

Abstract
The epithelial–mesenchymal transition (EMT) plays a critical role in embryonic development. EMT is also involved in cancer progression and metastasis and it is probable that a common molecular mechanism is shared by these processes. Cancer cells undergoing EMT can acquire invasive properties and enter the surrounding stroma, resulting in the creation of a favorable microenvironment for cancer progression and metastasis. Furthermore, the acquisition of EMT features has been associated with chemoresistance which could give rise to recurrence and metastasis after standard chemotherapeutic treatment. Thus, EMT could be closely involved in carcinogenesis, invasion, metastasis, recurrence, and chemoresistance. Research into EMT and its role in cancer pathogenesis has progressed rapidly and it is now hypothesized that novel concepts such as cancer stem cells and microRNA could be involved in EMT. However, the involvement of EMT varies greatly among cancer types, and much remains to be learned. In this review, we present recent findings regarding the involvement of EMT in cancer progression and metastasis and provide a perspective from clinical and translational viewpoints. (Cancer Sci 2009)
Development of distant metastases is the final stage of solid cancer progression and is responsible for the majority of cancer‐related deaths.( 1 ) Distant metastasis alone or with concurrent locoregional recurrence accounts for nearly 80% of all first relapses in women with breast cancer.( 2 ) While clinically of great importance, the biology of metastasis remains unsolved. The process of tumor metastasis consists of multiple steps, all of which are required to achieve tumor spreading.( 3 , 4 ) First, cancer cells escape from the primary tumor site. Next, cancer cells invade the tumor stroma and enter the blood circulation directly or the lymphatic system via intravasation. Most circulating cancer cells undergo apoptosis due to anoikis conditions.( 5 ) If cancer cells survive in circulation they may reach more suitable sites by attaching to endothelial cells and extravasating from the circulation into the surrounding tissues. Finally, distal colonization requires that cancer cells invade and grow in the new environment.
Recently, the concept of the epithelial–mesenchymal transition (EMT), as developed in the field of embryology, has been extended to cancer progression and metastasis.( 6 , 7 ) In vitro and experimental animal model data now support the role of EMT in metastasis, concepts supported by analyses of clinical samples. Indeed, the biology of EMT has been clarified in tumor samples through use of EMT‐associated markers, such as mesenchymal‐specific markers (i.e. vimentin and fibronectin),( 8 , 9 ) epithelial specific markers (i.e. E‐cadherin and cytokeratin),( 10 , 11 ) and transcription factors (i.e. SNAIL and SLUG).( 12 )
Most recently, several intriguing studies have described the novel mechanism underlying EMT activation. In the current study, we will discuss the role of small non‐coding RNA (microRNA) in regulating EMT‐related genes.( 13 , 14 , 15 ) Furthermore, Mani et al. disclosed that EMT could generate breast cancer cells with stem cell‐like characteristics.( 16 ) Here, we update and discuss recent progress in studies of EMT. These new data improve our understanding of the mechanisms of cancer progression and metastasis as well as therapy resistance. This new information may lead to development of novel clinical targets and improve the clinical management of cancer patients.
Involvement of EMT in Cancer Progression
In the 1980s, Greenburg and Hey first analyzed EMT‐associated changes in cell phenotype and mesenchymal states in adult and embryonic epithelia.( 17 ) EMT and the inverse process of mesenchymal–epithelial transition (MET) are major embryological mechanisms for tissue remodeling, as in gastrulation and segment formation.( 18 ) The process of EMT consists of multiple steps.( 19 , 20 ) First, cell–cell adhesion disintegrates with the loss of epithelial markers such as E‐cadherin and the gain of mesenchymal markers such as vimentin. Next, there is a loss of baso‐apical polarization and the acquisition of front‐rear polarization. Then, the cytoskeleton undergoes remodeling, with changes in cortical actin and actin stress fibers. Finally, cell‐matrix adhesion is altered, with activation of proteolytic enzymes such as matrix metalloproteases. Note that the process of metastasis in epithelial cancer also consists of multiple steps.( 3 , 4 ) That is, cells detach from the primary tumor and invade the surrounding tumor stroma. They subsequently enter into the circulation and reach new metastatic sites. Therefore, the process of EMT during cancer progression and metastasis closely resembles that observed in embryologic development. Accordingly, molecular analyses based on EMT in embryology have been applied to cancer progression.
In the 1990s, accumulating evidence indicated that EMT was associated with cancer progression.( 7 ) Indeed, these transformations may be associated with EMT‐related signal pathways during development.( 7 , 21 ) However, Boyer et al. stated that EMT during development depends on additional activities of distinct and specific signaling molecules which are highly controlled spatially and temporally, and which do not occur under normal circumstances. On the other hand, EMT in cancer progression could be due to autonomous oncogenic activation of signaling molecules without additional stimulation.( 22 ) Therefore, comparisons of EMT signaling pathways in embryological development and cancer progression may make it possible to identify novel pathways specific to cancer progression and to suggest new therapeutic strategies in cancer therapy.( 23 )
The Molecular Mechanism of EMT in Cancer Progression
Multiple complex signaling systems are required for induction of EMT because epithelial cells undergoing EMT must undergo both functional and morphologic changes. Studies of the crosstalk among the intracellular signal networks could help us to understand the mechanisms regulating EMT. Here, we discuss the regulation of representative molecules, E‐cadherin, a major EMT inducer, transforming growth factor‐β (TGF‐β) signal pathways, and microRNA regulation reported in recent studies (Fig. 1).
Figure 1.
