

Abstract
The anti‐apoptotic oncoproteins Bcl‐2 and Bcl‐xL play crucial roles in tumorigenesis and chemoresistance, and are thus therapeutic cancer targets. We searched for small molecules that disturbed the anti‐apoptotic function of Bcl‐2 or Bcl‐xL, and found vacuolar H+‐ATPase (V‐ATPase) inhibitors, such as bafilomycin A1 (BMA), that showed such activity. Bcl‐xL‐overexpressing Ms‐1 cells displayed resistance to anticancer drugs, but underwent apoptosis following treatment with a combination of V‐ATPase inhibitors at doses similar to those that caused inhibitory activities of V‐ATPase. We investigated the apoptosis mechanism induced by cotreatment of Bcl‐xL‐overexpressing Ms‐1 cells with BMA as a V‐ATPase inhibitor and taxol (TXL) as an anticancer drug. With BMA, TXL triggered mitochondrial membrane potential loss and cytochrome c release, whereas downstream caspase activation was not observed. In contrast, pronounced nuclear translocation of mitochondrial apoptosis‐inducing factor and endonuclease G, known as effectors of caspase‐independent apoptosis, was observed with BMA and TXL cotreatment. Moreover, depletion of apoptosis‐inducing factor and endonuclease G using each siRNA significantly rescued cells from BMA‐ and TXL‐induced apoptosis. Hence, the apoptosis‐inducing factor‐ and endonuclease G‐dependent pathway was critical for apoptosis induction by BMA and TXL cotreatment. Our data suggest that V‐ATPase inhibitors could not only suppress anti‐apoptotic Bcl‐2 nor Bcl‐xL but could also facilitate the caspase‐independent apoptotic pathway. V‐ATPase inhibition will be a promising therapeutic approach for Bcl‐2‐ or Bcl‐xL‐overexpressing malignancies. (Cancer Sci 2009)
Members of the Bcl‐2 family play important roles in cell fate determination.( 1 ) Due to their functions, these familial proteins are classified into two large groups: anti‐apoptotic proteins, such as Bcl‐2 and Bcl‐xL, and pro‐apoptotic proteins, such as Bax and Bak,( 2 , 3 ) and it is well known that anti‐ and pro‐apoptotic proteins balance out their opposing functions partly through heterodimerization.( 4 ) Early events in cells triggered to undergo apoptosis are often accompanied by transcriptional activation and post‐translational modification of pro‐apoptotic Bcl‐2 family proteins to connect the sequential decrease in MMP.( 5 ) These changes follow the opening of mitochondrial permeability transition pores, high‐conductance proteinaceous channels in the inner mitochondrial membrane, and then mitochondrial intermembrane space proteins that can trigger either a caspase‐dependent or caspase‐independent apoptotic pathway. One of the most important mitochondrial proteins that causes caspase‐dependent cell death is cytochrome c, which triggers caspase‐9 activation by binding to Apaf‐1 to form a procaspase‐9‐activating heptameric protein complex named the apoptosome, which proteolitically activates the executioner procaspase‐3 to induce apoptosis. Mitochondria have also been reported to contain the caspase‐independent death effectors AIF and EndoG, which are normally confined to the mitochondrial intermembrane space. However, in response to death stimuli, they translocate to the nucleus through the cytosol, where AIF induces chromatin condensation and large‐scale DNA fragmentation (50 kb),( 6 ) and EndoG causes oligonucleosomal DNA fragmentation,( 7 ) without caspase activation. Subsequent studies have demonstrated that EndoG catalyzes both high molecular weight DNA cleavage and oligonucleosomal DNA breakdown in a sequential fashion. Moreover, EndoG cooperates with exonuclease and DNase I to facilitate DNA processing.( 8 )
Anti‐apoptotic Bcl‐2 and Bcl‐xL are localized predominantly in the outer mitochondrial membrane and, through interacting pro‐apoptotic familial members, maintain mitochondrial membrane structural integrity during apoptotic stress, inhibiting egress of mitochondrial apoptogenic effectors.( 9 ) Bcl‐2 and Bcl‐xL are abnormally expressed in a number of malignancies, and consequent defects in the normal regulation of apoptosis frequently result in rendering them resistant to conventional chemotherapy; ( 10 , 11 ) therefore, the development of strategies to impair the function of oncogenic Bcl‐2 and Bcl‐xL is of great interest in cancer therapy. There has been a sustained effort to rationally design and synthesize compounds that recognize the surface pocket of Bcl‐2 or Bcl‐xL, through which Bcl‐2/Bcl‐xL interacts with pro‐apoptotic Bcl‐2 family members and mainly exerts its anti‐apoptotic function, and several compounds, such as HA14‐1,( 12 ) ABT‐737,( 13 ) and gossypol,( 14 ) have been discovered as Bcl‐2/Bcl‐xL inhibitors that bind Bcl‐2 and its relatives via the BH3 domain in the surface pocket. On the other hand, several lines of evidence clearly indicate that Bcl‐2/Bcl‐xL may function through interactions other than Bcl‐2 family members, including a protein kinase, a phosphatase, a chaperone, a cysteine protease, a protease‐interacting protein, and molecular function‐unknown proteins.( 15 ) We also found that mutant Bcl‐2, which cannot interact with pro‐apoptotic Bcl‐2 family members, could inhibit anticancer drug‐induced apoptosis.( 16 ) Therefore, development of a new type of small molecule that can inactivate the anti‐apoptotic function of Bcl‐2/Bcl‐xL by a mode of action distinct from other inhibitors that recognize the surface pocket of Bcl‐2/Bcl‐xL is required. Previously, we found a novel compound, incednine, of microbial origin using a cell‐based chemical–genetic screening system to discover small molecules that induce apoptosis in Ms‐1/Bcl‐xL cells only when combined with anticancer drugs.( 17 ) In the course of our continuous screening for other Bcl‐xL inhibitors, we found inhibitors of V‐ATPase as potent functional suppressors of Bcl‐xL.
