

Abstract
DNA microarray technology has revolutionized our understanding of the molecular basis of hepatocellular carcinoma (HCC), one of the most fatal human cancers with a high recurrence rate. Many researchers have used DNA microarray technology to reclassify HCC with respect to metastatic potential and to develop predictors for the outcome of HCC. However, developed predictors have reached the level only of small retrospective studies, and their current status is far from that required for clinical use. This is due to the lack of transparent data, the high cost and data instability associated with the high dimensionality of the technique, the infancy of bioinformatics, and the complicated nature of recurrent HCC. This comprehensive review summarizes: (i) class comparison studies to identify genes or pathways involved in HCC metastasis (ii) class discovery studies that have resulted in the identification of a new molecular subclass of HCC with respect to metastasis, and (iii) class prediction studies to develop multidimensional predictors for HCC outcome. We also discuss issues that need to be addressed so that the power of array‐based predictors can be estimated prospectively in large independent cohorts of HCC patients. (Cancer Sci 2008; 99: 659–665)
Hepatocellular carcinoma (HCC) is one of the most fatal cancers in humans, with an estimated 564 000 new cases worldwide in 2000. HCC represents a major international health problem because its incidence is exponentially increasing in many countries.( 1 ) Despite many advances in the treatment of HCC, the recurrence rate at 3 and 5 years after curative treatment exceeds 50% and 70%, respectively.( 2 , 3 ) Therefore, it is crucial to better understand the mechanisms involved in HCC recurrence and to provide effective therapies based on accurate outcome prediction. Many clinical staging systems have been applied to HCC patients( 4 , 5 ); however, there are limitations of these systems in the accurate prediction in individual patients. This problem has long frustrated hepatologists and pathologists. A robust predictive system for use in HCC patients is therefore necessary.
High‐dimensional array technology started a revolution in medical science upon the first publication of this technology in 1995.( 6 ) This high‐tech technology provides great promise with respect to genome‐wide searches for predictive molecular markers and has resulted in enhanced characterization of individual tumors with regard to metastatic potential compared to that provided by traditional clinicopathologic methods and single‐molecule systems.( 7 , 8 , 9 , 10 ) In the field of HCC research, Lau et al.( 11 ) used this technology to compare gene expression profiles of HCC and non‐HCC liver tissues. Since then, more than 300 HCC studies with use of DNA microarray technology, including many elegant works from Japan,( 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 ) have been published. Many researchers,( 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 ) have also developed array‐based predictors for metastasis, recurrence, and outcome of HCC; however, the high predictive accuracy of those systems,( 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 ) is likely to be limited to the individual cohorts tested. Thus far, the genes identified have shown little predictive value.
In this review, we highlighted genome‐wide studies on HCC metastasis; we classified them on the molecular basis of HCC metastasis into three major groups (i) class comparison (ii) class discovery, and (iii) class prediction, as proposed by Simon and colleagues.( 32 ) We then review the accomplishments of the three types of array studies. In particular, we focus on translational array studies that have developed a multidimensional predictor for HCC outcome.
Class comparison studies
Discovered metastasis‐related genes. Class comparison study is the analysis of gene expression in classes of specimens defined by criteria such as histopathologic features.( 32 ) The aim is to determine whether the expression profiles are different between the classes and, if so, to identify the feature genes as potential molecular targets for metastasizing cancer cells.( 32 ) Indeed, aberrations of many genes, such as RHOC,( 33 ) GRN,( 34 ) VIM,( 35 ) DLG7 (KIAA0008),( 36 ) HLA‐DRA,( 37 ) CLDN10,( 38 ) EFNA1,( 39 ) PDGFRA,( 40 ) Transcript AA454543,( 41 ) and NDRG1, ( 42 ) in HCC metastasis have been reported in class comparison DNA microarray studies. Among these genes,( 33 , 34 , 35 , 36 , 37 , 38 , 39 , 40 , 41 , 42 ) RHOC, VIM, DLG7, and CLDN10 function as cell invasion regulators, and GRN, PDGFRA, and NDRG1 function as cell growth regulators. HLA‐DRA is also involved in the immune response. The identification of genes with a broad range of function in HCC metastasis would allow for the control and/or prevention of metastasis. Other methods, such as differential display and nucleic acid subtraction, can also be used for this purpose. However, array technology makes it possible to search comprehensively for many relevant genes. Accordingly, another important task of class comparison study is to elucidate representative pathways or modules,( 43 ) that plays key roles in HCC metastasis. The identification of upstream regulation systems or gene networks of such metastasis‐related modules would lead to the development of more effective molecular targeting therapies for HCC.
Modules linked to venous invasion. Venous invasion (VI), particularly portal venous invasion (PVI), is a hallmark of the intrahepatic spread of HCC cells and of poor outcome.( 44 ) The presence of PVI is a statistically independent prognostic factor for cancer recurrence when the liver transplantation was applied to HCC patients that are beyond the Milan criteria.( 45 ) Thus, information regarding VI‐ or PVI‐related modules is important for the development of first‐line treatments involving the multiple metastatic cascades of HCC.
Ho et al.( 30 ) compared gene expression patterns between solitary HCC with VI and that without and identified 14 VI‐related genes, which were found to show good predictive value in an independent cohort of HCC patients. The 14 genes included TAF4B, SLC4A7, RAB38 and RYR1, most of which are related to cell growth. Chen et al.( 46 ) identified 91 genes for which expression levels were significantly correlated with the presence or absence of VI. That study showed up‐regulation of MMP14 and down‐regulation of two CYPs, ADAMTS1, and ITGA7 in HCC with VI. Okabe et al.( 47 ) identified 151 VI‐related genes, including 110 expressed sequence tags, RHOC, and two small GTPase‐related genes (ARHGAP8 and ARHGEF6). Consistent with that finding, another DNA microarray study showed that RHOC was up‐regulated in nodular HCC (NHCC) which possesses higher metastatic potential than solitary large HCC (SLHCC).( 33 ) Thus, the Rho‐related module plays an important role in VI of HCC.
