

Abstract
To identify the genomic profile and elucidate the pathogenesis of hepatoblastoma (HBL), the most common pediatric hepatic tumor, we performed high‐density genome‐wide single‐nucleotide polymorphism (SNP) microarray analyses of 17 HBL samples. The copy number analyzer for GeneChip® (CNAG) and allele‐specific copy number analysis using anonymous references (AsCNAR) algorithms enabled simple but sensitive inference of allelic composition without using paired normal DNA. Chromosomal aberrations were observed in 15 cases (88%). Gains in chromosomes 1q, 2 (or 2q), 8, 17q, and 20 and losses in chromosomes 4q and 11q were frequently identified. High‐grade amplifications were detected at 7q34, 14q11.2, and 11q22.2. Several types of deletions, except homozygous deletion, were identified. Most importantly, copy‐neutral loss of heterozygosity (uniparental disomy [UPD]) at 11p15 was detected in four of the 17 HBL samples. Insulin‐like growth factor II (IGF2) and H19 genes were located within this region. The methylated status of this region indicated the paternal origin of the UPD. The expression patterns of IGF2 and H19 were opposite between genes with and without the UPD. This difference in the expression patterns might influence the clinical features of HBL. (Cancer Sci 2008; 99: 564–570)
Hepatoblastoma (HBL) is the most common pediatric hepatic tumor predominantly observed in infants and children aged less than 3 years.( 1 , 2 , 3 ) The dramatic increase in the survival of patients that has been observed during the last three decades is mainly due to advances in the use of chemotherapy and surgical techniques.( 1 , 2 , 3 ) Currently, approximately 75% of children with HBL can be cured completely, although a large tumor, a multifocal tumor, and metastatic spread are all associated with a fatal outcome.( 3 ) The etiology of HBL remains unknown. Most HBL are sporadic; however, an association with prematurity or low birth weight,( 4 ) and genetic disorders such as familial adenomatous polyposis (FAP),( 5 ) or Beckwith–Wiedemann syndrome (BWS) has been documented.( 6 ) These findings imply that an alteration at 11p15, which is the critical region in BWS and critical to the wingless signaling pathway involving the adenomatosis polyposis coli (APC) gene that is constitutionally mutated in FAP patients,( 7 , 8 ) could also play a role in the genesis of sporadic HBL. Indeed, the loss of heterozygosity (LOH) at 11p15 and mutations in the APC and β‐catenin genes have also observed in some sporadic HBL.( 9 , 10 )
LOH and deletion of tumor suppressor genes are observed frequently in malignant cells and can be associated with the deregulation of cell fate and apoptosis.( 11 ) Similarly, amplification of the chromosomal regions can increase the expression of oncogenes during tumor progression. Conventional cytogenetic analyses of chromosomal aberrations in HBL performed using standard karyotyping,( 12 , 13 , 14 , 15 , 16 ) fluorescence in situ hybridization (FISH),( 17 , 18 , 19 , 20 ) and comparative genomic hybridization (CGH),( 21 , 22 ) have been reported. Although these analyses have identified several chromosomal aberrations in HBL, predominantly the gains in chromosomes 1q, 2, 8q, 17q, and 20 and the loss in chromosome 4q, the tumor‐associated genes of HBL involved in these genomic copy number (CN) alterations are yet to be identified.
In recent years, a high‐resolution genomic approach has been used for the systematic screening of chromosomal CN alterations. The availability of microarray‐based high‐density single‐nucleotide polymorphism (SNP) analysis allows a reproducible and rapid determination of genome‐wide alterations.( 23 , 24 , 25 ) The Affymetrix® GeneChip® platform, originally developed for large‐scale SNP typing, has a unique feature compared with array‐based CGH: it enables the genome‐wide detection of LOH in addition to extremely high‐resolution CN analysis of cancer genomes by using large numbers of SNP‐specific probes. The density, distribution, and allele specificity of SNP render them an excellent candidate for the high‐resolution analyses of LOH and CN alterations in cancer genomes.( 26 , 27 ) Conventionally, LOH analyses require the comparison of the genotypes of the tumor and its normal germline counterpart. However, for the analysis of cell line, xenograft, leukemia, and archival samples, paired normal DNA is often unavailable. In the absence of a paired normal DNA sample, LOH is inferred only based on the lower‐than‐expected frequencies of heterozygous SNP calls in the tumor samples. However, the low tumor content within the samples greatly hampers the sensitive detection of LOH due to increased heterozygous SNP calls. To overcome these difficulties with the current algorithms, we have recently developed novel algorithms (copy number analyzer for GeneChip®[CNAG] and allele‐specific copy number analysis using anonymous references [AsCNAR]) to analyze the allelic composition of cancer genomes based on the microarray data obtained from the GeneChip® platform.( 27 , 28 ) These algorithms calculate the allele‐specific CN independent of the availability of a paired control DNA, enabling the sensitive detection of both LOH and CN alterations in a wide spectrum of primary tumor specimens. The performance of the new algorithm was demonstrated by detecting the neutral CN LOH or uniparental disomy (UPD) in a large number of acute leukemia samples.( 28 )
In the present study, to identify the novel genomic alterations in sporadic HBL cases, we performed high‐resolution analyses of genome‐wide CN alterations such as gains, losses, allelic imbalances, and amplifications of small chromosomal regions. Due to the high resolution of the SNP arrays and the new algorithm AsCNAR, we could systematically identify several amplifications, deletions, and allelic imbalances, including the UPD.