The present study shows that V‐ATPase inhibitors have the ability to inhibit mitochondria‐protective functions of Bcl‐2/Bcl‐xL, which closely correlates with V‐ATPase inhibitory activity, thereby potentiating the activity of cytotoxic drugs in Bcl‐2/Bcl‐xL‐overexpressing tumor cells. Furthermore, V‐ATPase inhibitors could switch anticancer drug‐mediated apoptotic pathways from being caspase‐dependent to caspase‐independent in chemoresistant Bcl‐2/Bcl‐xL‐overexpressing tumor cells. Thus, our results show that inhibition of V‐ATPase would be a promising therapeutic approach for Bcl‐2/Bcl‐xL‐overexpressing malignancies.
Materials and Methods
Materials. BMA and camptothecin were purchased from Sigma Chemical Co. (St Louis, MO, USA). TXL and vinblastine were obtained from Wako Pure Chemical Industries (Tokyo, Japan). The pan‐caspase inhibitor z‐VAD‐fmk was obtained from EMD Chemicals (Darmstadt, Germany). Inostamycin and CMB were each isolated from the fermentation broth of the microorganisms that produce them.( 18 , 19 )
Isolation of DE. The culture broth (2 L) of producing strain was extracted with ethyl acetate (EtOAc) under basic conditions, and the extracts obtained were purified sequentially using silica gel columns and preparative reverse‐phase HPLC to obtain a small amount of pure active substance. Structure determination by UV/MS spectrometry and NMR spectra revealed the active compound to be DE.
Cell culture. The human SCLC Ms‐1 cell line and its stable transfectants with vector control, Bcl‐2, and Bcl‐xL (Ms‐1/neo, Ms‐1/Bcl‐2, and Ms‐1/Bcl‐xL)( 16 ) were cultured in RPMI‐1640 medium supplemented with 5% fetal bovine serum and antibiotics.
Acidic vesicular organelle staining. Cells grown on glass coverslips were treated with the indicated drugs for 4 h at 37°C, and then incubated with 3 µM acridine orange (Kanto Chemical Co., Tokyo, Japan) for 30 min. After three washes with Hanks’ solution, the cells were observed with a laser‐scanning confocal microscope (Olympus, Tokyo, Japan).
Analysis of MMP. To evaluate the status of MMP, cells were incubated with 2.5 µg/mL of 5,5′,6,6′‐tetrachloro‐1,1′,3,3′‐tetraethyl benzimidazolyl‐carbocyanine iodide (JC‐1; Wako Pure Chemical Industries) for 10 min at 37°C immediately before cytofluorometric analysis as previously described.( 20 )
Cell fractionation. Cell fractionation was carried out as described previously.( 21 , 22 ) Briefly, cells were resuspended in buffer A (20 mM Hepes‐KOH, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, and 1 mM DTT; pH 7.5) containing 250 mM sucrose. Cell homogenates were centrifuged at 1000 g for 10 min at 4°C to obtain nucleus‐enriched pellets and supernatants including cytosol and mitochondria. The pellets were washed twice with buffer A and lysed in buffer A supplemented with 0.5 M NaCl for 10 min at 4°C. This lysate was subsequently cleared by centrifugation at 15 000 g for 15 min at 4°C and the resulting supernatant was designated as the nuclear fraction. The supernatants including cytosol and mitochondria were centrifuged at 15 000 g for 60 min at 4°C and the resulting supernatants were collected and designated as the cytosolic fraction.
Western blotting. Western blotting was carried out as described previously.( 22 ) Cell lysates were separated by SDS‐PAGE and then subjected to immunoblotting using antibodies as follows: anticytochrome c (7H8.2C12; Pharmingen, Franklin Lakes, NJ, USA; mouse monoclonal IgG), anti‐AIF (E‐1; Santa Cruz, Santa Cruz, CA, USA; mouse monoclonal IgG), anti‐EndoG (3035‐0701; ProSci, Flint Place Poway, CA, USA; mouse monoclonal IgG), anti‐PARP (46D11; Cell Signaling, Danvers, MA, USA; mouse monoclonal IgG), antitubulin (T9026; Sigma Chemical Co., mouse monoclonal IgG) or anti‐Histone H1 (AE‐4; Santa Cruz, mouse monoclonal IgG).
siRNA transfection. siRNA double‐stranded oligonucleotides designed to interfere with the expression of AIF (sense antihuman AIF 5′‐AUAGCAUUGGGCAUCACCUUAACCC‐3′; Invitrogen, Carlsbad, CA, USA) or EndoG (sense antihuman EndoG 5′‐CAGGAUGUUUGGCACAAAGAGCAGC‐3′; Invitrogen), and non‐coding siRNA (Invitrogen) as a negative control were used. Reverse transfection was demonstrated by using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's instructions. Briefly, after being trypsinized, cells were resuspended in antibiotic‐free medium, and mixed with OPTI‐MEM (Gibco, Carlsbad, CA, USA) including 25 nM siRNA and Lipofectamine 2000. After incubation for 20 min at room temperature, cells were diluted with cultured medium and seeded into a 60‐mm dish. For the cell viability assay, siRNA‐transfected cells were reseeded into a 48‐well plate at 48 h after transfection. The silencing of AIF and EndoG was detected by measuring the expression of each protein just before drug treatment.