Considering that VI is not detected in well‐differentiated HCC but is frequently observed in moderately or less well‐differentiated HCC,( 48 ) Tsunedomi et al.( 49 ) focused their investigation on moderately differentiated hepatitis C virus (HCV)‐related HCC to minimize the bias of gene selection. That study identified 35 VI‐related genes including genes involved in apoptosis and the stress response (SGK, API5, and GADD45B), the cell cycle and cell proliferation (RAN and NUDC), oncogenesis (DDX1), and signal transduction (RHO6) (Suppl. Fig. S1). RHO6, a Rho GTPase family member, negatively regulates cell adhesion; therefore, control of Rho signaling may provide a promising treatment for the prevention of subsequent metastasis, as proposed by Okabe et al.( 47 ) Tsunedomi et al.( 49 ) also showed that transcription factor ID2 was decreased in VI‐positive HCC. Further study has shown that ID2 regulates the invasive potential of HCC cells via modulation of several matrix metalloproteinases (unpublished data, 2007).
Thus, class comparison studies performed with array technology have identified many genes related to VI in HCC. Unfortunately, there are few common VI‐related modules for HCC other than Rho module. Further studies are necessary to gain deeper insights with respect to these DNA array results.( 46 , 47 , 49 )
Modules linked to other metastatic aspects of HCC. Several class comparison studies have used a unique approach to gain deeper insight into HCC metastasis. Iizuka et al.( 50 ) compared gene expression patterns between HCC with extrahepatic recurrence (EHR) and that without EHR after curative surgery. The 46 identified EHR‐related genes included many cell adhesion‐related genes such as ITGA6, SPP1, DNMBP, CD44 and POSTN, all of which showed higher expression in HCC with EHR than in HCC without. It was noted that the molecular patterns of these 46 EHR‐related genes were quite distinct from those,( 24 , 37 ) of early intrahepatic recurrence (IHR)‐related genes identified previously, indicating that the metastatic processes of EHR and early IHR involve different molecular modules (Suppl. Figs S2 and S3).
As mentioned, Wang et al.( 33 ) compared gene expression patterns between NHCC, with higher metastatic potential, and SLHCC, with low metastatic potential, and identified RHOC as a gene up‐regulated in NHCC. In that study, levels of ITGA6 were also significantly greater in NHCC than in SLHCC. A recent class comparison study,( 50 ) showed that ITGA6 is up‐regulated in HCC with EHR compared to that without EHR. Thus, a high level of ITGA6 expression in HCC may be a strong predictor of the high metastatic potential.
Elucidation of differences in molecular expression pattern between primary HCC and intrahepatic metastasis (IM) may lead to the identification of the metastatic module and an understanding of multicentric hepatocarcinogenesis. Chen et al.( 46 ) defined primary HCC and IM on the basis of the clonality of nodules as determined by patterns of p53 mutation and hepatitis B virus (HBV) integration. They next investigated differences in gene expression patterns between primary HCC and IM in the same patients. Among 23 075 genes assayed, 90 showed differential expression levels between primary HCC and IM. These genes included BRMS1, CD53, and EMP3. They also identified decreased expression of genes (CYP2A7 and immunoglobulin genes, among others), involved in normal hepatocyte function in IM compared to primary HCC. Interestingly, the decreased expression of such hepatocyte‐specific genes has been observed in parallel with dedifferentiation grade in other genome‐wide studies.( 16 , 18 , 51 )
Modules linked to hallmark genes p53 and MET. p53 expression is a hallmark of cancer, and is related closely to the outcome of HCC.( 52 ) The class comparison study of Chen et al.( 46 ) showed increased expression levels of many cell growth‐related genes in HCC with nuclear accumulation of abnormal p53. Therefore, it is reasonable to assume that HCC with p53 mutation has a higher malignant potential than HCC with wild‐type p53. Okada et al.( 53 ) classified HCC into two subgroups according to p53 status, and identified many genes with promoter sequences that can be directly regulated by p53, most of which are related to cell growth and are up‐regulated in HCC with p53 mutation. Interestingly, a sample rearrangement study from the same institute showed that HCC with p53 mutation is more advanced than HCC with wild‐type p53.( 20 )
Kaposi‐Novak et al.( 31 ) studied the abnormalities of the MET oncogene that are responsible for hepatocarcinogenesis, and reclassified HCC into two subclasses according to poor or good outcome. This is an example of the application of a biochemical module, the MET‐related pathway, to a supervised learning‐based predictor for HCC outcome. The same group identified a new subclass of HCC by performing a cross‐comparison of rat, mouse, and human liver transcriptome data.( 54 ) This new subclass shared gene expression patterns with that of rat fetal hepatoblasts and showed poor prognosis compared to other subclasses of HCC. Interestingly, genes specific to hepatic oval cells distinguished this new HCC subclass from two other HCC subclasses, suggesting that the new subclass may arise from hepatic progenitor cells.
Class discovery studies
Class discovery study is one that reports a new HCC class based on the gene expression patterns and it is also fundamentally different from other two studies (class comparison and class prediction studies) in that no classes are predefined.( 32 ) An example of class discovery is the study by Alizadeh et al. that provided a new type of diffuse large B‐cell lymphoma by gene profiling.( 55 ) This type of study is exclusively performed using cluster analysis in an unsupervised learning manner. In the field of HCC research, this type is rare.
Breuhahn et al.( 56 ) reported that HCCs can be divided into two subgroups: group A, characterized by high‐level expression of interferon (IFN)‐regulated genes; and group B, which lacks induction of IFN‐regulated genes and apoptosis‐related genes. Interestingly, group A HCC showed down‐regulation of the IGF2 gene. Group B HCC was further subdivided into HCC with down‐regulation of the IGF2 gene and that with up‐regulation of the IGF2 gene. Unfortunately, the relation between patient outcome and subclass according to IFN and IGF2 expression patterns was not reported.
Class prediction studies
Published class prediction studies. Class prediction studies are translational studies performed with DNA microarray; the aims are to build a predictor with use of a classifier and to evaluate its performance on an independent sample set.( 32 ) This is an expanding area in HCC research.( 57 ) An initial class prediction study,( 24 ) for HCC recurrence was published 3 years after the report of Lau et al. ( 11 ) which applied DNA microarray technology to HCC research for the first time. Thereafter, various class prediction studies,( 25 , 26 , 27 , 28 , 29 , 30 , 31 , 58 , 59 ) using DNA microarray or other technologies have been performed (Table 1). Many studies ( 25, 26, 27, 30, 31, 58, 59 ) have used primary HCC tissues in the search for predictive gene signatures, although two DNA microarray studies used non‐cancerous liver tissues to predict the occurrence of de novo HCC,( 28 ) and recurrence.( 29 ) Among nine studies,( 25 , 26 , 27 , 28 , 29 , 30 , 31 , 58 , 59 ) four used oligonucleotide arrays, four used cDNA arrays, and one used polymerase chain reaction (PCR)‐based array. Two studies,( 29 , 58 ) adapted the array data to a quantitative reverse transcription (RT)‐PCR‐based predictive system, and a recent study,( 59 ) developed a multidimensional predictor based on proteomic analysis of early IHR of HCC (Table 1).