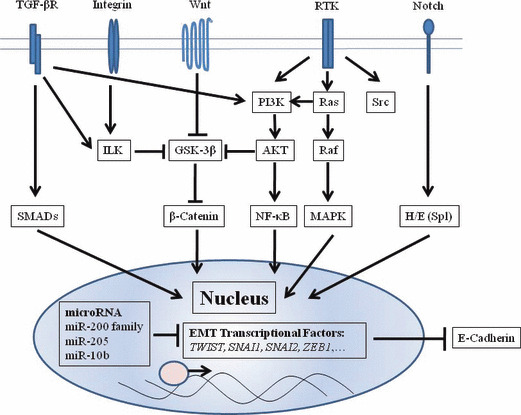
Depiction of signal pathways regulating the epithelial–mesenchymal transition (EMT). Selected signal pathways regulating E‐cadherin are schematized. Transforming growth factor (TGF)‐β signals toward the SMAD pathway or the PI3K/AKT axis. Wnt ligands block β‐catenin degradation. Excess β‐catenin enters the nucleus and upregulates SLUG and SNAIL transcription. In integrin signaling, overexpression of ILK leads to nuclear translocation of β‐catenin. Signals via RTK lead to EMT through the Ras‐Raf‐MAPK pathway or the PI3K/AKT pathway. AKT, serine/threonine kinase; GSK‐3β, glycogen synthase kinase‐3β; H/E (Spl), Hairy and enhancer of split; ILK, integrin‐linked kinase; MAPK, mitogen‐activated protein kinase; NF‐κB, nuclear factor‐κB; PI3K, phosphatidylinositol 3′ kinase; RTK, receptor tyrosine kinase; TGF‐βR, transforming growth factor‐β receptor.
E‐cadherin regulation. One of the characteristic findings in EMT is the loss of cell–cell adhesion with diminished expression of E‐cadherin. E‐cadherin, a calcium‐dependent transmembrane glycoprotein expressed in most epithelial tissues, constructs a tight junction which connects adjacent cells. The loss of E‐cadherin can lead to tumor progression, metastasis, and poorer prognosis in various human carcinomas.( 10 , 11 , 24 , 25 ) Genetic or epigenetic alterations cause a functional loss of E‐cadherin. For instance, mutations in E‐cadherin are found in diffuse gastric cancer( 26 ) and lobular breast carcinoma.( 27 ) In addition, hypermethylation of the E‐cadherin promoter region is found in various human carcinomas, resulting in frequent loss of E‐cadherin expression.( 28 , 29 ) Interestingly, Graff et al. proposed that the degree of methylation of the E‐cadherin promoter region during metastatic progression is unstable and heterogeneous.( 28 ) This finding suggests that the loss of E‐cadherin by methylation in a primary lesion may drive metastatic progression, indicating that EMT is involved in cancer metastasis. Besides genetic or epigenetic control, E‐cadherin is regulated by various signal networks, such as TGF‐β signaling and transcription factors as discussed in more detail below.
TGF‐β signaling. Miettinen et al. first revealed that TGF‐β induced EMT in normal mammary epithelial cells.( 30 ) In fact, TGF‐β is an important inducer of EMT in cancer progression. However, TGF‐β is well known to induce multiple responses in cancer progression.( 31 ) For example, loss of the TGF‐β signaling pathway results in the progression of cancer because TGF‐β is a strong growth inhibitor.( 32 ) Indeed, Hahn et al. reported that mutations in TGF‐β and Smad4 give rise to pancreatic cancer( 33 ) and colorectal cancer.( 34 ) On the other hand, TGF‐β can protect against apoptosis, and promote angiogenesis and immune suppression.( 35 ) TGF‐β induces EMT through multiple signal pathways, including direct phosphorylation of Smad 2 and Smad 3. As shown in Figure 1, TGF‐β also activates other EMT‐related signal pathways, including integrin, Notch, and Wnt signal pathways, all of which trigger EMT programs.
Transcription factors. Transcriptional repressors of E‐cadherin such as zinc finger proteins (ZEB1, ZEB2), bHLH protein (Twist), and the snail family of zinc finger proteins (Snail, Slug) are associated with EMT.( 36 , 37 , 38 , 39 , 40 ) As shown in Figure 1, various signal pathways such as TGF‐β,( 20 ) the Wnt cascade, and PI3K/AKT (phosphatidylinositol 3′ kinase–serine/threonine kinase) axis are connected with these transcriptional repressors of E‐cadherin.( 41 ) Recent studies have demonstrated that transcriptional repressors of E‐cadherin are regulated by microRNAs as described below. Several transcriptional factors such as Snail, Slug, and Twist are useful markers to predict prognosis in various human carcinomas (Table 1). Peinado et al. proposed that E‐cadherin repressors might participate in the process of EMT as follows. First, Snail and ZEB2 would initiate down‐regulation of E‐cadherin. Then, Slug and ZEB1 would maintain repression of E‐cadherin.( 42 ) However, the effect of E‐cadherin repressors on mesenchymal markers such as vimentin and N‐cadherin remains unsolved.
Table 1.