Results
V‐ATPase inhibitors overcome the anti‐apoptotic function of Bcl‐xL against anticancer drug‐induced apoptosis in Ms‐1/Bcl‐xL. In the course of screening for small molecules that inhibit the anti‐apoptotic function of Bcl‐xL from the cultured broth of microorganisms, we found that one fungus strain produced an active compound, which was isolated from extracts of fungal cultures following bioassay‐guided fractionation; both silica gel column chromatography and reverse‐phase HPLC were utilized. Spectroscopic analysis by NMR and mass spectrometry revealed this active compound to be a cyclic hexadepsipeptide, DE.( 23 ) DE was tested for its suppressive activity against the anti‐apoptotic function of Bcl‐xL. For this test, we used stable transfectants of human SCLC Ms‐1 cells, which overexpress Bcl‐xL compared with vector control Ms‐1/neo, as shown in Figure 1(a)/Ms‐1/Bcl‐xL cells displayed resistance to various types of anticancer drugs, such as camptothecin, inostamycin, TXL, and vinblastine, when compared to vector control Ms‐1/neo (Fig. 1b). However, Bcl‐xL‐mediated resistance to these anticancer drugs, each with a distinct mechanism of action, was overcome by subsequent treatment with DE (Fig. 2a). As DE is a well‐known inhibitor of V‐ATPase, we next examined whether other V‐ATPase inhibitors (BMA( 24 ) and CMB( 25 )) also suppressed the anti‐apoptotic function of Bcl‐xL. As shown in Figure 2(a), BMA and CMB showed similar effects to DE on anticancer drug‐induced apoptosis in Ms‐1/Bcl‐xL. As shown in Figure 2(b), similar results were obtained when Ms‐1/Bcl‐2, which stably overexpress Bcl‐2 (Fig. 1a), were used. On the other hand, V‐ATPase inhibitors did not show any synergistic effect on anticancer drug‐induced apoptosis in vector control Ms‐1/neo (Fig. 2c). These results clearly suggest that V‐ATPase inhibitors did not simply sensitize the cells to anticancer drugs but rather, could suppress the anti‐apoptotic function of Bcl‐2/Bcl‐xL.
Figure 1.
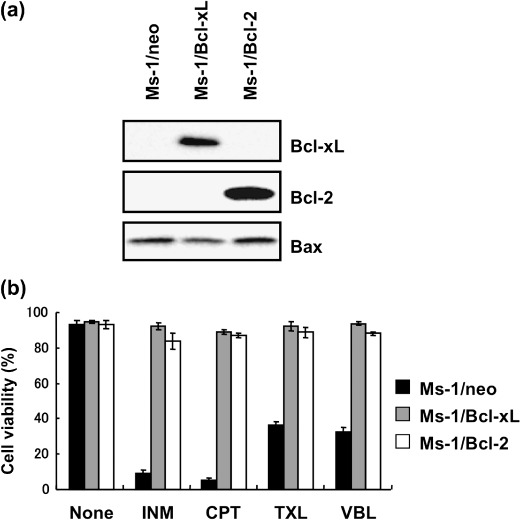
Bcl‐2/Bcl‐xL protected Ms‐1 cells from anticancer drug‐induced apoptosis. (a) Profiles of Bcl‐2 family protein expression in Ms‐1 clones. Cell lysates were immunoblotted with the antibody indicated. (b) Cells were treated with various anticancer drugs as indicated for 48 h. Cell viability was assessed by trypan blue dye exclusion assay. Values are the means of three samples: bars, SD. CPT, camptothecin, 3 µg/mL; INM, inostamycin, 0.1 µg/mL; TXL, taxol, 30 ng/mL; VBL, vinblastine, 10 ng/mL.
Figure 2.