Table 1.
Representative class prediction studies for outcome of hepatocellular carcinoma
| Aim of study | Strategy (tested gene number) | Sample source (Race) | Virus type | Cancer characteristics used to construct predictor | Algorithm used | Module or feature genes | Predictive accuracy | |
|---|---|---|---|---|---|---|---|---|
| Iizuka et al.( 24 ) | Prediction of early IHR within 1 years after curative surgery | oligonucleotide array (7070 genes) | primary HCC (Japanese) | HBV < HCV | early IHR within 1 year after surgery | Fisher linear classifier with 12 genes | Immune response | 25 (93%) of independent 27 samples |
| Ye et al.( 25 ) | Classificaton of HCC with IM and that without IM at surgery | cDNA microarray (9180 genes) | primary HCC (Chinese:Shanghai) | All HBV | IM at surgery | Compound covariate predictor with 153 genes | SPP1 | 16 (80%) of independent 20 samples |
| Kurokawa et al.( 26 ) | Prediction of early IHR within 2 years after curative surgery | PCR‐based array (3072 genes) | primary HCC (Japanese) | HBV < HCV | early IHR within 2 years after surgery | Weighted voting algorithm with 20 genes | E‐cadherin/Immune response | 29 (73%) of independent 40 samplesP = 0.008 (Max) |
| Lee et al.( 27 ) | Prediction of overall survival after surgery | oligonucleotide array (21 329 genes) | primary HCC (Chinese/Belgian) | HBV > HCV | long‐ and short‐term survival | Five‐type predictors with 406 genes | Proliferation regulator | in independent 44 samples |
| Okamoto et al.( 28 ) | Prediction of the risk of multicentric de novo HCC after surgery | cDNA microarray (12 814 genes) | non‐cancerous liver (Japanese) | All HCV | Single nodular HCC versus multicentric HCC | Prediction score with 36 genes | FPS/FES proto‐oncogene | 30 (75%) of 40 training samples |
| Budhu et al.( 29 ) | Prediction of recurrence during 3 years follow‐up | cDNA microarray/QRT‐PCR (9180 genes) | non‐cancerous liver (Chinese:Shanghai) | Almost HBV | Venous invasion or extrahepatic metastasis | Prediction analysis of microarray with 17 genes | Inflammation/Immune response | 87 (92%) of independent 95 samplesP = 0.028 |
| Ho et al.( 30 ) | Prediction of recurrence after curative surgery | cDNA microarray (13 564 genes) | primary HCC (Chinese:Taiwan) | HBV = HCV | Venous invasion | k‐Nearest Neighbor classifier with 14 genes | Proliferation regulator MAGE family | in independent 35 samplesP = 0.00015 (Max) |
| Kaposi‐Novak et al.( 31 ) | Prediction of overall survival after surgery | oligonucleotide array (21 329 genes) | primary HCC (Chinese/Belgian) | HBV > HCV | Presence or absence of Met signature | Six‐type predictors with 111 genes | MET | in 61 samples† |
| Somura et al.( 58 ) | Prediction of early IHR within 1 years after curative surgery | oligonucleotide array/QRT‐PCR (7070 genes) | primary HCC (Japanese) | HBV < HCV | early IHR within 1 year after surgery | Fisher linear classifier with 3 genes | HLA‐DRA/DDX17/LAPTM5 | 35 (81%) of independent 43 samples |
| Yokoo et al.( 59 ) | Prediction of early IHR within 6 months after curative surgery | Proteomics with 2D‐DIGE | primary HCC (Japanese) | HBV < HCV | early IHR within 6 months after surgery | Support vector machine with 23 spots (proteins) | Proliferation regulator/Antioxidant | 12 (92%) of independent 13 samples |
FES/FPS, feline sarcoma; HBV, hepatitis B virus; HCV, hepatitis C virus; HCC, hepatocellular carcinoma; IHR: intrahepatic recurrence; IM: intrahepatic metastasis; MAGE, melanoma antigen; MET, met proto‐oncogene; QRT‐PCR: quantitative real‐time reverse transcription‐polymerase chain reaction; SPP, secreted phosphoprotein; 2D‐DIGE: two‐dimensional fluorescence difference gel electrophoresis.
When Met‐regulated genes were selected, an information from both test and training sample was used.
Significance of sample labels for predictive systems. All of these studies use a training‐validation approach in which a classifier (i.e. predictor) is built in silico on the basis of information from training samples, and its predictive power is evaluated on independent test samples. Usually, the procedure is performed in a supervised‐learning manner in which the independency between training and test samples is critical.( 32 ) One study,( 28 ) reported the accuracy of a predictor based only on a training sample set. In another study,( 31 ) information from both training and test samples was used in selecting genes to be integrated into a predictor.
The term ‘sample labels’ used in a training‐validation studies refers to the characteristics of each cancer (i.e. recurrence vs non‐recurrence), which are also involved in the metastatic cascades that we would like to analyze. To obtain an accurate predictor, the sample must be precisely labeled.( 60 ) Which sample label is most suitable for HCC outcome? It is likely that the use of sample label depends on individual cohorts. Japanese researchers,( 24 , 26 , 58 , 59 ) frequently use early IHR, which is defined as recurrent liver tumors detected from 6 months to 2 years after surgery, as a sample label (Table 1). This can be explained largely by the fact that the majority of Japanese HCC cases are attributable to HCV infection, which preferentially causes multicentric de novo HCC (i.e. late IHR) after treatment; this must be distinguished from early IHR due to metastasis.( 61 ) Therefore, early IHR would be an ideal sample label in a cohort containing predominantly HCV‐related HCC. However, it has limitations from the standpoint of diagnostic accuracy; the definition of early IHR is mostly based on clinicopathologic findings rather than on molecular or genetic findings, true IHR due to metastasis can appear even 3 years after surgery, and an accurate classification of early IHR or non‐recurrence does not always lead to precise outcome prediction because recurrence can appear in distant organs regardless of early IHR, IHR due to de novo HCC can appear within 1 year after surgery,( 62 ) and the outcome also depends on liver function itself.