Materials and Methods
Patients and samples. We obtained 17 primary HBL samples at the time of diagnosis from five patients treated at the Gunma Children's Medical Center and 12 patients treated at different institutes in Japan, including Saitama Children's Medical Center. No patient had received chemo‐ and/or radiotherapy before the biopsy of the primary tumors. After obtaining informed consent from the parents and approval for the study from the institutional review board of each institute, all the HBL samples were subjected to genomic DNA extraction using the QIAamp DNA Mini Kit (Qiagen, Chatsworth, CA, USA) according to the manufacturer's instructions. Total RNA was extracted from the frozen stocked tumors using the Isogen reagent (Nippon Gene, Osaka, Japan), according to the manufacturer's instructions. The total RNA was reverse transcribed to synthesize cDNA using the Ready‐To‐Go T‐Primed First‐Strand Kit (GE Healthcare Bio‐Sciences, Piscataway, NJ, USA).
SNP array analysis. The array experiments were performed according to the standard protocol of Affymetrix® GeneChip® Mapping 50K XbaI Array (Affymetrix, Inc., Santa Clara, CA, USA). In brief, the total genomic DNA (250 ng from each sample) was first digested with a restriction enzyme (XbaI). The digested DNA was then ligated to an appropriate adapter that recognized the four cohesive base pair (bp) overhangs, and polymerase chain reaction (PCR) amplification was performed using a single primer that recognized the adapter sequence. After fragmentation with DNase I, the PCR products were labeled with a biotinylated nucleotide analog using terminal deoxynucleotidyl transferase, and the labeled products were hybridized to the GeneChip® Human Mapping 50K Array for 17 h. Subsequently, the arrays were washed, stained, and scanned.
The genotype calls and the intensity of the SNP probes were determined using GeneChip Operation software (GCOS; Affymetrix, Inc.). The SNP CN and chromosomal regions with gains or losses were individually evaluated using the CNAG( 27 ) and AsCNAR algorithms,( 28 ) which enabled an accurate determination of allele‐specific CN as well as the sensitive detection of LOH even in the presence of normal cell contamination of up to 70–80% without requiring constitutive DNA (Fig. 1; http://www.genome.umin.jp).
Figure 1.
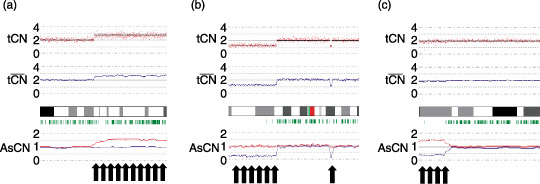
Representative results of the allele‐specific copy number analysis using anonymous references (AsCNAR) program with regard to copy number (CN) alterations detected in our series at particular loci, such as (a) gain (b) chromosomal loss, and (c) uniparental disomy (UPD), which have not been detected using conventional algorithms. The red dots indicate the raw CN plot for each single‐nucleotide polymorphism (SNP), and the blue lines indicate the local mean CN of five SNP. The vertical green bar indicates the heterozygous SNP calls.
Validation of CN alterations using the interphase FISH. We performed FISH to validate the CN status obtained using the SNP array analysis. FISH probes were prepared using the BAC clones RP11‐185M22, RP11‐80P10, and RP11‐86M15. Each BAC DNA was purified, and 100 ng of the clone was labeled with digoxigenin‐dUTP using random primers; these labeled clones were used as probes for FISH analysis by following the established protocols.( 29 , 30 )
Quantitative real‐time PCR and reverse transcription (RT)‐PCR. Real‐time quantitative PCR (RQ‐PCR) and real‐time quantitative RT‐PCR (RQ‐RT‐PCR) analyses were carried out to quantify the relative CN of several amplifications in the HBL samples and the expression levels of the defender against cell death 1 (DAD1), EPH receptor B6 (EphB6), ErbB4, insulin‐like growth factor II (IGF2), and H19 genes using a Power SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA) with an ABI prism 7700 real‐time PCR detection system (Applied Biosystems). The primer pairs were designed using PrimerExpress software (Applied Biosystems) and synthesized by Invitrogen (Carlsbad, CA, USA).The primer sets used for the RQ‐PCR experiments are listed in Table 1. Data were captured using Sequence Detection software (version 1.7a; Applied Biosystems). For each primer pair, a standard curve was generated from five‐fold serial dilution from approximately 50–80 pg of control DNA from a healthy individual. The amounts of genomic DNA and cDNA used in each test and the reference marker for all HBL samples were calculated using the appropriate standard curve. Normalization was performed using the β‐actin gene as the internal control.
Table 1.
Primers used for polymerase chain reaction (PCR) analyses
| Gene | Primer forward | Primer reverse |
|---|---|---|
| (Genomic RQ‐PCR) | ||
| EphB6 | GGACTGCAACTGAACGTCAA | TCTGGAAAGGAAGCAAAGGA |
| DAD1 | GTTATGTCGGCGTCGGTAGT | GTCCCCACGAGGAGACAGTA |
| (RQ‐RT‐PCR) | ||
| ERBB4 | AACAGCAGTACCGAGCCTTG | CCAGAGGCAGGTAACGAAAC |
| DAD1 | CGAGCCTTTGCTGATTTTCT | TCCAATAAGCTGCCATCTCC |
| IGF2 | CTCTCCGTGCTGTTCTCTCC | TATCGGGAAATGAGGTCAGC |
| H19 | GAAGGAGGTTTAGGGGATCG | TTGCTCTTTCTGCCTGGAAC |
| (Bisulfite PCR/RQ‐PCR) | ||
| H19DMR (Methylated) | GGTACGGTTTTTTTAGGTTTATGTC | ACCCCTACAACCTCCTTACTACG |
| H19DMR (Unmethylated) | TATGGTTTTTTTAGGTTTATGTTGG | ACCCCTACAACCTCCTTACTACAC |
Primers and probes were designed using Primer Express software and MethPrimer software. RQ‐PCR, real‐time quantitative PCR; RQ‐RT‐PCR, real‐time quantitative reverse transcription‐PCR.