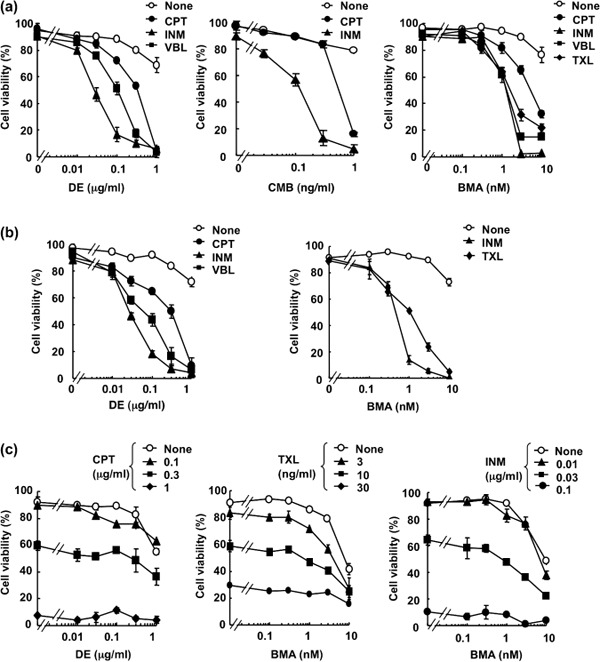
Vacuolar H+‐ATPase (V‐ATPase) inhibitors overcome anti‐apoptotic Bcl‐2/Bcl‐xL against anticancer drugs. Human small cell lung carcinoma Ms‐1 cell lines transfected with (a) Bcl‐xL or (b) Bcl‐2 were treated with destruxin E (DE), bafilomycin A1 (BMA), or concanamycin B (CMB) in combination with various anticancer drugs as indicated for 48 h. CPT, camptothecin, 3 µg/mL; INM, inostamycin, 0.1 µg/mL; TXL, taxol, 30 ng/mL; VBL, vinblastine, 10 ng/mL. (c) Ms‐1/neo cells were treated with DE or BMA in combination with various concentrations of CPT, TXL, or INM for 48 h. Cell viability was assessed by trypan blue dye exclusion assay. Values are the means of three samples; bars, SD.
Inhibition of V‐ATPase is required for BMA‐mediated inhibition of Bcl‐xL function. Next we examined whether the suppressive activity of Bcl‐2/Bcl‐xL by V‐ATPase inhibitors was due to inhibition of V‐ATPase activity. The low pH of intracellular acidic organelles, such as lysosomes, is known to be maintained by V‐ATPase, and this acidification is easily detected as orange fluorescence when cells are stained with acridine orange, an acidophilic weak base fluorescent reagent.( 26 ) When Ms‐1/Bcl‐xL were incubated with acridine orange, intracellular organelles were stained with orange fluorescence, whereas the orange fluorescence in these organelles almost completely disappeared upon treatment with BMA at 3 nM for 4 h (Fig. 3a), indicating that this concentration of BMA inhibited V‐ATPase in our cell culture system. Moreover, this concentration of BMA is comparable to that for the inhibition of Bcl‐xL function in Ms‐1/Bcl‐xL (Fig. 2a). Similar results were obtained with DE‐ and CMB‐treated cells (Fig. 3b and data not shown). These findings strongly suggest that V‐ATPase inhibitors modulate the anti‐apoptotic function of Bcl‐xL through their inherent ability to inhibit V‐ATPase activity.
Figure 3.
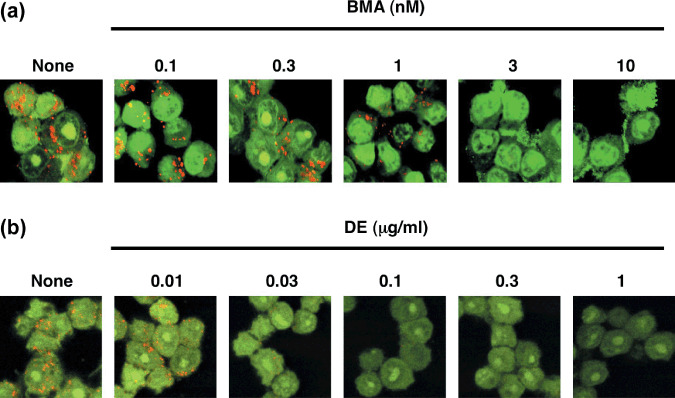
Effect of vacuolar H+‐ATPase (V‐ATPase) inhibitors on acidification of intracellular acidic organelles in human small cell lung carcinoma Ms‐1 cell lines transfected with Bcl‐xL (Ms‐1/Bcl‐xL). Ms‐1/Bcl‐xL cells were treated with the indicated concentrations of (a) bafilomycin A1 (BMA) and (b) destruxin E (DE) for 4 h, and then stained with 3 µM acridine orange for 30 min. After washing, the cells were observed under a laser‐scanning confocal microscope.
Treatment with BMA and TXL leads to dissipation of MMP in Ms‐1/Bcl‐xL. We next examined the underlying mechanism of apoptosis induced by combination treatment of V‐ATPase inhibitors and anticancer drugs in Ms‐1/Bcl‐xL using BMA and TXL respectively. Dissipation of mitochondrial integrity is one of the early events leading to apoptosis; therefore, we first evaluated the effect of BMA on MMP in Ms‐1/Bcl‐xL by using a mitochondrial probe, JC‐1, at 16 h under conditions where the cells had not yet seriously undergone apoptosis (Fig. 4a). As shown in Figure 4(b), although treatment of Ms‐1/Bcl‐xL with TXL or BMA alone did not affect MMP, a significant decrease in MMP was observed at 16 h following cotreatment with TXL and BMA. These data suggested that BMA suppressed the function of Bcl‐xL protecting mitochondria, and allowed TXL to induce MMP loss.
Figure 4.