By contrast, in areas,( 25 , 27 , 29 , 30 , 31 ) in which HBV infection is endemic, researchers use sample labels other than early IHR for the outcome prediction of HCC (Table 1). Sample labels include IM at surgery,( 25 ) patient survival,( 27 ) and VI.( 29 ) These sample labels are also unstable. For example, IM at surgery cannot be correctly diagnosed without examination of tumor cell origin, such as clonality, as proposed by Cheung et al.( 51 ) Patient survival is largely affected by liver function or status and postsurgical treatment as opposed to tumor factors,( 2 ) suggesting that accurate prediction of patient survival by gene profiling alone of the primary HCC site is due to chance. VI is also correlated with HCC outcome,( 2 , 47 ); however, some proportion of HCC patients without detectable VI have a poor outcome.
The work of Mas et al.,( 63 ) while not class prediction study, is a fascinating approach to distinguish completely true recurrence due to metastasis from de novo HCC. They profiled signature genes in HCCs from HCV‐infected patients undergoing liver transplantation (LT) and found 10, including the IFN‐regulated genes STAT1 and OAS1, for which expression differed significantly between patients with recurrence and those without. It will be interesting to confirm whether the identified gene signatures work well in predicting recurrence in patients undergoing hepatectomy as well as in LT patients in an independent cohort.
There are limitations in using predictors made with only one from among several sample labels if the goal is to individualize outcome of HCC patients. We therefore must prepare several sets of predictors and sample labels to correspond to each of various modes of HCC recurrence. However, it should be noted that any sample label used will have some pitfalls and must be used with caution. We propose that at least four sets of sample labels and corresponding predictors are needed to individualize HCC outcome. The four are early IHR, EHR, de novo HCC, and drug response of individual HCCs (Fig. 1). Both primary HCC and non‐cancerous liver tissues may be required. There are a few DNA microarray studies,( 64 , 65 ) of the response of HCC cells or HCC tissue to 5‐fluorouracil, IFN‐α, or a combination, which are widely used clinically. Further effort must be devoted to identifying drug response‐related signature genes.
Figure 1.
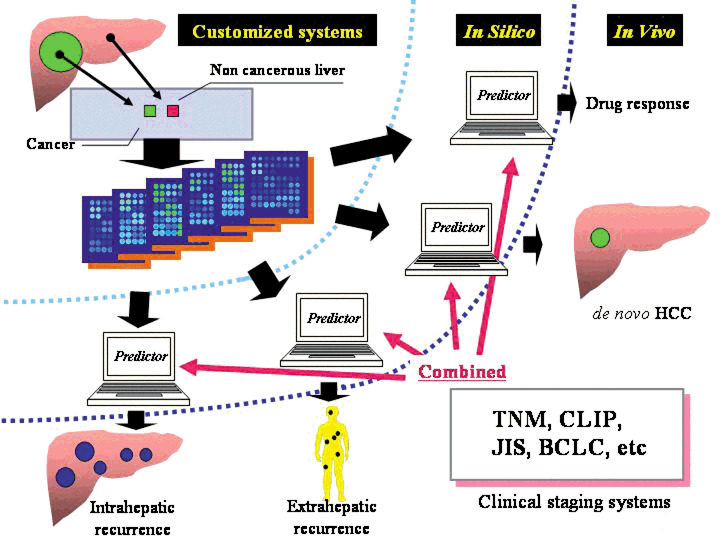
Customized predictive systems for outcome of hepatocellular carcinoma patients. BCLC, Barcelona clinic liver cancer staging system; CLIP, Cancer of the Liver Italian Program; HCC, hepatocellular carcinoma; JIS, Japan integrated staging; TNM, tumor node metastasis.
Lack of overlap of predictive genes identified at various institutes. In Table 1, we can easily identify a lack of overlap of individual predictive genes. Ein‐Dor et al.( 66 ) used probably approximately correct (PAC) sorting to calculate the quality of predictive gene lists, and found that thousands of training samples are needed for cancer outcome prediction. This means that no reproducible results can be obtained from microarray studies using hundreds of samples. It is therefore reasonable that there are no overlaps between predictive signatures from different studies with the same goal (Table 1). There are likely more predictive genes required to design accurate predictors. Conversely, many genes may have the same ability to predict outcome. Such genes may also form a module. Intriguingly, our recent resampling study,( 67 ) showed that many HLA class II genes are predictive of early IHR in artificial cohorts consisting of 1000 HCC samples that reproduce virtually the geographic distribution pattern of HBV and HCV in six representative geographic regions. This means that the predictive power of an immune‐related module such as HLA class II is independent of infection patterns of hepatitis virus types. This study is an in silico simulation of our previous DNA array data,( 67 ); more predictive modules for HCC recurrence would be identified if meta‐analysis of the published array data was performed. For this purpose, the published array data must be available and described precisely with transparency. If possible, these data should be standardized according to minimum information about a microarray experiment.( 68 )
To build a predictor that can be applied to the daily clinical use, both ‘accuracy’ and ‘simplicity’ are required. All the studies,( 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 , 58 , 59 ) in Table 1 used the bulk liver tissues without a laser microdissection technology to develop an easy‐to‐use simple system. This might also account for another possibility of the lack of overlapping feature genes among institutes. Dual information from purified cancer and non‐cancer cells would be required to gain deeper insights in HCC metastasis and to identify more common feature genes or modules for the robust predictor.
Factors affecting predictive accuracy. The predictive accuracy of predictors ranges from 73% to 93% (mean, 84%) (Table 1). The number of genes used in predictors and test samples range from 3 to 406 (mean, 79.5) and from 13 to 95 (mean, 41.8), respectively (Table 1). There is no association between predictive accuracy and background factors such as the number of genes used in predictors or test samples, publication year, or impact factor of journal in eligible seven studies (Suppl. Fig. S4). It is considered that oligonucleotide arrays are more advantageous than cDNA arrays because of the decreased possibility of probe mix‐ups.( 69 ) However, the array type used is unlikely to affect predictive performance (Table 1).