Sodium bisulfite modification and methylation‐specific PCR. The genomic DNA from the tumor samples was treated with sodium bisulfite as described previously.( 31 ) Briefly, 1 µg of DNA was denatured with sodium hydroxide and modified with sodium bisulfite. The modified DNA was then purified with the Wizard® DNA Clean‐Up System (Promega, Madison, WI, USA), precipitated with ethanol, resuspended in Tris‐EDTA (TE) buffer (pH 8.0), and either used immediately or stored at –20°C until use. The bisulfite‐modified DNA was amplified with primer pairs for the methylated and unmethylated complete sequences upstream of the H19 promoter CpG islands in the HBL samples with UPD in 11p15. The primer pairs were designed using MethPrimer software,( 32 ) and synthesized by Invitrogen. The primers for methylation‐specific DNA and unmethylation‐specific primers are listed in Table 1. Normal lymphocyte DNA was used as the control. PCR was carried out in a 25 µL reaction volume using Ex Taq Hot Start Version (TaKaRa Bio Inc., Kyoto, Japan). The PCR conditions were as follows: 1 cycle at 95°C for 10 min; followed by 35 cycles of 94°C for 30 s, 60°C for 30 s, 72°C for 2 min; and a final extension at 72°C for 5 min. The PCR products were separated on 3% agarose gels and visualized under UV illumination after ethidium bromide staining. To quantify the ratio of the methylation status, we also carried out the methylation‐specific RQ‐PCR analysis.
Results
Detection of CN alterations in HBL samples. We investigated 17 HBL samples obtained from the sporadic cases of HBL by using the Affymetrix® GeneChip® 50K XbaI Mapping Array. Although these specimens did not contain paired control DNA and had varying degrees of normal tissue contamination, the genomic alterations were accurately determined in most specimens by our CNAG/AsCNAR program (Fig. 1). The real CN and LOH status was inferred from the observed signal ratios of the tumor to the reference, based on the hidden Markov models implemented in the CNAG/AsCNAR program; these are summarized in Fig. 2. The CN data were validated at a number of SNP sites using FISH analysis of the cell nuclei extracted from the HBL samples (Fig. 3). The CN data obtained using the FISH analyses were consistent with those obtained using SNP mapping.
Figure 2.

Overview of the DNA copy number (CN) gains and losses detected in 17 hepatoblastoma (HBL) samples. A gain is indicated by the red bar above the chromosome ideogram, and a loss is indicated by the green bar under the chromosome ideogram. Each horizontal line represents an aberration detected in a single tumor.
Figure 3.
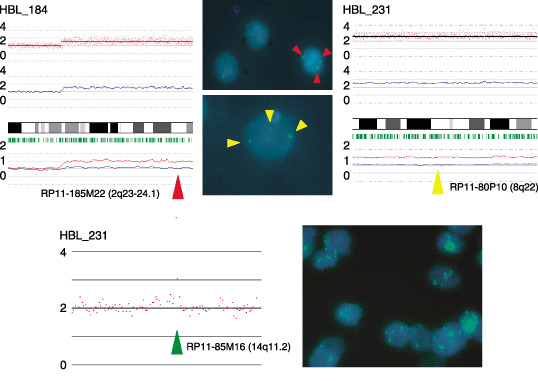
Representative results of array analysis of hepatoblastoma (HBL) samples (HBL_184 and HBL_231). Fluorescent in situ hybridization analysis with BAC probes confirmed the detected changes. We detected three signals from chromosomes 2q and 8q. At the high‐amplification region of chromosome 14q, three and more signals were detected.
Numerical chromosomal aberrations were observed in 15 HBL samples (88%), excluding two HBL samples (HBL_22 and HBL_250). These 15 cases had variable degrees of CN gains and losses; however, the gains including the amplifications were more frequent than the losses (Table 2 and Fig. 2). Total or partial gains in chromosomes 1q and 2 were the most frequent aberrations detected in eight of the 17 patients (47%). The gain in chromosome 8 was the second most frequent aberration detected in five of the 17 samples (29%). The gains in chromosomes 17q and 20 were observed in 24% of the cases (four of 17 cases). The LOH in chromosomes 4q and 11q was observed in three (18%) and two (12%) of the 17 samples, respectively. However, these regions were usually large, and we could not determine the presence or absence of alterations in specific genes within these regions.
Table 2.