Epithelial–mesenchymal transition (EMT)‐associated markers in clinical samples predict patient prognosis
| EMT‐associated gene | Characteristics | Cancer types | Reference (author) |
|---|---|---|---|
| Epithelial marker | |||
| E‐cadherin | Type I cell–cell adhesion glycoprotein | Breast cancer | Gould Rothberg and Bracken(25) |
| Gastric cancer | Chan et al. (24) | ||
| Colorectal cancer | Doridi et al. ( 84 ) | ||
| Claudin‐1 | Tight junctions restrict lateral diffusion of lipids and membrane proteins | Lung cancer | Chao et al. ( 85 ) |
| Renal cell carcinoma | Fritzsche et al. ( 86 ) | ||
| Ovarian carcinoma | Kleinberg et al. ( 87 ) | ||
| Mesenchymal marker | |||
| Vimentin | Intermediate filaments represent a third class of cytoskeletal elements | Breast cancer | Thomas et al. ( 88 ) |
| Lung cancer | AI‐Saad et al. ( 89 ) | ||
| Gastric cancer | Utsunomiya et al. ( 90 ) | ||
| N‐cadherin | Type I cell–cell adhesion glycoprotein | Esophageal cancer | Yoshinaga et al. ( 91 ) |
| Lung cancer | Nakashima et al. ( 92 ) | ||
| Urothelial tumor | Lascombe et al. ( 93 ) | ||
| Fibronectin | High‐molecular weight extracellular matrix glycoprotein | Bladder tumor | Mutlu et al. ( 94 ) |
| Colorectal cancer | Inufusa et al. ( 95 ) | ||
| Ovarian carcinoma | Franke et al. ( 96 ) | ||
| Transcription factor | |||
| Snail | Zinc finger transcriptional repressor | Adenocortical carcinoma | Waldmann et al. ( 97 ) |
| Esophageal cancer | Natsugoe et al. ( 98 ) | ||
| Hepatocellular carcinoma | Miyoshi et al. ( 99 ) | ||
| Slug | Zinc finger transcriptional repressor | Lung cancer | Shih et al. ( 100 ) |
| Colorectal cancer | Shioiri et al. ( 101 ) | ||
| Esophageal cancer | Uchikado et al. ( 102 ) | ||
| Twist | Basic helix‐loop‐helix transcription factors | Cervical cancer | Shibata et al. ( 103 ) |
| Ovarian carcinoma | Hosono et al. ( 104 ) | ||
| Breast cancer | Martin et al. ( 105 ) | ||
Regulation of EMT by microRNA. Recent studies of small non‐coding RNAs are shedding light on the regulation of gene expression and proteins in metastasis. It was shown that miR‐10b overexpression is associated with invasiveness and metastatic potential.( 43 ) miR‐10b is overexpressed in metastatic breast cancer, and up‐regulated by EMT transcription factor Twist. Recent independent studies revealed that the miR‐200 family (miR‐200a, miR‐200b, miR‐200c, miR‐141, and miR‐429) and miR‐205 play critical roles in regulating EMT, targeting the E‐cadherin repressors ZEB1 and ZEB2.( 13 , 15 ) Gibbons et al. found that metastasis‐prone tumor cells established from metastatic lung adenocarcinoma (with evidence of mutant K‐ras and p53) could transit reversibly between epithelial and mesenchymal states, a property that was regulated by the miR‐200 family.( 44 ) Furthermore, two recent independent studies showed that members of the miR‐200 family can induce the EMT process and regulate the sensitivity to epidermal growth factor receptor (EGFR) in bladder cancer cells and to gemcitabine in pancreatic cancer cells.( 45 , 46 ) As for regulating TGF‐β, microRNAs related to TGF‐β signaling such as miR‐155 and miR‐29a have been identified in breast cancer tissues.( 47 , 48 ) It is important to identify microRNAs involved in EMT to elucidate up‐stream regulators of various known signal pathways.
Microenvironment and EMT
The tumor microenvironment is composed of the extracellular matrix (ECM), cancer‐associated fibroblasts, myofibroblasts, immune cells, and soluble factors required for cancer progression and metastasis. Interaction among cancer cells in the tumor microenvironment can induce EMT by auto‐ and/or paracrine secretion of mediators such as growth factors, cytokines, and ECM proteins.( 21 ) Media conditioned by cultures of cancer‐associated fibroblast induce EMT in breast cancer cells.( 49 ) In a comparison of the central areas of primary colorectal cancer and corresponding metastases, nuclear β‐catenin was found in dedifferentiated mesenchyme‐like tumor cells at the invasive front and it was localized to the membrane and cytoplasm.( 50 ) This study suggested that the tumor microenvironment may induce or maintain EMT (Fig. 2). For instance, cancer‐associated fibroblasts may be supplied from cancer cells undergoing EMT.( 51 ) Similarly, oral squamous cancer cells can directly induce a myofibroblastic phenotype via secretion of TGF‐β. TGF‐β signaling by stromal myofibroblast can induce secretion of hepatocyte growth factor (HGF) which promotes cancer cell proliferation and invasion.( 52 )
Figure 2.
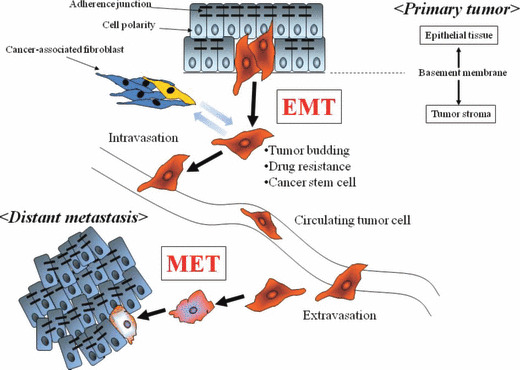
The epithelial–mesenchymal transition (EMT) and mesenchymal–epithelial transition (MET) are involved in cancer metastasis. Cancer cells undergoing EMT in a primary tumor disseminate through the fragmented basement membrane and acquire the characteristics of drug resistance and cancer stem cells. They can be recognized in tumor buds in histological specimens. EMT cells invade into tumor stroma and enter the circulation, allowing transport to distant organs. At metastatic sites, solitary cancer cells form the new metastatic focus through MET.
Drug Resistance and EMT
Cells undergoing EMT become invasive and develop resistance to anticancer agents (Fig. 2). In fact, EMT can be induced by anticancer agents, and stress conditions such as exposure to radiation and hypoxic conditions.( 53 , 54 ) Up‐regulation of TWIST was associated with cellular resistance to paclitaxel in human nasopharyngeal, bladder, ovarian, and prostate cancers.( 55 ) In colorectal cancer, stable oxaliplatin‐resistant cells established by chronic exposure to oxaliplatin can acquire the ability to migrate and invade with phenotypic changes resembling EMT (spindle‐cell shape, loss of polarity, intercellular separation, and pseudopodia formation).( 56 ) In pancreatic and ovarian cancer, stable cell lines resistant to gemcitabine and paclitaxel established by continuous exposure can undergo EMT with increased expression of Snail and Twist, EMT‐regulatory transcription factors.( 57 , 58 )
Various types of molecularly targeted agents have been developed and used against many carcinomas with or without combination of traditional anticancer agents, leading to improved clinical outcome and survival rate.( 59 , 60 ) However, EMT reportedly confers resistance to these targeted agents. For example, lung cancer cell lines having undergone EMT, expressing vimentin and/or fibronectin, were insensitive to the growth inhibitory effects of EGFR kinase inhibition (erotinib) in vitro and in xenografts( 61 ) as well as other EGFR inhibitors such as gefitinib and cetuximab.( 62 , 63 ) We have often encountered patients who have suffered relapses after drug treatment, even when the tumors were initially highly sensitive. Thus, EMT can lead to resistance to multiple drugs and permit rapid progression of the tumor. These clinical findings may be attributed to the inherent characteristics of EMT. Clarifying the correlation between EMT and drug resistance may help clinicians select an optimal anticancer drug treatment.