As summarized in Table 1, various algorithms have been used to build individual classifiers. The question is which classifier is the most robust in predicting HCC outcome. Generally, Fisher linear classifier (FLC) is one of distribution‐dependent parametric classifiers. Artificial neural network and support vector machine (SVM) classifiers are non‐parametric classifiers that work in a distribution‐independent manner. The latter is widely applicable to various situations in predictive oncology. Iizuka et al.( 24 ) showed that an FLC with 12 genes was superior to an SVM classifier with 50 genes with respect to the predictive accuracy in the same cohort. However, according to Lee et al. ( 27 ) there were no differences in predictive accuracy among FLC, SVM, compound covariate predictor, nearest centroid, and nearest neighbor classifiers. Intriguingly, using the same series of Lee et al. ( 27 ) Kaposi‐Novak et al.( 31 ) showed that a nearest neighbor 3 classifier was the most robust in predicting overall survival among the six classifiers tested. These results indicate that the predictive power of individual classifiers depends on study design, such as aim of prediction and sample size, and is not always stable. Therefore, it is important to establish the best way to select a suitable classifier for data. However, this concept may result in a limitation in constructing uniform classifiers for a same goal.
Rather than the classifier design, the gene selection method may be critical for a robust predictive system.( 70 ) In particular, single‐pass gene selection is insufficient for the selection of robust remarkable genes for prediction with small sample sizes similar to those used in DNA microarray experiment.( 70 ) This concept was proposed in the 1970s,( 71 ) and confirmed in the 1990s in the field of pattern recognition.( 70 ) To address this issue, we applied a cross‐validation (CV) method to outcome prediction in HCC.( 24 ) With the CV approach, we obtain a virtual artificial training sample set in which we can select robust genes with the ability to minimize the error rate. Moreover, our previous exhaustive search,( 24 ) examined combinations of genes that yield the most robust classifier. Many class prediction studies,( 25 , 26 , 27 , 29 , 30 , 31 , 59 ) listed in Table 1 used the CV test. However, the application is limited to predictor design but not to gene selection. Identification of the best way to select feature genes that work well in a virtual large cohort with much variation will allow us to build robust predictors. Our work is still preliminary; however, a three‐gene predictor constructed by a data‐driven procedure showed an accuracy of >80% in predicting early IHR in an independent HCC sample set.( 58 )
Major bottlenecks for the routine clinical use of multidimensional predictors. As mentioned, to develop a robust predictor as a tool for clinical decision making (i) sample label (ii) availability of data to search for predictive signature genes, and (iii) gene selection (feature selection) methods must be reconsidered in a class prediction study of HCC outcome. Use of array data from class comparison or discovery studies will allow for the construction of a robust predictor. Of course, prior to addressing these issues, we must address others that can affect the quality of array data, such as sample quality (ribonucleic acid quality), probe sequence and labeling, hybridization procedure including dye effect, scanning process, and array slide conditions such as surface coating. Addressing these issues will enhance the reliability and reproducibility of data within arrays and between institutes. Clinically, the next tasks are to address limiting factors, such as high cost and high dimensionality, for the routine use of array‐based systems.
Breast cancer may be the only example for which the above problems of array technology have been addressed. A recent study by Glas et al.( 72 ) showed that a 70‐gene breast cancer outcome signature obtained from previous high‐dimensional array data,( 7 ) can be translated into a customized mini‐array format and that the new array works well in predicting outcomes in newly enrolled breast cancer patients. Translation of a 12‐predictive gene signature for HCC to the customized mini‐array format is underway in our laboratory. If a low dimensional, inexpensive, and easy‐to‐use predictor based on a customized mini‐array format shows high predictive power in independent large HCC series, we could individualize, but not stratify, the outcome of HCC patients on the basis of the four‐type predictors in combination with various clinicopathologic staging systems, as illustrated in Figure 1.
Conclusions
The potential of array‐based multidimensional predictors to outperform traditional clinical parameters is fascinating. The number of such array‐oriented studies will increase exponentially. However, the current multidimensional array systems are far from routine clinical use for individualizing the outcome of HCC patients. We have reached a point where it is clear what we should do prior to the application of DNA array technology to daily clinical use. Future challenges are to identify a small subset of highly predictive signature genes for HCC outcome, to establish a cheaper easy‐to‐use predictor, and to validate the clinical efficacy of the predictor prospectively on a larger cohort of HCC patients.
Supporting information
Fig. S1. Map of module related to venous invasion. Relationships from literature databases between 35 up‐ and down‐regulated genes responsible for venous invasion (Ref. 49) were extracted and analyzed by using the Genomatix software (http://www.genomatix.de/). The redder and bluer boxes are the higher up‐regulated and lower down‐regulated genes, respectively. Green color indicates the presence of binding motifs in the promoter of the potential transcriptional target.
Fig. S2. Map of module related to early intrahepatic recurrence. Relationships from literature databases between 46 up‐ and down‐regulated genes responsible for early intrahepatic recurrence (Refs. 24 and 37) were extracted and analyzed by using the Genomatix software (http://www.genomatix.de/). The redder and bluer boxes are the higher up‐regulated and lower down‐regulated genes, respectively. Green color indicates the presence of binding motifs in the promoter of the potential transcriptional target.
Fig. S3. Map of module related to extrahepatic recurrence. Relationships from literature databases between 46 up‐ and down‐regulated genes responsible for extrahepatic recurrence (Ref. 50) were extracted and analyzed by using the Genomatix software (http://www.genomatix.de/). The redder and bluer boxes are the higher up‐regulated and lower down‐regulated genes, respectively. Green color indicates the presence of binding motifs in the promoter of the potential transcriptional target.
Fig. S4. Performance of predictors in eligible seven studies. A, Relation between predictive accuracy, number of gene used, and number of test sample. B, Relation between predictive accuracy, publication year, and journal impact factor. The journal impact factors are based on Science Citation Index (SCI ®), which is originally produced by the Institute for Scientific Information (ISI) in 2005.
Please note: Blackwell Publishing are not responsible for the content or functionality of any supplementary materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Acknowledgments
This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology (No. 18390366, no. 17591406 and Knowledge Cluster Initiative); the Venture Business Laboratory of Yamaguchi University; the New Energy and Industrial Technology Development Organization (Grant number: 03A02018a).
Grant sponsors: the Ministry of Education, Culture, Sports, Science and Technology (No. 18390366, no. 17591406 and Knowledge Cluster Initiative); the Venture Business Laboratory of Yamaguchi University; the New Energy and Industrial Techno‐logy Development Organization (Grant number: 03A02018a).
