cycle progression in human promyelocytic leukemia HL‐60 cells by MCS‐C2, novel cyclin‐dependent kinase inhibitor. J Microbiol Biotechnol 2003; 13: 607–12. [Google Scholar]
- 18. Kim MK, Cho YH, Kim JM et al. Induction of apoptosis in human leukemia cells by MCS‐C2 via caspase‐dependent Bid cleavage and cytochrome c release. Cancer Lett 2005; 223: 239–47. [DOI] [PubMed] [Google Scholar]
- 19. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2−ΔΔCT method. Methods 2001; 25: 402–8. [DOI] [PubMed] [Google Scholar]
- 20. Kaufmann SH. Induction of endonucleolytic DNA cleavage in human acute myelogenous leukemia cells by etoposide, camptothecin, and other cytotoxic anticancer drugs: a cautionary note. Cancer Res 1989; 49: 5870–8. [PubMed] [Google Scholar]
- 21. Hickmann JA. Apoptosis induced by anticancer drugs. Cancer Metastasis Rev 1992; 11: 121–39. [DOI] [PubMed] [Google Scholar]
- 22. Tewari M, Quan LT, O’Rourke K et al. Yama/CPP3β, a mammalian homolog of CED‐3 is a crmA‐inhibitable protease that cleaves the death substrate poly (ADP‐ribose) polymerase. Cell 1995; 81: 801–9. [DOI] [PubMed] [Google Scholar]
- 23. Verhagen AM, Ekert PG, Pakusch M et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 2000; 102: 43–53. [DOI] [PubMed] [Google Scholar]
- 24. Fiscella M, Ullrich SJ, Zambrano N et al. Mutation of the serine 15 phosphorylation site of human p53 reduces the ability of p53 to inhibit cell cycle progression. Oncogene 1993; 8: 1519–28. [PubMed] [Google Scholar]
- 25. Shieh SY, Ahn J, Tamai K, Taya Y, Prives C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage‐inducible sites. Genes Dev 2000; 14: 289–300. [PMC free article] [PubMed] [Google Scholar]
- 26. Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage‐induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 1997; 91: 325–34. [DOI] [PubMed] [Google Scholar]
- 27. Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature 1997; 387: 296–9. [DOI] [PubMed] [Google Scholar]
- 28. Kohn KW. Molecular interaction map of the mammalian cell cycle control and DNA repair systems. Mol Biol Cell 1999; 10: 2703–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Puri PL, Maclachlan TK, Levrero M, Giordano A. The intrinsic cell cycle: from yeast to mammals. In: Stein GS, Baserga R, Giordano A, Denhardt DT, eds. The Molecular Basis of Cell Cycle and Growth Control. New York: Wiley, 1999; 15–79. [Google Scholar]
- 30. Kundsen ES, Wang JY. Dual mechanism for the inhibition of E2F binding to Rb by cyclin‐dependent kinase‐mediated Rb phosphorylation. Mol Cell Biol 1997; 17: 5771–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lim HY, Kim MK, Cho YH, Kim JM, Lim YH, Lee CH. Inhibition of cell cycle progression and induction of apoptosis in HeLa cells by HY558‐1, a novel CDK inhibitor isolated from Penicillium minioluteum F558. J Microbiol Biotechnol 2004; 14: 978–84. [Google Scholar]
- 32. Lee CH, Lim HY, Kim MK et al. Isolation and biological properties of novel cell cycle inhibitor, HY558, isolated from Penicillium minioluteum F558. J Microbiol Biotechnol 2002; 12: 470–5. [Google Scholar]
- 33. Sedlak TW, Oltvai ZN, Yang E et al. Multiple Bcl‐2 family members demonstrate selective dimerizations with Bax. Proc Natl Acad Sci USA 1995; 92: 7834–8. [DOI] [PMC free article] [PubMed] [Google Scholar]