Cancer Stem Cells and EMT
Cancer researchers have recently found a minor fraction of cells (cancer stem cells [CSC]) with the ability to self‐renew and give rise to differentiated tumor cells. CSC have been identified in breast, colon, and pancreatic cancer.( 64 , 65 , 66 ) CSC as well as cells undergoing EMT are considered to be more resistant to toxic injuries and chemoradiation therapy than differentiated daughter cells.( 67 , 68 ) Furthermore, cancer cells under hypoxic conditions acquire the properties of CSC.( 69 , 70 ) Even though evidence indicates a relationship between EMT and cancer cells with the traits of stemness,( 71 ) CSC are rare in whole tumor tissues.( 68 , 72 ) However, it remains controversial among pathologists whether CSC as well as cells undergoing EMT exist in human cancer tissues.( 73 ) Intriguingly, Mani et al. initially disclosed that immortalized human mammary epithelial cells (HMLEs) undergoing EMT are CSC‐like as characterized by their CD44high/CD24low phenotype.( 16 ) These investigators induced EMT in HMLEs by ectopic expression of Twist or Snail, known inducers of EMT. The cells undergoing EMT acquired a fibroblastoid mesenchymal appearance. Furthermore, Mani et al. observed down‐regulation of epithelial markers such as E‐cadherin and up‐regulation of mesenchymal markers such as N‐cadherin, vimentin, and fibronectin. They also noted a CD44high/CD24low expression pattern associated with human breast CSCs. Furthermore, they revealed that the cells undergoing EMT had the properties of CSC, including self‐renewal and the capacity to form mammospheres. These findings suggest that EMT may play a role in the development of CSC and properties of invasiveness, metastasis, recurrence, and chemoresistance (Fig. 2).
Clinical Significance of EMT
EMT‐associated markers in clinical samples and their effects on prognosis are summarized in Table 1. Most EMT‐associated markers have been identified in histological specimens. However, the existence of EMT cells in clinical specimens has been challenged.( 74 ) In response, Voulgari et al. suggested that the controversy between experimental and clinical studies is due to the ‘spatial’ and ‘temporal’ heterogeneity of EMT (Fig. 3).( 19 ) Cells undergoing EMT may gain metastatic potential but may constitute only a small proportion of the total population of tumor cells. Tumor budding is commonly observed in clinical practice, and it consists of a single cancer cell or small cell cluster at the invasive front of tumor tissues. Indeed, cancer cells in tumor buds have down‐regulated E‐cadherin( 75 ) and have characteristics of CSC.( 76 ) Therefore, identification of cancer cells undergoing EMT in clinical specimens is difficult for pathologists.
Figure 3.
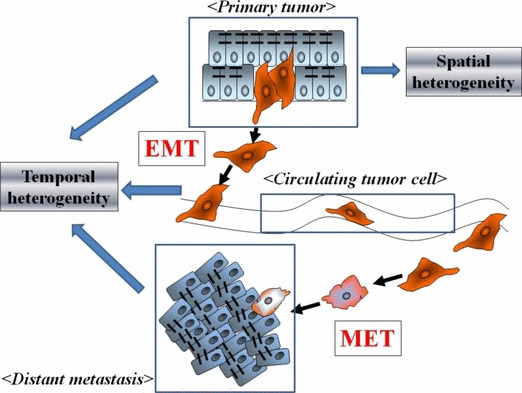
Spatial and temporal heterogeneity of the epithelial–mesenchymal transition (EMT). Cancer cells undergoing EMT are expected to be only a small proportion of primary tumor tissues. EMT cells transported to metastatic sites are expected to undergo and mesenchymal–epithelial transition (MET). Therefore, the spatial and temporal heterogeneity of EMT/MET severely restricts the ability of pathologists to detect cancer cells undergoing EMT in histological sections.
The temporal heterogeneity of EMT (and the reverse, MET) is readily explained. MET is observed in vitro following addition of bone morphogenetic protein 7 (BMP7), removal of an EMT‐inducer such as TGF‐β, and establishment of hypoxic conditions.( 54 , 77 ) A similar process may occur at metastatic sites which require cancer cells to recover the expression of E‐cadherin for cell adhesion. The phenotypes of metastatic specimens are often compared with primary specimens to confirm the diagnosis by hematoxylin–eosin staining. The presence of the same cancer cell characteristics or phenotypes in both primary and metastatic lesions can provide the diagnosis of cancer metastasis. Therefore, the occurrence of MET could make it difficult to prove that EMT, a transient phenomenon that involves only a minority of cells, has occurred in human cancer specimens. However, EMT‐associated genes obviously are useful as predictive biomarkers (Table 1). Clinical verification of EMT will require advanced techniques such as in vivo imaging.
Treatments Targeting EMT
As shown in Figure 1, EMT‐related pathways provide targets for therapy. For instance, inhibition of integrin‐linked kinase (ILK) increases the sensitivity of mesenchymal cells to EGFR‐target therapy in hepatocellular carcinoma.( 63 ) In in vitro studies, Src kinase inhibitors effectively inhibit the growth of cells undergoing EMT.( 78 ) Furthermore, the inhibition of hedgehog signaling can prevent pancreatic cancer cells from acquiring tumor‐initiating property and undergoing EMT.( 79 , 80 )
RNA interference and microRNA are new technologies in drug development. For instance, silencing of Snail by shRNA induced MET and reduced in vivo tumor growth.( 81 ) As for microRNA, Krutzfeldt et al. disclosed that specific silencers of endogenous miRNAs, antagomirs, are powerful tools to silence specific miRNAs in vivo. ( 82 ) Therefore, microRNAs associated with EMT such as the miR‐10b and miR‐200 family could be exploited as therapeutic strategies in the future.