References
- 1. Parkin DM, Bray FI, Devesa SS. Cancer burden in the year 2000. The global picture. Eur J Cancer 2001; 37: S4–66. [DOI] [PubMed] [Google Scholar]
- 2. Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet 2003; 362: 1907–17. [DOI] [PubMed] [Google Scholar]
- 3. Okada S, Shimada K, Yamamoto J et al . Predictive factors for postoperative recurrence of hepatocellular carcinoma. Gastroenterology 1994; 106: 1618–24. [DOI] [PubMed] [Google Scholar]
- 4. Kudo M, Chung H, Osaki Y. Prognostic staging system for hepatocellular carcinoma (CLIP score): its value and limitations, and a proposal for a new staging system, the Japan Integrated Staging Score (JIS score). J Gastroenterol 2003; 38: 207–15. [DOI] [PubMed] [Google Scholar]
- 5. Sala M, Forner A, Varela M, Bruix J. Prognostic prediction in patients with hepatocellular carcinoma. Semin Liver Dis 2005; 25: 171–80. [DOI] [PubMed] [Google Scholar]
- 6. Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 1995; 270: 467–70. [DOI] [PubMed] [Google Scholar]
- 7. Van De Vijver MJ, He YD, Van't Veer LJ et al . A gene‐expression signature as a predictor of survival in breast cancer. N Engl J Med 2002; 347: 1999–2009. [DOI] [PubMed] [Google Scholar]
- 8. Rosenwald A, Wright G, Chan W et al . The use of molecular profiling to predict survival after chemotherapy for diffuse large B cell lymphoma. N Engl J Med 2002; 346: 1937–47. [DOI] [PubMed] [Google Scholar]
- 9. Tomida S, Koshikawa K, Yatabe Y et al . Gene expression‐based, individualized outcome prediction for surgically treated lung cancer patients. Oncogene 2004; 23: 5360–70. [DOI] [PubMed] [Google Scholar]
- 10. Ohira M, Oba S, Nakamura Y et al . Expression profiling using a tumor‐specific cDNA microarray predicts the prognosis of intermediate risk neuroblastomas. Cancer Cell 2005; 7: 337–50. [DOI] [PubMed] [Google Scholar]
- 11. Lau WY, Lai PB, Leung MF et al . Differential gene expression of hepatocellular carcinoma using cDNA microarray analysis. Oncol Res 2000; 12: 59–69. [DOI] [PubMed] [Google Scholar]
- 12. Kawai HF, Kaneko S, Honda M, Shirota Y, Kobayashi K. alpha‐fetoprotein‐producing hepatoma cell lines share common expression profiles of genes in various categories demonstrated by cDNA microarray analysis. Hepatology 2001; 33: 676–91. [DOI] [PubMed] [Google Scholar]
- 13. Shirota Y, Kaneko S, Honda M, Kawai HF, Kobayashi K. Identification of differentially expressed genes in hepatocellular carcinoma with cDNA microarrays. Hepatology 2001; 33: 832–40. [DOI] [PubMed] [Google Scholar]
- 14. Hanafusa T, Yumoto Y, Nouso K et al . Reduced expression of insulin‐like growth factor binding protein‐3 and its promoter hypermethylation in human hepatocellular carcinoma. Cancer Lett 2002; 176: 149–58. [DOI] [PubMed] [Google Scholar]
- 15. Naiki T, Nagaki M, Shidoji Y et al . Analysis of gene expression profile induced by hepatocyte nuclear factor 4alpha in hepatoma cells using an oligonucleotide microarray. J Biol Chem 2002; 277: 14011–19. [DOI] [PubMed] [Google Scholar]
- 16. Midorikawa Y, Tsutsumi S, Taniguchi H et al . Identification of genes associated with dedifferentiation of hepatocellular carcinoma with expression profiling analysis. Jpn J Cancer Res 2002; 93: 636–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Iizuka N, Oka M, Yamada‐Okabe H et al . Comparison of gene expression profiles between hepatitis B virus‐ and hepatitis C virus‐infected hepatocellular carcinoma by oligonucleotide microarray data based on a supervised learning method. Cancer Res 2002; 62: 3939–44. [PubMed] [Google Scholar]
- 18. Chuma M, Sakamoto M, Yamazaki K et al . Expression profiling in multistage hepatocarcinogenesis: identification of HSP70 as a molecular marker of early hepatocellular carcinoma. Hepatology 2003; 37: 198–207. [DOI] [PubMed] [Google Scholar]
- 19. Tamori A, Yamanishi Y, Kawashima S et al . Alteration of gene expression in human hepatocellular carcinoma with integrated hepatitis B virus DNA. Clin Cancer Res 2005; 11: 5821–6. [DOI] [PubMed] [Google Scholar]
- 20. Iizuka N, Oka M, Yamada‐Okabe H et al . Self‐organizing‐map‐based molecular signature representing the development of hepatocellular carcinoma. FEBS Lett 2005; 579: 1089–100. [DOI] [PubMed] [Google Scholar]
- 21. Honma N, Genda T, Matsuda Y et al . MEK/ERK signaling is a critical mediator for integrin‐induced cell scattering in highly metastatic hepatocellular carcinoma cells. Lab Invest 2006; 86: 687–96. [DOI] [PubMed] [Google Scholar]
- 22. Tsujimura K, Asamoto M, Suzuki S, Hokaiwado N, Ogawa K, Shirai T. Prediction of carcinogenic potential by a toxicogenomic approach using rat hepatoma cells. Cancer Sci 2006; 97: 1002–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chiba T, Kita K, Zheng YW et al . Side population purified from hepatocellular carcinoma cells harbors cancer stem cell‐like properties. Hepatology 2006; 44: 240–51. [DOI] [PubMed] [Google Scholar]
- 24. Iizuka N, Oka M, Yamada‐Okabe H et al . Oligonucleotide microarray for prediction of early intrahepatic recurrence of hepatocellular carcinoma after curative resection. Lancet 2003; 361: 923–9. [DOI] [PubMed] [Google Scholar]
- 25. Ye QH, Qin LX, Forgues M et al . Predicting hepatitis B virus‐positive metastatic hepatocellular carcinomas using gene expression profiling and supervised machine learning. Nat Med 2003; 9: 416–23. [DOI] [PubMed] [Google Scholar]
- 26. Kurokawa Y, Matoba R, Takemasa I et al . Molecular‐based prediction of early recurrence in hepatocellular carcinoma. J Hepatol 2004; 41: 284–91. [DOI] [PubMed] [Google Scholar]
- 27. Lee JS, Chu IS, Heo J et al . Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology 2004; 40: 667–76. [DOI] [PubMed] [Google Scholar]