Chromosomal aberrations in 17 primary hepatoblastoma (HBL) samples
| Sample | Copy number gain | Copy number loss | Uniparental disomy |
|---|---|---|---|
| HBL_4 | 1q, 2q34, 5p13.1, 17q23.3‐qter | 3p13‐pter, 3q13.11, 6q14.1‐qter, 11q23.1‐qter | Not detected |
| HBL_7 | 1q, 2, 8, 14q11.2, 20 | not detected | 11p15.4‐pter |
| HBL_8 | 7q34 | not detected | Not detected |
| HBL_9 | 1q, 2q14.1‐qter, 6p, 7, 14q11.2 | 6p12.1, 9p21.1 | Not detected |
| HBL_12 | 8q11.23, 10q21.3, 10q26.13, 14q11.2, 22q13.31 | 7q35 | Not detected |
| HBL_14 | 2p16.3‐p22.3, 2p23.1, 2q11.2‐q14.1, 2q33.1‐q34, 3p21.33‐p22.1, 3p24.2, 3p25.1, 3p25.2, 4q32.2‐q32.3, 5p13.2, 6q14.3‐q16.1, 7q, 11p15.1, 10p13‐pter, 11q22.2‐q22.3, 12p13.2‐pter, 14q23.3‐q31.1, 15q22.31‐q26.2, 16p12.3, 20p11.23 | 1q31.1‐qter, 2p12‐14, 3, 4q, 5p14.1‐pter, 5q32‐qter, 6p12.13‐pter, 6q11.1, 6q25.1‐qter, 8, 9, 12p11.1‐13.1, 17q24.3, 18p11.21‐11.32, 18q21.1‐qter, 19, 22 | Not detected |
| HBL_22 | Not detected | Not detected | Not detected |
| HBL_27 | 1q, 2q24.2‐24.3, 7q34, 14q11.2 | 4q32.3‐qter, 16p12.1 | Not detected |
| HBL_28 | 3p26.1, 7q34, 14q11.2, 20 | 2p24.1 | 11p14.3‐pter |
| HBL_34 | 1q, 2, 7q34, 14q11.2, 17 | 4q34.1‐qter | Not detected |
| HBL_36 | 1q32.1‐qter | 1p13.3‐pter, 4q21.22‐qter, 5p13.1 | Not detected |
| HBL_37 | 1q, 2, 5, 6, 7q34, 8, 10, 12, 14, 14q11.2, 15, 16q22.1‐pter, 17, 19, 20 | Not detected | 11p15.2‐pter, 16q22.2‐qter |
| HBL_184 | 2q14.2‐qter, 3p24.3, 4q33, 10p14, 11p14.3, 14q11.2 | Not detected | Not detected |
| HBL_185 | 6p, 21q21.2 | Not detected | Not detected |
| HBL_231 | 8, 14q11.2, 19, 20 | Not detected | 11p15.4‐pter |
| HBL_246 | 1q, 2, 5, 6, 8, 10, 12, 13, 16, 17, 19, 20, 21, 22 | Not detected | 4, 9 |
| HBL_250 | Not detected | Not detected | Not detected |
High‐grade amplification and common deletion. High‐grade amplifications are of particular interest because they may indicate the loci of oncogenes. The regions with high‐grade amplification were defined as segments with at least five SNP loci with an inferred CN of >5. High‐grade amplifications of 7q34 and 14q11.2 were observed in five (29%) and nine (53%) HBL samples, respectively. For the validation of the amplifications observed using the SNP array, FISH analysis and genomic RQ‐PCR were performed. To determine the genes that are potentially affected at 14q11.2, several genes localized at the 14q11.2 chromosomal region with overlap or proximity to the BAC‐RP11‐85M16 were examined using the UCSC browser (http://www.genome.ucsc.edu). Genes that map to these regions include EphB6 and DAD1, which are identified as the negative regulators of apoptosis. These two genes were subjected to RQ‐PCR. FISH analysis with RP11‐85M16 BAC clone probe showed multiple signals, confirming CN gains at 14q11.2 (Fig. 3). Further, in RQ‐PCR analysis, the CN gain of EphB6 and DAD1 was evident in all samples that showed high‐grade amplification in SNP array (data not shown). Other high‐grade amplifications are listed in Table 3. The size of these amplicons was typically less than 1 Mb, and the possible genes present in these regions are summarized in the same table. All these candidate genes, except MMP7, have not been reported previously with regard to HBL.( 33 )
Table 3.
High‐grade amplifications in hepatoblastoma (HBL) samples
| Cytoband | Implicated region (base pairs) | Candidate target genes in the region | |
|---|---|---|---|
| Start–end | Size | ||
| 2q34 | 211 193 864–212 239 181 | 1 045 318 | ErbB4 |
| 3p25.2 | 11 888 124–12 876 175 | 988 052 | RAF1 |
| 7q34 | 141 721 559–142 076 238 | 354 680 | EphB6 |
| 11q22.2‐q22.3 | 101 394 973–102 830 195 | 1 435 223 | MMP1, 7, 20 |
| 14q11.2 | 21 426 631–22 130 392 | 703 762 | DAD1 |
Homozygous deletions are also of particular interest because they may indicate a tumor suppressor gene. However, homozygous deletions were not identified in any sample.