Furthermore, the tumor microenvironment, which contributes to the maintenance of EMT, could be targeted. A small‐interfering RNA targeted at TGF‐β reportedly reduces metastasis in vivo,( 83 ) and this observation could be applied to TGF‐β secreted by tumor stroma. Note that reducing EMT could also lessen the occurrence of anticancer drug resistance and thereby improve the efficacy of conventional therapy. To eradicate cancer cells effectively and cause minimal toxicity to normal cells, further studies are required to define the molecular differences between EMT in embryological development and that in cancer progression.
Perspectives
During the past few decades, an increasing number of studies have shown that EMT is associated with cancer progression, metastasis, and drug resistance. Furthermore, improved understanding of microRNAs and cancer stem cells will clarify the processes underlying EMT. Current understanding of traditional signal pathways coupled with these new concepts could accelerate progress in cancer research. However, the multimodal nature of these complex pathways presents formidable challenges to researchers attempting to inhibit the onset of EMT. Finally, the clinical evidence supporting the role of EMT in cancer progression is still relatively weak. Thus, better methods for EMT detection in patient samples are needed.
Acknowledgments
This work was supported by the following grants and foundations: CREST, Japan Science and Technology Agency (JST); Japan Society for the Promotion of Science (JSPS) Grant‐in‐Aid for Scientific Research, grant numbers 20390360, 20591547, 20790960, 21591644, 21791295, 21791297, 215921014, and 21679006.
References
- 1. Weigelt B, Peterse JL, Van‘t Veer LJ. Breast cancer metastasis: markers and models. Nat Rev Cancer 2005; 5: 591–602. [DOI] [PubMed] [Google Scholar]
- 2. Citron ML, Berry DA, Cirrincione C et al. Randomized trial of dose‐dense versus conventionally scheduled and sequential versus concurrent combination chemotherapy as postoperative adjuvant treatment of node‐positive primary breast cancer: first report of Intergroup Trial C9741/Cancer and Leukemia Group B Trial 9741. J Clin Oncol 2003; 21: 1431–9. [DOI] [PubMed] [Google Scholar]
- 3. Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer 2002; 2: 563–72. [DOI] [PubMed] [Google Scholar]
- 4. Woodhouse EC, Chuaqui RF, Liotta LA. General mechanisms of metastasis. Cancer 1997; 80: 1529–37. [DOI] [PubMed] [Google Scholar]
- 5. Mehes G, Witt A, Kubista E, Ambros PF. Circulating breast cancer cells are frequently apoptotic. Am J Pathol 2001; 159: 17–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer 2009; 9: 265–73. [DOI] [PubMed] [Google Scholar]
- 7. Thiery JP. Epithelial‐mesenchymal transitions in tumour progression. Nat Rev Cancer 2002; 2: 442–54. [DOI] [PubMed] [Google Scholar]
- 8. Ngan CY, Yamamoto H, Seshimo I et al. Quantitative evaluation of vimentin expression in tumour stroma of colorectal cancer. Br J Cancer 2007; 96: 986–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Raymond WA, Leong AS. Vimentin – a new prognostic parameter in breast carcinoma? J Pathol 1989; 158: 107–14. [DOI] [PubMed] [Google Scholar]
- 10. Dorudi S, Sheffield JP, Poulsom R, Northover JM, Hart IR. E‐cadherin expression in colorectal cancer. An immunocytochemical and in situ hybridization study. Am J Pathol 1993; 142: 981–6. [PMC free article] [PubMed] [Google Scholar]
- 11. Kowalski PJ, Rubin MA, Kleer CG. E‐cadherin expression in primary carcinomas of the breast and its distant metastases. Breast Cancer Res 2003; 5: R217–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer 2007; 7: 415–28. [DOI] [PubMed] [Google Scholar]
- 13. Gregory PA, Bert AG, Paterson EL et al. The miR‐200 family and miR‐205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 2008; 10: 593–601. [DOI] [PubMed] [Google Scholar]
- 14. Korpal M, Lee ES, Hu G, Kang Y. The miR‐200 family inhibits epithelial‐mesenchymal transition and cancer cell migration by direct targeting of E‐cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem 2008; 283: 14910–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Park SM, Gaur AB, Lengyel E, Peter ME. The miR‐200 family determines the epithelial phenotype of cancer cells by targeting the E‐cadherin repressors ZEB1 and ZEB2. Genes Dev 2008; 22: 894–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mani SA, Guo W, Liao MJ et al. The epithelial‐mesenchymal transition generates cells with properties of stem cells. Cell 2008; 133: 704–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Greenburg G, Hay ED. Epithelia suspended in collagen gels can lose polarity and express characteristics of migrating mesenchymal cells. J Cell Biol 1982; 95: 333–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hay ED. The mesenchymal cell, its role in the embryo, and the remarkable signaling mechanisms that create it. Dev Dyn 2005; 233: 706–20. [DOI] [PubMed] [Google Scholar]
- 19. Voulgari A, Pintzas A. Epithelial‐mesenchymal transition in cancer metastasis: mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochim Biophys Acta 2009; 1796: 75–90. [DOI] [PubMed] [Google Scholar]
- 20. Zavadil J, Bottinger EP. TGF‐beta and epithelial‐to‐mesenchymal transitions. Oncogene 2005; 24: 5764–74. [DOI] [PubMed] [Google Scholar]
- 21. Moustakas A, Heldin CH. Signaling networks guiding epithelial‐mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci 2007; 98: 1512–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boyer B, Valles AM, Edme N. Induction and regulation of epithelial‐mesenchymal transitions. Biochem Pharmacol 2000; 60: 1091–9. [DOI] [PubMed] [Google Scholar]
- 23. Yang J, Weinberg RA. Epithelial‐mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell 2008; 14: 818–29. [DOI] [PubMed] [Google Scholar]