- 28. Okamoto M, Utsunomiya T, Wakiyama S et al . Specific gene‐expression profiles of noncancerous liver tissue predict the risk for multicentric occurrence of hepatocellular carcinoma in hepatitis C virus‐positive patients. Ann Surg Oncol 2006; 13: 947–54. [DOI] [PubMed] [Google Scholar]
- 29. Budhu A, Forgues M, YeQH et al . Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell 2006; 10: 99–111. [DOI] [PubMed] [Google Scholar]
- 30. Ho MC, Lin JJ, Chen CN et al . A gene expression profile for vascular invasion can predict the recurrence after resection of hepatocellular carcinoma: a microarray approach. Ann Surg Oncol 2006; 13: 1474–84. [DOI] [PubMed] [Google Scholar]
- 31. Kaposi‐Novak P, Lee JS, Gomez‐Quiroz L, Coulouarn C, Factor VM, Thorgeirsson SS. Met‐regulated expression signature defines a subset of human hepatocellular carcinomas with poor prognosis and aggressive phenotype. J Clin Invest 2006; 116: 1582–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Simon R, Radmacher MD, Dobbin K, McShane LM. Pitfalls in the use of DNA microarray data for diagnostic and prognostic classification. J Natl Cancer Inst 2003; 95: 14–8. [DOI] [PubMed] [Google Scholar]
- 33. Wang W, Yang LY, Huang GW et al . Genomic analysis reveals RhoC as a potential marker in hepatocellular carcinoma with poor prognosis. Br J Cancer 2004; 90: 2349–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cheung ST, Wong SY, Leung KL et al . Granulin‐epithelin precursor overexpression promotes growth and invasion of hepatocellular carcinoma. Clin Cancer Res 2004; 10: 7629–36. [DOI] [PubMed] [Google Scholar]
- 35. Hu L, Lau SH, Tzang CH et al . Association of Vimentin overexpression and hepatocellular carcinoma metastasis. Oncogene 2004; 23: 298–302. [DOI] [PubMed] [Google Scholar]
- 36. Zhao L, Qin LX, Ye QH et al . KIAA0008 gene is associated with invasive phenotype of human hepatocellular carcinoma – a functional analysis. J Cancer Res Clin Oncol 2004; 130: 719–27. [DOI] [PubMed] [Google Scholar]
- 37. Matoba K, Iizuka N, Gondo T et al . Tumor HLA‐DR expression linked to early intrahepatic recurrence of hepatocellular carcinoma. Int J Cancer 2005; 115: 231–40. [DOI] [PubMed] [Google Scholar]
- 38. Cheung ST, Leung KL, Ip YC et al . Claudin‐10 expression level is associated with recurrence of primary hepatocellular carcinoma. Clin Cancer Res 2005; 11: 551–6. [PubMed] [Google Scholar]
- 39. Iida H, Honda M, Kawai HF et al . Ephrin‐A1 expression contributes to the malignant characteristics of (alpha)‐fetoprotein producing hepatocellular carcinoma. Gut 2005; 54: 843–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang T, Sun HC, Xu Y et al . Overexpression of platelet‐derived growth factor receptor alpha in endothelial cells of hepatocellular carcinoma associated with high metastatic potential. Clin Cancer Res 2005; 11: 8557–63. [DOI] [PubMed] [Google Scholar]
- 41. Cheung ST, Ho JC, Leung KL et al . Transcript AA454543 is a novel prognostic marker for hepatocellular carcinoma after curative partial hepatectomy. Neoplasia 2005; 7: 91–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chua MS, Sun H, Cheung ST et al . Overexpression of NDRG1 is an indicator of poor prognosis in hepatocellular carcinoma. Mod Pathol 2007; 20: 76–83. [DOI] [PubMed] [Google Scholar]
- 43. Segal E, Shapira M, Regev A et al . Module networks: identifying regulatory modules and their condition‐specific regulators from gene expression data. Nat Genet 2003; 34: 166–76. [DOI] [PubMed] [Google Scholar]
- 44. Vauthey JN, Lauwers GY, Esnaola NF et al . Simplified staging for hepatocellular carcinoma. J Clin Oncol 2002; 20: 1527–36. [DOI] [PubMed] [Google Scholar]
- 45. Kim YS, Lim HK, Rhim H, Lee WJ, Joh JW, Park CK. Recurrence of hepatocellular carcinoma after liver transplantation. patterns and prognostic factors based on clinical and radiologic features. AJR. Am J Roentgenol 2007; 189: 352–8. [DOI] [PubMed] [Google Scholar]
- 46. Chen X, Cheung ST, So S et al . Gene expression patterns in human liver cancers. Mol Biol Cell 2002; 13: 1929–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Okabe H, Satoh S, Kato T et al . Genome‐wide analysis of gene expression in human hepatocellular carcinomas using cDNA microarray: identification of genes involved in viral carcinogenesis and tumor progression. Cancer Res 2001; 61: 2129–37. [PubMed] [Google Scholar]
- 48. Kojiro M. Pathological evolution of early hepatocellular carcinoma. Oncology 2002; 62: 43–7. [DOI] [PubMed] [Google Scholar]
- 49. Tsunedomi R, Iizuka N, Yamada‐Okabe H et al . Identification of ID2 associated with invasion of hepatitis C virus‐related hepatocellular carcinoma by gene expression profile. Int J Oncol 2006; 29: 1445–51. [PubMed] [Google Scholar]
- 50. Iizuka N, Tamesa T, Sakamoto K, Miyamoto T, Hamamoto Y, Oka M. Different molecular pathways determining extrahepatic and intrahepatic recurrences of hepatocellular carcinoma. Oncol Rep 2006; 16: 1137–42. [PubMed] [Google Scholar]
- 51. Cheung ST, Chen X, Guan XY et al . Identify metastasis‐associated genes in hepatocellular carcinoma through clonality delineation for multinodular tumor. Cancer Res 2002; 62: 4711–21. [PubMed] [Google Scholar]
- 52. Hsu HC, Tseng HJ, Lai PL, Lee PH, Peng SY. Expression of p53 gene in 184 unifocal hepatocellular carcinomas: association with tumor growth and invasiveness. Cancer Res 1993; 53: 4691–4. [PubMed] [Google Scholar]
- 53. Okada T, Iizuka N, Yamada‐Okabe H et al . Gene expression profile linked to p53 status in hepatitis C virus‐related hepatocellular carcinoma. FEBS Lett 2003; 555: 583–90. [DOI] [PubMed] [Google Scholar]
- 54. Lee JS, Heo J, Libbrecht L et al . A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat Med 2006; 12: 410–16. [DOI] [PubMed] [Google Scholar]