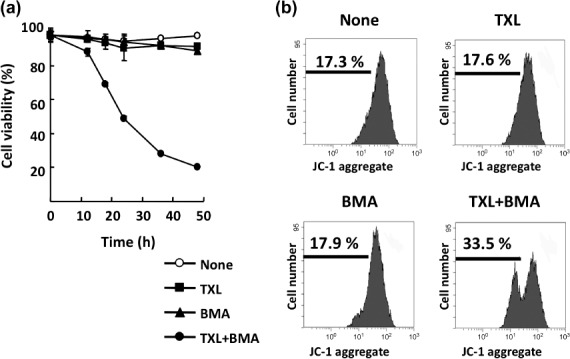
Co‐treatment with bafilomycin A1 (BMA) and taxol (TXL) leads to dissipation of mitochondrial membrane potential in human small cell lung carcinoma Ms‐1 cell lines transfected with Bcl‐xL (Ms‐1/Bcl‐xL). (a) Ms‐1/Bcl‐xL were cotreated with BMA (3 nM) and TXL (30 ng/mL) for the indicated time. Cell viability was assessed by trypan blue dye exclusion assay. Values are the means of three samples; bars, SD. (b) Ms‐1/Bcl‐xL cells were cotreated with BMA (3 nM) and TXL (30 ng/mL) for 16 h. Collected cells were stained with JC‐1 and analyzed by flow cytometry.
Caspase activation is not required in BMA‐ and TXL‐induced cell death. Loss of MMP often reflects an increase in mitochondrial outer membrane permeability, which results in the release of proteins that trigger cell death.( 27 ) The most well‐known apoptotic factor released from permeabilized mitochondria is the respiratory component cytochrome c. As shown in Figure 5(a), TXL (30 ng/mL) induced the release of cytochrome c into the cytosolic fraction in vector control Ms‐1/neo. In Ms‐1/Bcl‐xL, TXL‐induced cytochrome c release was observed only when cells were treated with BMA. Upon release into the cytosol, cytochrome c activates Apaf‐1 and triggers the caspase cascade. One of the key events in this pathway is caspase‐3 activation, leading to oligonucleosomal DNA fragmentation.( 28 , 29 ) As expected from its effect on cytochrome c release, TXL significantly induced the activation of caspase‐3 in Ms‐1/neo as judged from PARP cleavage; however, it was noteworthy that cotreatment of Ms‐1/Bcl‐xL with TXL and BMA did not activate caspase‐3 under the condition where cytochrome c release was observed (Fig. 5a). Moreover, pretreatment with the pan‐caspase inhibitor z‐VAD‐fmk could suppress cell death induced by TXL in Ms‐1/neo, but not by BMA and TXL cotreatment in Ms‐1/Bcl‐xL (Fig. 5b). Similar results were obtained when other anticancer drugs, such as inostamycin, were combined (Fig. 5b). These results clearly suggest that caspase‐3 activation was not involved in the process of cell death induced by BMA and TXL cotreatment in Ms‐1/Bcl‐xL (see ‘Discussion’).
Figure 5.
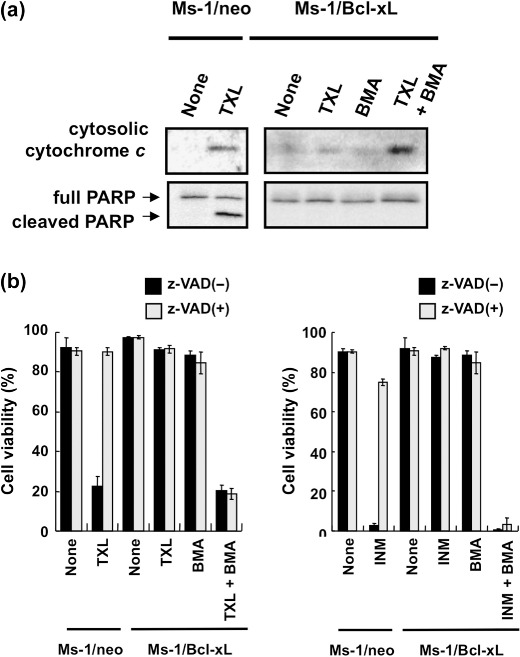
Caspase activation is not required for cell death by cotreatment with bafilomycin A1 (BMA) and taxol (TXL). (a) Human small cell lung carcinoma Ms‐1 cell lines transfected with vector control (Ms‐1/neo) or Bcl‐xL (Ms‐1/Bcl‐xL) were treated with BMA (3 nM) and/or TXL (30 ng/mL) for 18 h. Cytochrome c in the cytosolic fraction and Poly(ADP‐ribose) polymerase (PARP) in the whole lysate were immunoblotted with each antibody. (b) Left, Ms‐1/neo and Ms‐1/Bcl‐xL were treated with BMA (3 nM) and/or TXL (30 ng/mL) in the presence or absence of z‐VAD‐fmk (100 µM) for 48 h; right, Ms‐1/neo and Ms‐1/Bcl‐xL were treated with BMA (3 nM) and/or inostamycin (INM) (0.1 µg/mL) in the presence or absence of z‐VAD‐fmk (100 µM) for 48 h. Cell viability was assessed by trypan blue dye exclusion assay. Values are the means of three samples; bars, SD.