- 24. Chan AO, Chu KM, Lam SK et al. Soluble E‐cadherin is an independent pretherapeutic factor for long‐term survival in gastric cancer. J Clin Oncol 2003; 21: 2288–93. [DOI] [PubMed] [Google Scholar]
- 25. Gould Rothberg BE, Bracken MB. E‐cadherin immunohistochemical expression as a prognostic factor in infiltrating ductal carcinoma of the breast: a systematic review and meta‐analysis. Breast Cancer Res Treat 2006; 100: 139–48. [DOI] [PubMed] [Google Scholar]
- 26. Becker KF, Atkinson MJ, Reich U et al. E‐cadherin gene mutations provide clues to diffuse type gastric carcinomas. Cancer Res 1994; 54: 3845–52. [PubMed] [Google Scholar]
- 27. Berx G, Cleton‐Jansen AM, Nollet F et al. E‐cadherin is a tumour/invasion suppressor gene mutated in human lobular breast cancers. EMBO J 1995; 14: 6107–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Graff JR, Herman JG, Lapidus RG et al. E‐cadherin expression is silenced by DNA hypermethylation in human breast and prostate carcinomas. Cancer Res 1995; 55: 5195–9. [PubMed] [Google Scholar]
- 29. Yoshiura K, Kanai Y, Ochiai A, Shimoyama Y, Sugimura T, Hirohashi S. Silencing of the E‐cadherin invasion‐suppressor gene by CpG methylation in human carcinomas. Proc Natl Acad Sci U S A 1995; 92: 7416–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Miettinen PJ, Ebner R, Lopez AR, Derynck R. TGF‐beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J Cell Biol 1994; 127: 2021–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bierie B, Moses HL. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer 2006; 6: 506–20. [DOI] [PubMed] [Google Scholar]
- 32. Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med 2000; 342: 1350–8. [DOI] [PubMed] [Google Scholar]
- 33. Hahn SA, Schutte M, Hoque AT et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996; 271: 350–3. [DOI] [PubMed] [Google Scholar]
- 34. Miyaki M, Iijima T, Konishi M et al. Higher frequency of Smad4 gene mutation in human colorectal cancer with distant metastasis. Oncogene 1999; 18: 3098–103. [DOI] [PubMed] [Google Scholar]
- 35. Derynck R, Akhurst RJ, Balmain A. TGF‐beta signaling in tumor suppression and cancer progression. Nat Genet 2001; 29: 117–29. [DOI] [PubMed] [Google Scholar]
- 36. Batlle E, Sancho E, Franci C et al. The transcription factor snail is a repressor of E‐cadherin gene expression in epithelial tumour cells. Nat Cell Biol 2000; 2: 84–9. [DOI] [PubMed] [Google Scholar]
- 37. Comijn J, Berx G, Vermassen P et al. The two‐handed E box binding zinc finger protein SIP1 downregulates E‐cadherin and induces invasion. Mol Cell 2001; 7: 1267–78. [DOI] [PubMed] [Google Scholar]
- 38. Eger A, Aigner K, Sonderegger S et al. DeltaEF1 is a transcriptional repressor of E‐cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene 2005; 24: 2375–85. [DOI] [PubMed] [Google Scholar]
- 39. Hajra KM, Chen DY, Fearon ER. The SLUG zinc‐finger protein represses E‐cadherin in breast cancer. Cancer Res 2002; 62: 1613–8. [PubMed] [Google Scholar]
- 40. Yang J, Mani SA, Donaher JL et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004; 117: 927–39. [DOI] [PubMed] [Google Scholar]
- 41. Larue L, Bellacosa A. Epithelial‐mesenchymal transition in development and cancer: role of phosphatidylinositol 3’ kinase/AKT pathways. Oncogene 2005; 24: 7443–54. [DOI] [PubMed] [Google Scholar]
- 42. Peinado H, Portillo F, Cano A. Transcriptional regulation of cadherins during development and carcinogenesis. Int J Dev Biol 2004; 48: 365–75. [DOI] [PubMed] [Google Scholar]
- 43. Ma L, Teruya‐Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA‐10b in breast cancer. Nature 2007; 449: 682–8. [DOI] [PubMed] [Google Scholar]
- 44. Gibbons DL, Lin W, Creighton CJ et al. Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR‐200 family expression. Genes Dev 2009; 23: 2140–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Adam L, Zhong M, Choi W et al. miR‐200 expression regulates epithelial‐to‐mesenchymal transition in bladder cancer cells and reverses resistance to epidermal growth factor receptor therapy. Clin Cancer Res 2009; 15: 5060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li Y, VandenBoom TG 2nd, Kong D et al. Up‐regulation of miR‐200 and let‐7 by natural agents leads to the reversal of epithelial‐to‐mesenchymal transition in gemcitabine‐resistant pancreatic cancer cells. Cancer Res 2009; 69: 6704–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gebeshuber CA, Zatloukal K, Martinez J. miR‐29a suppresses tristetraprolin, which is a regulator of epithelial polarity and metastasis. EMBO Rep 2009; 10: 400–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kong W, Yang H, He L et al. MicroRNA‐155 is regulated by the transforming growth factor beta/Smad pathway and contributes to epithelial cell plasticity by targeting RhoA. Mol Cell Biol 2008; 28: 6773–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lebret SC, Newgreen DF, Thompson EW, Ackland ML. Induction of epithelial to mesenchymal transition in PMC42‐LA human breast carcinoma cells by carcinoma‐associated fibroblast secreted factors. Breast Cancer Res 2007; 9: R19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Brabletz T, Jung A, Reu S et al. Variable beta‐catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc Natl Acad Sci U S A 2001; 98: 10356–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Radisky DC, Kenny PA, Bissell MJ. Fibrosis and cancer: do myofibroblasts come also from epithelial cells via EMT? J Cell Biochem 2007; 101: 830–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lewis MP, Lygoe KA, Nystrom ML et al. Tumour‐derived TGF‐beta1 modulates myofibroblast differentiation and promotes HGF/SF‐dependent invasion of squamous carcinoma cells. Br J Cancer 2004; 90: 822–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jung JW, Hwang SY, Hwang JS, Oh ES, Park S, Han IO. Ionising radiation induces changes associated with epithelial‐mesenchymal transdifferentiation and increased cell motility of A549 lung epithelial cells. Eur J Cancer 2007; 43: 1214–24. [DOI] [PubMed] [Google Scholar]