- 55. Alizadeh AA, Eisen MB, Davis RE et al . Distinct types of diffuse large B‐cell lymphoma identified by gene expression profiling. Nature 2000; 403: 503–11. [DOI] [PubMed] [Google Scholar]
- 56. Breuhahn K, Vreden S, Haddad R et al . Molecular profiling of human hepatocellular carcinoma defines mutually exclusive interferon regulation and insulin‐like growth factor II overexpression. Cancer Res 2004; 64: 6058–64. [DOI] [PubMed] [Google Scholar]
- 57. Bruix J, Boix L, Sala M, Llovet JM. Focus on hepatocellular carcinoma. Cancer Cell 2004; 5: 215–19. [DOI] [PubMed] [Google Scholar]
- 58. Somura H, Iizuka N, Tamesa T et al . A three‐gene predictor for early intrahepatic recurrence of hepatocellular carcinoma after curative hepatectomy. Oncol Rep 2008; 19: 489–95. [PubMed] [Google Scholar]
- 59. Yokoo H, Kondo T, Okano T et al . Protein expression associated with early intrahepatic recurrence of hepatocellular carcinoma after curative surgery. Cancer Sci 2007; 98: 665–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Iizuka N, Hamamoto Y, Oka M. Predicting individual outcomes in hepatocellular carcinoma. Lancet 2004; 364: 1837–9. [DOI] [PubMed] [Google Scholar]
- 61. Oikawa T, Ojima H, Yamasaki S, Takayama T, Hirohashi S, Sakamoto M. Multistep and multicentric development of hepatocellular carcinoma. histological analysis of 980 resected nodules. J Hepatol 2005; 42: 225–9. [DOI] [PubMed] [Google Scholar]
- 62. Sakon M, Umeshita K, Nagano H et al . Clinical significance of hepatic resection in hepatocellular carcinoma: analysis by disease‐free survival curves. Arch Surg 2000; 135: 1456–9. [DOI] [PubMed] [Google Scholar]
- 63. Mas VR, Fisher RA, Archer KJ et al . Genes associated with progression and recurrence of hepatocellular carcinoma in hepatitis C patients waiting and undergoing liver transplantation: preliminary results. Transplantation 2007; 83: 973–81. [DOI] [PubMed] [Google Scholar]
- 64. Kurokawa Y, Matoba R, Nagano H et al . Molecular prediction of response to 5‐fluorouracil and interferon‐alpha combination chemotherapy in advanced hepatocellular carcinoma. Clin Cancer Res 2004; 10: 6029–38. [DOI] [PubMed] [Google Scholar]
- 65. Moriyama M, Hoshida Y, Kato N et al . Genes associated with human hepatocellular carcinoma cell chemosensitivity to 5‐fluorouracil plus interferon‐alpha combination chemotherapy. Int J Oncol 2004; 25: 1279–87. [PubMed] [Google Scholar]
- 66. Ein‐Dor L, Zuk O, Domany E. Thousands of samples are needed to generate a robust gene list for predicting outcome in cancer. Proc Natl Acad Sci USA 2006; 103: 5923–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Uchimura S, Iizuka N, Tamesa T, Miyamoto T, Hamamoto Y, Oka M. Resampling based on geographic patterns of hepatitis virus infection reveals a common gene signature for early intrahepatic recurrence of hepatocellular carcinoma. Anticancer Res 2007; 27: 3323–30. [PubMed] [Google Scholar]
- 68. Brazma A, Hingamp P, Quackenbush J et al . Minimum information about a microarray experiment (MIAME)‐toward standards for microarray data. Nat Genet 2001; 29: 365–71. [DOI] [PubMed] [Google Scholar]
- 69. Hardiman G. Microarray platforms‐comparisons and contrasts. Pharmacogenomics 2004; 5: 487–502. [DOI] [PubMed] [Google Scholar]
- 70. Iizuka N, Hamamoto Y, Oka M. Prediction of cancer outcome with microarrays. Lancet 2005; 365: 1683–4. [DOI] [PubMed] [Google Scholar]
- 71. Kanal L. Patterns in pattern‐recognition‐1968–74. IEEE Transactions Information Theory 1974; 20: 697–722. [Google Scholar]
- 72. Glas AM, Floore A, Delahaye LJ et al . Converting a breast cancer microarray signature into a high‐throughput diagnostic test. BMC Genomics 2006; 7: 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Map of module related to venous invasion. Relationships from literature databases between 35 up‐ and down‐regulated genes responsible for venous invasion (Ref. 49) were extracted and analyzed by using the Genomatix software (http://www.genomatix.de/). The redder and bluer boxes are the higher up‐regulated and lower down‐regulated genes, respectively. Green color indicates the presence of binding motifs in the promoter of the potential transcriptional target.
Fig. S2. Map of module related to early intrahepatic recurrence. Relationships from literature databases between 46 up‐ and down‐regulated genes responsible for early intrahepatic recurrence (Refs. 24 and 37) were extracted and analyzed by using the Genomatix software (http://www.genomatix.de/). The redder and bluer boxes are the higher up‐regulated and lower down‐regulated genes, respectively. Green color indicates the presence of binding motifs in the promoter of the potential transcriptional target.
Fig. S3. Map of module related to extrahepatic recurrence. Relationships from literature databases between 46 up‐ and down‐regulated genes responsible for extrahepatic recurrence (Ref. 50) were extracted and analyzed by using the Genomatix software (http://www.genomatix.de/). The redder and bluer boxes are the higher up‐regulated and lower down‐regulated genes, respectively. Green color indicates the presence of binding motifs in the promoter of the potential transcriptional target.
Fig. S4. Performance of predictors in eligible seven studies. A, Relation between predictive accuracy, number of gene used, and number of test sample. B, Relation between predictive accuracy, publication year, and journal impact factor. The journal impact factors are based on Science Citation Index (SCI ®), which is originally produced by the Institute for Scientific Information (ISI) in 2005.
Please note: Blackwell Publishing are not responsible for the content or functionality of any supplementary materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item