Involvement of the AIF‐ and EndoG‐mediated pathway in BMA‐ and TXL‐induced Ms‐1/Bcl‐xL cell death. Next we investigated whether AIF and EndoG were involved in Ms‐1/Bcl‐xL cell death induced by BMA and TXL cotreatment. Because AIF and EndoG are considered to exert their effects in the nucleus, we first examined the subcellular distribution of AIF and EndoG. Twenty‐hour cotreatment of Ms‐1/Bcl‐xL with BMA and TXL resulted in the appearance of AIF and EndoG in the nuclear‐enriched fraction (Fig. 6a), whereas each treatment alone was not sufficient to stimulate the translocation of AIF and EndoG to the nucleus, suggesting that translocation of mitochondrial AIF and EndoG to the nucleus required both the functional inhibition of Bcl‐xL by BMA and releasing activity of AIF and EndoG by TXL. To further confirm the contribution of AIF and EndoG to death of Ms‐1/Bcl‐xL cotreated with BMA and TXL, we used RNA interference to downregulate the protein expression of AIF and/or EndoG. Transfection with AIF and/or EndoG siRNA for 72 h resulted in a substantial decrease in AIF or EndoG protein levels in Ms‐1/Bcl‐xL, whereas these effects were not observed in control experiments with non‐coding siRNA sequences (Fig. 6b). Cell viability was then assessed upon cotreatment with BMA and TXL in siRNA‐transfected Ms‐1/Bcl‐xL. As shown in Figure 6(c), decreased expression of AIF and/or EndoG by their specific siRNA significantly restored the cell viability loss induced by BMA and TXL, whereas control siRNA did not. Thus, AIF‐ and EndoG‐dependent apoptotic pathways were critical for cell death in Bcl‐xL‐overexpressing cells induced by cotreatment with BMA and TXL, and caspases were dispensable for the occurrence of these types of cell death.
Figure 6.
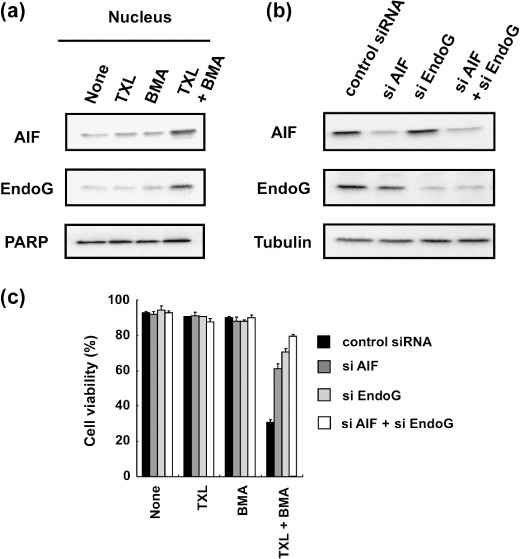
Involvement of apoptosis‐inducing factor (AIF) and endonuclease G (EndoG) in cell death by cotreatment with bafilomycin A1 (BMA) and taxol (TXL) in human small cell lung carcinoma Ms‐1 cell lines transfected with Bcl‐xL (Ms‐1/Bcl‐xL). (a) Ms‐1/Bcl‐xL cells were treated BMA (3 nM) and/or TXL (30 ng/mL) for 20 h. AIF and EndoG in the nucleic fraction were immunoblotted with each antibody. Poly(ADP‐ribose) polymerase (PARP) was immunoblotted as a loading control. (b) Ms‐1/Bcl‐xL cells were transfected with siRNA as indicated for 72 h. Cell lysate was immunoblotted with anti‐AIF and anti‐EndoG antibodies. Tubulin was immunoblotted as a loading control. (c) siRNA‐transfected cells were treated with BMA (3 nM) and/or TXL (30 ng/mL) for 36 h. Cell viability was assessed by trypan blue dye exclusion assay. Values are the means of three samples; bars, SD.
At present, it is less clear whether AIF‐ and EndoG‐dependent apoptotic pathways are also activated in anticancer drug‐treated Ms‐1/neo. To clarify this point, we examined the effect of TXL, BMA, and TXL and BMA cotreatment on MMP and its downstream components cytochrome c or AIF and EndoG in Ms‐1/neo. Upon treatment of either TXL alone or TXL and BMA cotreatment in Ms‐1/neo, MMP loss (data not shown), the resultant release of mitochondrial cytochrome c into the cytosol (Fig. 7a), and downstream caspase activation (data not shown) were observed. AIF and EndoG were predominantly observed in the cytosol but not in the nucleus following TXL treatment of Ms‐1/neo, whereas cotreatment of Ms‐1/neo with TXL and BMA led to nuclear translocation of mitochondrial AIF and EndoG (Fig. 7a). Interestingly, we did not observe simultaneous accumulation of AIF/EndoG in cytosol, suggesting that AIF/EndoG was quickly translocated to the nucleus just after being released from mitochondria in the presence of BMA. Furthermore, TXL‐induced apoptosis was completely blocked by the pan‐caspase inhibitor z‐VAD‐fmk, but not by AIF/EndoG siRNA. On the other hand, cell death induced by TXL and BMA cotreatment was significantly rescued only when cells were treated with both AIF/EndoG siRNA and z‐VAD‐fmk (Fig. 7b). These results suggest that the caspase‐dependent pathway acted solely in TXL‐treated Ms‐1/neo; however, in the presence of BMA, both caspase‐dependent and ‐independent pathways worked in a complementary style.