- 54. Yang MH, Wu MZ, Chiou SH et al. Direct regulation of TWIST by HIF‐1alpha promotes metastasis. Nat Cell Biol 2008; 10: 295–305. [DOI] [PubMed] [Google Scholar]
- 55. Wang X, Ling MT, Guan XY et al. Identification of a novel function of TWIST, a bHLH protein, in the development of acquired taxol resistance in human cancer cells. Oncogene 2004; 23: 474–82. [DOI] [PubMed] [Google Scholar]
- 56. Yang AD, Fan F, Camp ER et al. Chronic oxaliplatin resistance induces epithelial‐to‐mesenchymal transition in colorectal cancer cell lines. Clin Cancer Res 2006; 12: 4147–53. [DOI] [PubMed] [Google Scholar]
- 57. Kajiyama H, Shibata K, Terauchi M et al. Chemoresistance to paclitaxel induces epithelial‐mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. Int J Oncol 2007; 31: 277–83. [PubMed] [Google Scholar]
- 58. Shah AN, Summy JM, Zhang J, Park SI, Parikh NU, Gallick GE. Development and characterization of gemcitabine‐resistant pancreatic tumor cells. Ann Surg Oncol 2007; 14: 3629–37. [DOI] [PubMed] [Google Scholar]
- 59. Hurwitz H, Fehrenbacher L, Novotny W et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004; 350: 2335–42. [DOI] [PubMed] [Google Scholar]
- 60. Piccart‐Gebhart MJ, Procter M, Leyland‐Jones B et al. Trastuzumab after adjuvant chemotherapy in HER2‐positive breast cancer. N Engl J Med 2005; 353: 1659–72. [DOI] [PubMed] [Google Scholar]
- 61. Thomson S, Buck E, Petti F et al. Epithelial to mesenchymal transition is a determinant of sensitivity of non‐small‐cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res 2005; 65: 9455–62. [DOI] [PubMed] [Google Scholar]
- 62. Frederick BA, Helfrich BA, Coldren CD et al. Epithelial to mesenchymal transition predicts gefitinib resistance in cell lines of head and neck squamous cell carcinoma and non‐small cell lung carcinoma. Mol Cancer Ther 2007; 6: 1683–91. [DOI] [PubMed] [Google Scholar]
- 63. Fuchs BC, Fujii T, Dorfman JD et al. Epithelial‐to‐mesenchymal transition and integrin‐linked kinase mediate sensitivity to epidermal growth factor receptor inhibition in human hepatoma cells. Cancer Res 2008; 68: 2391–9. [DOI] [PubMed] [Google Scholar]
- 64. Al‐Hajj M, Wicha MS, Benito‐Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A 2003; 100: 3983–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Li C, Heidt DG, Dalerba P et al. Identification of pancreatic cancer stem cells. Cancer Res 2007; 67: 1030–7. [DOI] [PubMed] [Google Scholar]
- 66. O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007; 445: 106–10. [DOI] [PubMed] [Google Scholar]
- 67. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature 2001; 414: 105–11. [DOI] [PubMed] [Google Scholar]
- 68. Sagar J, Chaib B, Sales K, Winslet M, Seifalian A. Role of stem cells in cancer therapy and cancer stem cells: a review. Cancer Cell Int 2007; 7: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Axelson H, Fredlund E, Ovenberger M, Landberg G, Pahlman S. Hypoxia‐induced dedifferentiation of tumor cells – a mechanism behind heterogeneity and aggressiveness of solid tumors. Semin Cell Dev Biol 2005; 16: 554–63. [DOI] [PubMed] [Google Scholar]
- 70. Platet N, Liu SY, Atifi ME et al. Influence of oxygen tension on CD133 phenotype in human glioma cell cultures. Cancer Lett 2007; 258: 286–90. [DOI] [PubMed] [Google Scholar]
- 71. Prindull G. Hypothesis: cell plasticity, linking embryonal stem cells to adult stem cell reservoirs and metastatic cancer cells? Exp Hematol 2005; 33: 738–46. [DOI] [PubMed] [Google Scholar]
- 72. Ishii H, Iwatsuki M, Ieta K et al. Cancer stem cells and chemoradiation resistance. Cancer Sci 2008; 99: 1871–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Christiansen JJ, Rajasekaran AK. Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer Res 2006; 66: 8319–26. [DOI] [PubMed] [Google Scholar]
- 74. Garber K. Epithelial‐to‐mesenchymal transition is important to metastasis, but questions remain. J Natl Cancer Inst 2008; 100: 232–3. 9. [DOI] [PubMed] [Google Scholar]
- 75. Masaki T, Goto A, Sugiyama M et al. Possible contribution of CD44 variant 6 and nuclear beta‐catenin expression to the formation of budding tumor cells in patients with T1 colorectal carcinoma. Cancer 2001; 92: 2539–46. [DOI] [PubMed] [Google Scholar]
- 76. Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Opinion: migrating cancer stem cells – an integrated concept of malignant tumour progression. Nat Rev Cancer 2005; 5: 744–9. [DOI] [PubMed] [Google Scholar]
- 77. Rees JR, Onwuegbusi BA, Save VE, Alderson D, Fitzgerald RC. In vivo and in vitro evidence for transforming growth factor‐beta1‐mediated epithelial to mesenchymal transition in esophageal adenocarcinoma. Cancer Res 2006; 66: 9583–90. [DOI] [PubMed] [Google Scholar]
- 78. Finn RS, Dering J, Ginther C et al. Dasatinib, an orally active small molecule inhibitor of both the src and abl kinases, selectively inhibits growth of basal‐type/”triple‐negative” breast cancer cell lines growing in vitro. Breast Cancer Res Treat 2007; 105: 319–26. [DOI] [PubMed] [Google Scholar]