CN neutral LOH (UPD). LOH can be more sensitively detected with the CNAG/AsCNAR algorithms by evaluating the allele‐specific CN than from the grossly reduced heterozygous SNP calls, particularly when the SNP shows no CN losses. The UPD regions were identified in five of the 17 samples. In four samples (HBL_7, HBL_28, HBL_37, and HBL_231), 11p15 was the common UPD region (Fig. 4a). Other UPD regions were observed within chromosomes 4, 9, and 16q22 (Table 2). The candidate target genes that map to the UPD region located within 11p15 include IGF2 and H19. Methylation‐specific PCR analysis was performed for the four HBL samples having UPD within 11p15 to identify the origin of the amplified allele. The methylation status of the differential methylated region (DMR) of H19 is shown in Fig. 4b. Hypermethylation of the H19 DMR was detected in all HBL samples having UPD within 11p15; however, normal lymphocyte DNA showed the mosaic methylation pattern. In general, the H19 DMR is hypermethylated on the paternal allele and hypomethylated on the maternally expressed allele in humans. This indicates that the UPD within this region is considered to be derived from the paternal allele. Furthermore, a low expression level of the non‐methylated allele was also observed; methylation‐specific RQ‐PCR analysis revealed that the ratio of the methylation status ranged from 66.6% to 91.8%.
Figure 4.
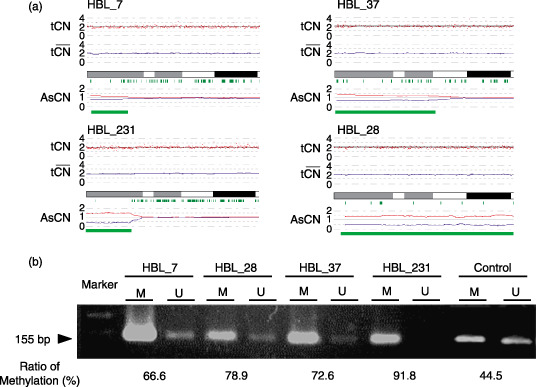
(a) Copy numbers (CN) of chromosome 11p in four hepatoblastoma (HBL) samples with uniparental disomy (UPD). Although complete CN alterations are not observed, UPD is clearly predicted based on the allele‐specific CN alterations (green lines). (b) Methylation‐specific polymerase chain reaction (PCR) analysis of the H19 differential methylated region (DMR). Modified DNA was amplified with primer pairs for methylated and unmethylated complete sequences of the H19 DMR. H19 DMR hypermethylation was detected in all HBL samples; however, normal lymphocyte DNA exhibited the mosaic methylation pattern. The results of quantitative real‐time methylation‐specific PCR analysis are shown below the image depicting the results of electrophoresis.
Expression analyses using RQ‐RT‐PCR. In order to examine the impact of the abovementioned amplifications and UPD on gene expression, we measured the expression levels of four genes (DAD1, ErbB4, IGF2, and H19) through RQ‐RT‐PCR (Fig. 5). Normal liver total RNA served as the non‐neoplastic reference and control. HBL_184 and HBL_231 for which RNA were available showed a high expression of the ErbB4 gene. However, the expression of DAD1 was down‐regulated in both these samples. The IGF2 and H19 genes were oppositely expressed between HBL_184 and HBL_231, having UPD within 11p15.
Figure 5.
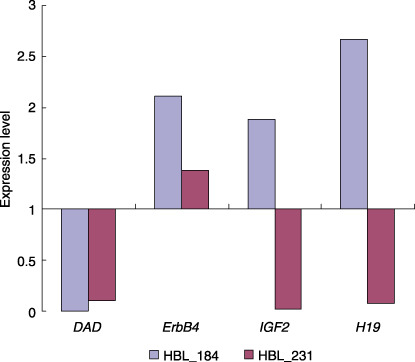
The results of the expression levels of four genes (defender against cell death 1 [DAD1], EPH receptor B6 [EphB6], ErbB4, insulin‐like growth factor II [IGF2], and H19 genes) through real‐time quantitative reverse transcription‐polymerase chain reaction (RQ‐RT‐PCR) analyses.
Discussion
The present study represents the application of the SNP array technology for the genome‐wide analysis of CN aberrations in HBL. Several recent studies and our previous research have demonstrated that this technology provided a unique opportunity to assess the DNA CN alterations and LOH simultaneously throughout the entire genome.( 24 , 25 , 26 , 27 , 29 ) As shown in the present analysis, the use of high‐resolution SNP arrays improved the ability to identify structural chromosomal aberrations in cancer cells and detect genes affected by these aberrations. Additionally, high‐density SNP array analysis with the CN analyzer software can also facilitate the identification of allelic imbalances such as copy‐neutral LOH in the absence of a paired normal DNA reference.
The aberrations in chromosomes 1q, 2, 8, and 20 have been noted as the most commonly occurring aberrations in all previous reports,( 21 , 22 ) as well as in the present study. In the present study, the most frequently detected aberrations were gains in chromosomes 1q and 2 (or 2q), observed in approximately 50% of the cases.
Trisomy in chromosome 1q is a well‐known alteration in HBL.( 34 ) Similar 1q imbalances have also been described in other pediatric neoplastic disorders such as lymphoma,( 35 ) Wilms’ tumor,( 36 ) and sarcoma,( 37 ) indicating that these aberrations are related to tumor progression. The candidate genes in 1q included the NTRK1, ABL2, CD34, DAP3 (death receptor protein‐3), and caspase‐3 genes.( 38 ) The anomalies in chromosome 2, which almost always result in gains in 2q, are also common in HBL. These imbalances are also commonly found in embryonal rhabdomyosarcoma and other pediatric tumors related to BWS. Translocation involving the PAX3 gene located in 2q35 has been suggested to play a crucial role in the pathogenesis of alveolar rhabdomyosarcoma.( 39 ) Based on this, a genetic link has been suggested between HBL and alveolar rhabdomyosarcoma. The role of the PAX3 gene in the pathogenesis of HBL is yet to be determined. Additionally, the 2q24–32 region contains several genes that may also have an oncogenic potential. These include a serine/threonine kinase receptor, ITRAF, FRZB, a secreted antagonist of WNT signaling, and BRCA1‐associated RING domain 1 (BARD1) genes. However, no specific gene has been identified in the previous,( 21 , 22 ) and present studies.