Figure 7.
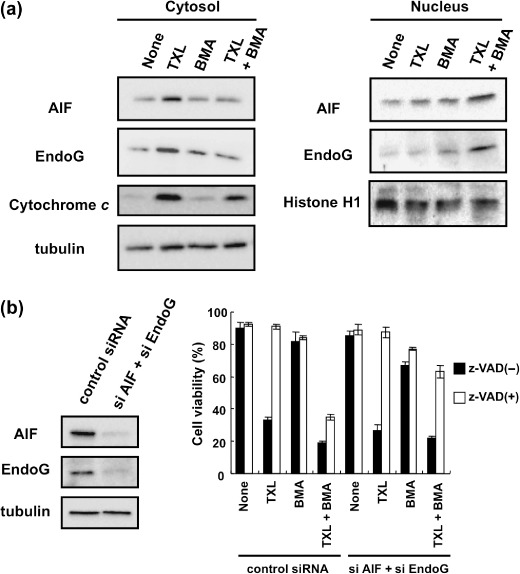
Bafilomycin A1 (BMA) promoted translocation to the nucleus of apoptosis‐inducing factor (AIF) and endonuclease G (EndoG) in Ms‐1/neo cells. (a) Ms‐1/neo cells were treated with taxol (TXL) for 20 h. AIF and EndoG in the cytosolic or nucleic fractions were immunoblotted with each antibody. Tubulin and histone H1 were immunoblotted as loading controls. (b) Left, Human small cell lung carcinoma Ms‐1 cell lines transfected with vector control (Ms‐1/neo) were transfected with siRNA as indicated for 72 h. Cell lysate was immunoblotted with anti‐AIF and anti‐EndoG antibodies. Tubulin was immunoblotted as a loading control; right, siRNA‐transfected Ms‐1/neo cells were treated with BMA (3 nM) and/or TXL (30 ng/mL) in the presence or absence of z‐VAD‐fmk (100 µM) for 48 h. Cell viability was assessed by trypan blue dye exclusion assay. Values are the means of three samples; bars, SD.
Apoptotic pathways induced by TXL and/or BMA in Ms‐1/neo and Ms‐1/Bcl‐xL are summarized in Figure 8.
Figure 8.
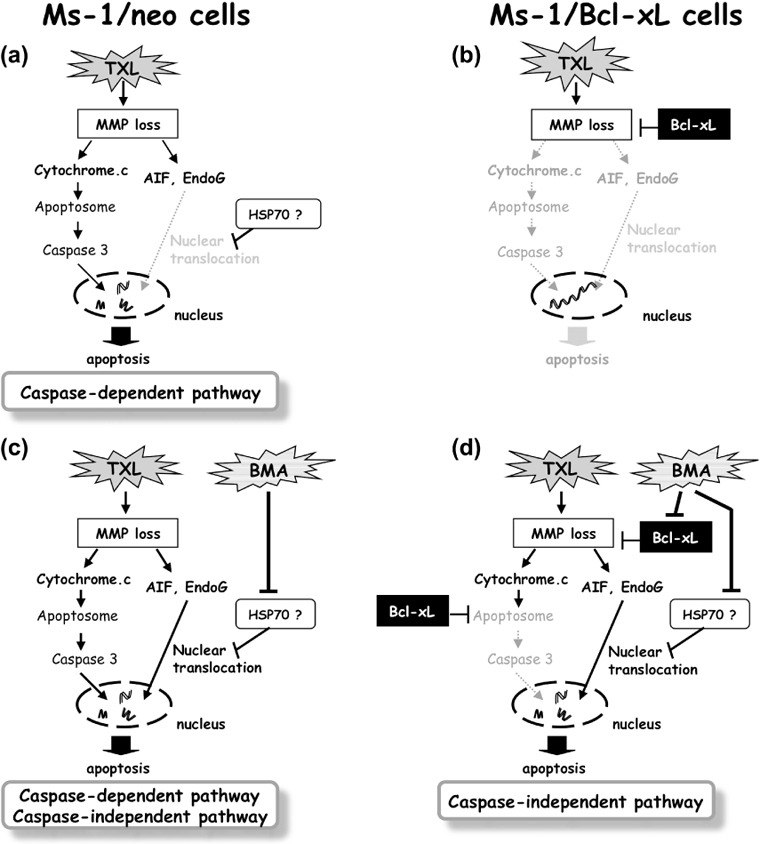
Schematic illustration of the apoptosis pathway. (a) Under normal conditions, taxol (TXL) induces mitochondrial membrane potential (MMP) loss and apoptosis in a caspase‐dependent manner. (b) Bcl‐xL overexpression completely blocks TXL‐induced cell death by the prevention of MMP loss and subsequent release of both cytochrome c and AIF/EndoG. (c) In the presence of bafilomycin A1 (BMA), TXL can induce apoptosis by both caspase‐dependent and ‐independent pathways as the situation demands. (d) BMA predominantly suppresses the mitochondria gatekeeping function of Bcl‐xL. In this situation, TXL can induce caspase‐independent apoptosis in human small cell lung carcinoma Ms‐1 cell lines transfected with Bcl‐xL (Ms‐1/Bcl‐xL) (via AIF/EndoG translocation to the nucleus).