- 79. Feldmann G, Fendrich V, McGovern K et al. An orally bioavailable small‐molecule inhibitor of Hedgehog signaling inhibits tumor initiation and metastasis in pancreatic cancer. Mol Cancer Ther 2008; 7: 2725–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Feldmann G, Dhara S, Fendrich V et al. Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: a new paradigm for combination therapy in solid cancers. Cancer Res 2007; 67: 2187–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Olmeda D, Jorda M, Peinado H, Fabra A, Cano A. Snail silencing effectively suppresses tumour growth and invasiveness. Oncogene 2007; 26: 1862–74. [DOI] [PubMed] [Google Scholar]
- 82. Krutzfeldt J, Rajewsky N, Braich R et al. Silencing of microRNAs in vivo with ‘antagomirs’. Nature 2005; 438: 685–9. [DOI] [PubMed] [Google Scholar]
- 83. Moore LD, Isayeva T, Siegal GP, Ponnazhagan S. Silencing of transforming growth factor‐beta1 in situ by RNA interference for breast cancer: implications for proliferation and migration in vitro and metastasis in vivo. Clin Cancer Res 2008; 14: 4961–70. [DOI] [PubMed] [Google Scholar]
- 84. Dorudi S, Hanby AM, Poulsom R, Northover J, Hart IR. Level of expression of E‐cadherin mRNA in colorectal cancer correlates with clinical outcome. Br J Cancer 1995; 71: 614–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Chao YC, Pan SH, Yang SC, et al. Claudin‐1 is a metastasis suppressor and correlates with clinical outcome in lung adenocarcinoma. Am J Respir Crit Care Med 2009; 179: 123–33. [DOI] [PubMed] [Google Scholar]
- 86. Fritzsche FR, Oelrich B, Johannsen M, et al. Claudin‐1 protein expression is a prognostic marker of patient survival in renal cell carcinomas. Clin Cancer Res 2008; 14: 7035–42. [DOI] [PubMed] [Google Scholar]
- 87. Kleinberg L, Holth A, Trope CG, Reich R, Davidson B. Claudin upregulation in ovarian carcinoma effusions is associated with poor survival. Hum Pathol 2008; 39: 747–57. [DOI] [PubMed] [Google Scholar]
- 88. Thomas PA, Kirschmann DA, Cerhan JR, et al. Association between keratin and vimentin expression, malignant phenotype, and survival in postmenopausal breast cancer patients. Clin Cancer Res 1999; 5: 2698–703. [PubMed] [Google Scholar]
- 89. Al‐Saad S, Al‐Shibli K, Donnem T, Persson M, Bremnes RM, Busund LT. The prognostic impact of NF‐kappaB p105, vimentin, E‐cadherin and Par6 expression in epithelial and stromal compartment in non‐small‐cell lung cancer. Br J Cancer 2008; 99: 1476–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Utsunomiya T, Yao T, Masuda K, Tsuneyoshi M. Vimentin‐positive adenocarcinomas of the stomach: co‐expression of vimentin and cytokeratin. Histopathology 1996; 29: 507–16. [DOI] [PubMed] [Google Scholar]
- 91. Yoshinaga K, Inoue H, Utsunomiya T, et al. N‐cadherin is regulated by activin A and associated with tumor aggressiveness in esophageal carcinoma. Clin Cancer Res 2004; 10: 5702–7. [DOI] [PubMed] [Google Scholar]
- 92. Nakashima T, Huang C, Liu D, et al. Neural‐cadherin expression associated with angiogenesis in non‐small‐cell lung cancer patients. Br J Cancer 2003; 88: 1727–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lascombe I, Clairotte A, Fauconnet S, et al. N‐cadherin as a novel prognostic marker of progression in superficial urothelial tumors. Clin Cancer Res 2006; 12: 2780–7. [DOI] [PubMed] [Google Scholar]
- 94. Mutlu N, Turkeri L, Emerk K. Analytical and clinical evaluation of a new urinary tumor marker: bladder tumor fibronectin in diagnosis and follow‐up of bladder cancer. Clin Chem Lab Med 2003; 41: 1069–74. [DOI] [PubMed] [Google Scholar]
- 95. Inufusa H, Nakamura M, Adachi T, et al. Localization of oncofetal and normal fibronectin in colorectal cancer. Correlation with histologic grade, liver metastasis, and prognosis. Cancer 1995; 75: 2802–8. [DOI] [PubMed] [Google Scholar]
- 96. Franke FE, Von Georgi R, Zygmunt M, Munstedt K. Association between fibronectin expression and prognosis in ovarian carcinoma. Anticancer Res 2003; 23: 4261–7. [PubMed] [Google Scholar]
- 97. Waldmann J, Feldmann G, Slater EP, et al. Expression of the zinc‐finger transcription factor Snail in adrenocortical carcinoma is associated with decreased survival. Br J Cancer 2008; 99: 1900–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Natsugoe S, Uchikado Y, Okumura H, et al. Snail plays a key role in E‐cadherin‐preserved esophageal squamous cell carcinoma. Oncol Rep 2007; 17: 517–23. [PubMed] [Google Scholar]
- 99. Miyoshi A, Kitajima Y, Kido S, et al. Snail accelerates cancer invasion by upregulating MMP expression and is associated with poor prognosis of hepatocellular carcinoma. Br J Cancer 2005; 92: 252–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Shih JY, Tsai MF, Chang TH, et al. Transcription repressor slug promotes carcinoma invasion and predicts outcome of patients with lung adenocarcinoma. Clin Cancer Res 2005; 11: 8070–8. [DOI] [PubMed] [Google Scholar]
- 101. Shioiri M, Shida T, Koda K, et al. Slug expression is an independent prognostic parameter for poor survival in colorectal carcinoma patients. Br J Cancer 2006; 94: 1816–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Uchikado Y, Natsugoe S, Okumura H, et al. Slug Expression in the E‐cadherin preserved tumors is related to prognosis in patients with esophageal squamous cell carcinoma. Clin Cancer Res 2005; 11: 1174–80. [PubMed] [Google Scholar]
- 103. Shibata K, Kajiyama H, Ino K, et al. Twist expression in patients with cervical cancer is associated with poor disease outcome. Ann Oncol 2008; 19: 81–5. [DOI] [PubMed] [Google Scholar]
- 104. Hosono S, Kajiyama H, Terauchi M, et al. Expression of Twist increases the risk for recurrence and for poor survival in epithelial ovarian carcinoma patients. Br J Cancer 2007; 96: 314–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Martin TA, Goyal A, Watkins G, Jiang WG. Expression of the transcription factors snail, slug, and twist and their clinical significance in human breast cancer. Ann Surg Oncol 2005; 12: 488–96. [DOI] [PubMed] [Google Scholar]