The losses in chromosomes 4q and 11q were comprehensively observed. In hepatocellular carcinoma (HCC) cells, Wong et al. demonstrated a growth advantage following the loss in the 4q arm.( 40 ) In HCC, 4q21–q22 and 4q35 have been identified as commonly deleted regions, and allelic losses in 4q35 have been associated with a larger tumor size and an aggressive histological tumor type.( 41 ) Previous studies have not reported a significant correlation between HBL with loss in the distal 4q arm and prognosis, but the underlying oncogenic event might be due to the loss of a gene on the distal 4q arm.
Many minimal regions of amplification and deletion were detected using high‐density SNP arrays, although homozygous deletion was not identified in any sample. The SNP loci located in 7q34 and 14q11.2 were found to be highly amplified in sporadic HBL samples. The candidate genes at these loci are EphB6, DAD1, and BCL‐like 2 (BCL2L2) genes that encode the proteins associated with the execution of cell apoptosis. Gains as well as high amplifications in this region have not been reported previously; however, such an observation will be of particular interest for the discovery of oncogenes involved in the pathogenesis of HBL.
The UPD regions were identified in five of the 17 samples. This is chiefly important because UPD is being particularly considered as a possible mechanism of tumor initiation. During tumorigenesis, UPD is believed to arise due to a mitotic recombination caused by a rare crossover event during mitotic cell division. The products of mitotic recombination are the regions of the genome exhibiting UPD, and both the genomic regions originate from the same parent. We could identify a common UPD on chromosome 11p that is reminiscent of BWS with paternal UPD; in this case, the loss of function of the 11p15 maternal alleles through various mechanisms may be the critical event associated with tumorigenesis and BWS.( 42 ) BWS is a neonatal overgrowth syndrome that predisposes an individual to cancer,( 6 ) and the importance of the maternally active locus in chromosome 11p15 in tumorigenesis is supported by the finding that the loss of imprinted allele and paternal duplication leads to tissue overgrowth and subsequent tumor development. Methylation analysis was performed for the four HBL samples having UPD within 11p15, and hypermethylation of H19 DMR was detected in all four HBL samples. Because H19 DMR was hypermethylated on the paternal allele and hypomethylated on the maternally expressed allele in humans, we consider that the UPD within 11p15 was of paternal origin.
Two candidate genes, namely, IGF2 and H19, are located within the telomeric region of chromosome 11p15.5 and have opposite imprinting patterns.( 43 ) In the majority of human tissues, IGF2 is expressed only from the paternal allele, whereas H19 is transcribed only from the maternal allele. H19 is an untranslated gene but has been suggested to function as a tumor suppressor.( 44 ) In fetal and adult organs, the transcriptionally silent H19 allele was extensively hypermethylated throughout the entire gene and its promoter. On the maternally expressed H19 allele, H19 DMR is unmethylated and can bind to the CTCF protein. On the paternal H19 allele, H19 DMR is highly methylated. This not only prevents the expression of the imprinted paternal H19 alleles but also blocks the binding of the CTCF protein.( 43 ) In general, the outcome of UPD with losses of the 11p15 maternal alleles in HBL is the same as that of the loss of imprinting on the inactivated, imprinted, and maternally expressed genes in BWS. Weksberg et al. proposed a dual pathway model for tumor development in BWS, wherein methylation defects at H19 and/or IGF2 in 11p15 were found to play a role in Wilms’ and HBL tumorigenesis.( 45 ) The combined loss of expressions in various 11p15‐imprinted genes may contribute to tumorigenesis.
In the present study, we identified that the expression patterns of IGF2 and H19 were opposite between genes with and without the UPD in 11p15. This difference in the expression patterns might influence the clinical features of HBL. Further prospective studies are required to reveal any potential correlations between specific LOH and clinical outcomes.
In summary, the analysis of LOH and CN alterations using the SNP microarray in HBL samples revealed significant areas of allelic imbalance. We hypothesize that UPD, in addition to allelic imbalance, constitutes a novel genetic mechanism involved in tumorigenesis. Therefore, detailed characterizations such as functional studies should be conducted to elucidate the significance of the regions detected in this study, many of which may contain the candidate tumor suppressor genes and oncogenes involved in the pathogenesis of HBL.
Acknowledgments
This work was supported in part by a Grant‐in‐Aid for Cancer Research from the Ministry of Health, Labour and Welfare of Japan, a Grant‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and a grant from the 21st Century COE Program from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- 1. Roebuck DJ, Perilongo G. Hepatoblastoma: an oncological review. Pediatr Radiol 2006; 36: 183–6. [DOI] [PubMed] [Google Scholar]
- 2. Tiao GM, Bobey N, Allen S et al . The current management of hepatoblastoma: a combination of chemotherapy, conventional resection, and liver transplantation. J Pediatr 2005; 146: 204–11. [DOI] [PubMed] [Google Scholar]
- 3. Schnater JM, Kohler SE, Lamers WH, Von Schweinitz D, Aronson DC. Where do we stand with hepatoblastoma? A review. Cancer 2003; 98: 668–78. [DOI] [PubMed] [Google Scholar]
- 4. Ikeda H, Matsuyama S, Tanimura M. Association between hepatoblastoma and very low birth weight: a trend or a chance? J Pediatr 1997; 130: 557–60. [DOI] [PubMed] [Google Scholar]
- 5. Hughes LJ, Michels VV. Risk of hepatoblastoma in familial adenomatous polyposis. Am J Med Genet 1992; 43: 1023–5. [DOI] [PubMed] [Google Scholar]
- 6. DeBaun MR, Tucker MA. Risk of cancer during the first four years of life in children from The Beckwith–Wiedemann Syndrome Registry. J Pediatr 1998; 132: 398–400. [DOI] [PubMed] [Google Scholar]
- 7. Fukuzawa R, Hata J, Hayashi Y, Ikeda H, Reeve AE. Beckwith–Wiedemann syndrome‐associated hepatoblastoma: wnt signal activation occurs later in tumorigenesis in patients with 11p15.5 uniparental disomy. Pediatr Dev Pathol 2003; 6: 299–306. [DOI] [PubMed] [Google Scholar]
- 8. Little MH, Thomson DB, Hayward NK, Smith PJ. Loss of alleles on the short arm of chromosome 11 in a hepatoblastoma from a child with Beckwith–Wiedemann syndrome. Hum Genet 1988; 79: 186–9. [DOI] [PubMed] [Google Scholar]
- 9. Albrecht S, Von Schweinitz D, Waha A, Kraus JA, Von Deimling A, Pietsch T. Loss of maternal alleles on chromosome arm 11p in hepatoblastoma. Cancer Res 1994; 54: 5041–4. [PubMed] [Google Scholar]
- 10. Oda H, Imai Y, Nakatsuru Y, Hata J, Ishikawa T. Somatic mutations of the APC gene in sporadic hepatoblastomas. Cancer Res 1996; 56: 3320–3. [PubMed] [Google Scholar]
- 11. Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature 1998; 396: 643–9. [DOI] [PubMed] [Google Scholar]
- 12. Yeh YA, Rao PH, Cigna CT, Middlesworth W, Lefkowitch JH, Murty VV. Trisomy 1q, 2, and 20 in a case of hepatoblastoma: possible significance of 2q35‐q37 and 1q12‐q21 rearrangements. Cancer Genet Cytogenet 2000; 123: 140–3. [DOI] [PubMed] [Google Scholar]
- 13. Nagata T, Mugishima H, Shichino H et al . Karyotypic analyses of hepatoblastoma. Report of two cases and review of the literature suggesting chromosomal loci responsible for the pathogenesis of this disease. Cancer Genet Cytogenet 1999; 114: 42–50. [DOI] [PubMed] [Google Scholar]
- 14. Sainati L, Leszl A, Stella M et al . Cytogenetic analysis of hepatoblastoma: hypothesis of cytogenetic evolution in such tumors and results of a multicentric study. Cancer Genet Cytogenet 1998; 104: 39–44. [DOI] [PubMed] [Google Scholar]
- 15. Tonk VS, Wilson KS, Timmons CF, Schneider NR. Trisomy 2, trisomy 20, and del (17p) as sole chromosomal abnormalities in three cases of hepatoblastoma. Genes Chromosomes Cancer 1994; 11: 199–202. [DOI] [PubMed] [Google Scholar]
- 16. Park JP, Ornvold KT, Brown AM, Mohandas TK. Trisomy 2 and 19, and tetrasomy 1q and 14 in hepatoblastoma. Cancer Genet Cytogenet 1999; 115: 86–7. [DOI] [PubMed] [Google Scholar]
- 17. Balogh E, Swanton S, Kiss C, Jakab ZS, Secker‐Walker LM, Olah E. Fluorescence in situ hybridization reveals trisomy 2q by insertion into 9p in hepatoblastoma. Cancer Genet Cytogenet 1998; 102: 148–50. [DOI] [PubMed] [Google Scholar]
- 18. Sainati L, Leszl A, Surace C, Perilongo G, Rocchi M, Basso G. Fluorescence in situ hybridization improves cytogenetic results in the analysis of hepatoblastoma. Cancer Genet Cytogenet 2002; 134: 18–20. [DOI] [PubMed] [Google Scholar]
- 19. Surace C, Leszl A, Perilongo G, Rocchi M, Basso G, Sainati L. Fluorescent in situ hybridization (FISH) reveals frequent and recurrent numerical and structural abnormalities in hepatoblastoma with no informative karyotype. Med Pediatr Oncol 2002; 39: 536–9. [DOI] [PubMed] [Google Scholar]
- 20. Parada LA, Limon J, Iliszko M et al . Cytogenetics of hepatoblastoma: further characterization of 1q rearrangements by fluorescence in situ hybridization: an international collaborative study. Med Pediatr Oncol 2000; 34: 165–70. [DOI] [PubMed] [Google Scholar]
- 21. Hu J, Wills M, Baker BA, Perlman EJ. Comparative genomic hybridization analysis of hepatoblastomas. Genes Chromosomes Cancer 2000; 27: 196–201. [DOI] [PubMed] [Google Scholar]
- 22. Weber RG, Pietsch T, Von Schweinitz D, Lichter P. Characterization of genomic alterations in hepatoblastomas. A role for gains on chromosomes 8q and 20 as predictors of poor outcome. Am J Pathol 2000; 157: 571–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Janne PA, Li C, Zhao X et al . High‐resolution single‐nucleotide polymorphism array and clustering analysis of loss of heterozygosity in human lung cancer cell lines. Oncogene 2004; 23: 2716–26. [DOI] [PubMed] [Google Scholar]
- 24. Huang J, Wei W, Zhang J et al . Whole genome DNA copy number changes identified by high density oligonucleotide arrays. Hum Genomics 2004; 1: 287–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Peiffer DA, Le JM, Steemers FJ et al . High‐resolution genomic profiling of chromosomal aberrations using Infinium whole‐genome genotyping. Genome Res 2006; 16: 1136–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhao X, Li C, Paez JG et al . An integrated view of copy number and allelic alterations in the cancer genome using single nucleotide polymorphism arrays. Cancer Res 2004; 64: 3060–71. [DOI] [PubMed] [Google Scholar]
- 27. Nannya Y, Sanada M, Nakazaki K et al . A robust algorithm for copy number detection using high‐density oligonucleotide single nucleotide polymorphism genotyping arrays. Cancer Res 2005; 65: 6071–9. [DOI] [PubMed] [Google Scholar]
- 28. Yamamoto G, Nannya Y, Kato M et al . Highly sensitive method for genomewide detection of allelic composition in nonpaired, primary tumor specimens by use of affymetrix single‐nucleotide‐polymorphism genotyping microarrays. Am J Hum Genet 2007; 81: 114–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wong KK, Tsang YT, Shen J et al . Allelic imbalance analysis by high‐density single‐nucleotide polymorphic allele (SNP) array with whole genome amplified DNA. Nucleic Acids Res 2004; 32: e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Trask BJ. Fluorescence in situ hybridization: applications in cytogenetics and gene mapping. Trends Genet 1991; 7: 149–54. [DOI] [PubMed] [Google Scholar]
- 31. Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation‐specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA 1996; 93: 9821–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics 2002; 18: 1427–31. [DOI] [PubMed] [Google Scholar]
- 33. Koch A, Waha A, Hartmann W et al . Elevated expression of Wnt antagonists is a common event in hepatoblastomas. Clin Cancer Res 2005; 11: 4295–304. [DOI] [PubMed] [Google Scholar]
- 34. Douglass EC, Green AA, Hayes FA, Etcubanas E, Horowitz M, Wilimas JA. Chromosome 1 abnormalities: a common feature of pediatric solid tumors. J Natl Cancer Inst 1985; 75: 51–4. [PubMed] [Google Scholar]
- 35. Kaneko Y, Variakojis D, Kluskens L, Rowley JD. Lymphoblastic lymphoma: cytogenetic, pathologic, and immunologic studies. Int J Cancer 1982; 30: 273–9. [DOI] [PubMed] [Google Scholar]
- 36. Kaneko Y, Kondo K, Rowley JD, Moohr JW, Maurer HS. Further chromosome studies on Wilms’ tumor cells of patients without aniridia. Cancer Genet Cytogenet 1983; 10: 191–7. [DOI] [PubMed] [Google Scholar]
- 37. Nilsson M, Meza‐Zepeda LA, Mertens F, Forus A, Myklebost O, Mandahl N. Amplification of chromosome 1 sequences in lipomatous tumors and other sarcomas. Int J Cancer 2004; 109: 363–9. [DOI] [PubMed] [Google Scholar]
- 38. Kissil JL, Kimchi A. Assignment of death associated protein 3 (DAP3) to human chromosome 1q21 by in situ hybridization. Cytogenet Cell Genet 1997; 77: 252. [DOI] [PubMed] [Google Scholar]
- 39. Turc‐Carel C, Lizard‐Nacol S, Justrabo E, Favrot M, Philip T, Tabone E. Consistent chromosomal translocation in alveolar rhabdomyosarcoma. Cancer Genet Cytogenet 1986; 19: 361–2. [DOI] [PubMed] [Google Scholar]
- 40. Wong N, Lai P, Lee SW et al . Assessment of genetic changes in hepatocellular carcinoma by comparative genomic hybridization analysis: relationship to disease stage, tumor size, and cirrhosis. Am J Pathol 1999; 154: 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bando K, Nagai H, Matsumoto S et al . Identification of a 1‐cM region of common deletion on 4q35 associated with progression of hepatocellular carcinoma. Genes Chromosomes Cancer 1999; 25: 284–9. [PubMed] [Google Scholar]
- 42. Koufos A, Hansen MF, Copeland NG, Jenkins NA, Lampkin BC, Cavenee WK. Loss of heterozygosity in three embryonal tumours suggests a common pathogenetic mechanism. Nature 1985; 316: 330–4. [DOI] [PubMed] [Google Scholar]
- 43. Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM. CTCF mediates methylation‐sensitive enhancer‐blocking activity at the H19/Igf2 locus. Nature 2000; 405: 486–9. [DOI] [PubMed] [Google Scholar]
- 44. Zhang Y, Shields T, Crenshaw T, Hao Y, Moulton T, Tycko B. Imprinting of human H19: allele‐specific CpG methylation, loss of the active allele in Wilms tumor, and potential for somatic allele switching. Am J Hum Genet 1993; 53: 113–24. [PMC free article] [PubMed] [Google Scholar]
- 45. Weksberg R, Nishikawa J, Caluseriu O et al . Tumor development in the Beckwith–Wiedemann syndrome is associated with a variety of constitutional molecular 11p15 alterations including imprinting defects of KCNQ1OT1. Hum Mol Genet 2001; 10: 2989–3000. [DOI] [PubMed] [Google Scholar]