Discussion
Ever since their implication in multidrug chemoresistance in tumors, anti‐apoptotic Bcl‐2 family proteins have become primary molecular targets in oncology. In part because of the key role of Bcl‐2 and Bcl‐xL in inhibiting apoptosis coupled with its ability to bind pro‐apoptotic Bcl‐2 family members, recent efforts to develop their inhibitors have focused on the discovery of small molecules targeting their binding pockets. However, their actual molecular functions have yet to be elucidated, and the present challenge has been to search for small molecules that reveal the molecular mechanisms of Bcl‐2 and Bcl‐xL. Here we showed that V‐ATPase inhibitors potentiated the cytotoxicity of anticancer drugs in Bcl‐2/Bcl‐xL‐overexpressing tumor cells but not in vector control cells. Moreover, we demonstrated that dysfunction of Bcl‐2/Bcl‐xL by V‐ATPase inhibitors was closely correlated to their V‐ATPase inhibitory activities.
V‐ATPase is composed of a peripheral domain (V1) containing eight subunits and an integral domain (Vo) containing six subunits, and has an important role in the control of intracellular pH and extracellular pH through Vo‐mediated proton pumping driven by ATP hydrolysis within V1 and the sequential rotation of a central rotary domain of Vo.( 30 ) Upregulation or overexpression of V‐ATPase is frequently observed in several types of solid tumors and the causative acidic microenvironment offers an advantage in tumor progression, chemoresistance, and metastatic behavior.( 31 , 32 , 33 ) Recently, it has been shown that inhibition of V‐ATPase by knockdown of the subunit ATP6L of V‐ATPase using siRNA significantly decreases the growth and metastasis of human hepatocellular carcinoma xenografts.( 34 ) Therefore, V‐ATPase is now a remarkable target for novel anticancer therapy, and inhibitors of V‐ATPase have recently inspired great interest as a new type of therapeutic opportunity. Indeed, several papers have reported that V‐ATPase inhibitors show antitumor effects in vitro and in vivo. However, so far, the suppressive effects of V‐ATPase inhibitors on the anti‐apoptotic function of Bcl‐2/Bcl‐xL have not been reported; therefore, this is the first report to find that V‐ATPase inhibitors induce the dysfunction of anti‐apoptotic Bcl‐2/Bcl‐xL. Furthermore, V‐ATPase inhibitors inhibit neither the binding capacity of Bcl‐xL to Bax, nor decrease the expression levels of Bcl‐xL (data not shown). These results clearly suggest that V‐ATPase inhibitors can inactivate the anti‐apoptotic function of Bcl‐2/Bcl‐xL by a distinct mode of action from other inhibitors that recognize the surface pocket of Bcl‐2/Bcl‐xL. Given that V‐ATPase inhibitors could induce intracellular acidification, one possible explanation of how V‐ATPase inhibitors modulate anti‐apoptotic Bcl‐2 and Bcl‐xL is that a change in cellular pH may directly or indirectly cause the alternation of pro‐ or anti‐apoptotic protein functions. In fact, some studies have shown that changes in intracellular pH often result in significant alteration of cellular kinetics; staurosporine‐ or tumor necrosis factor (TNF)‐mediated changes in intracellular pH activates Bax, a pro‐apoptotic Bcl‐2 family protein.( 35 ) Because V‐ATPase inhibitor‐mediated changes in intracellular pH did not affect the interaction of Bcl‐xL with Bax, further investigation of proteins involved in a change of cellular pH will provide valuable information for clarifying the molecular mechanism of Bcl‐2/Bcl‐xL.
In this study, we also demonstrated the apoptotic pathway induced by TXL or TXL and BMA cotreatment in Ms‐1/neo and Ms‐1/Bcl‐xL. In Ms‐1/neo, TXL induced mitochondrial cytochrome c and AIF/EndoG release, resulting in cytochrome c‐dependent and/or AIF/EndoG‐dependent apoptosis due to a combination of BMA. In Ms‐1/Bcl‐xL cells, V‐ATPase inhibitors suppress the function of Bcl‐xL protecting mitochondria, and allow anticancer drugs to induce the release of both caspase‐dependent and ‐independent apoptotic effectors from mitochondria; nevertheless, BMA and TXL cotreated Ms‐1/Bcl‐xL underwent apoptosis only through the caspase‐independent pathway. These results might be explained by findings in other reports that Bcl‐xL can physically associate with Apaf‐1 and caspase‐9, and inhibit cytochrome c‐dependent apoptosome formation and subsequent caspase maturation.( 36 ) If this is the case, BMA could predominantly suppress the protective function of Bcl‐xL on mitochondria but not suppress the inhibitory function of Bcl‐xL on apoptosome formation.
In addition, whereas AIF/EndoG released from mitochondria by TXL treatment of Ms‐1/neo were largely observed in the cytosol, AIF/EndoG was predominantly detected in the nucleus in the presence of BMA. A similar result was obtained when Ms‐1/Bcl‐xL cells were used. These results suggest that cytosolic AIF/EndoG does not spontaneously translocate to the nucleus. Recently, it has been shown that HSP70 can interact with both AIF and EndoG, and is linked to the regulation of their activity.( 37 , 38 ) It also appears that genetic downregulation of HSP70 facilitates the induction of caspase‐independent cell death in human breast cancer (MCF)‐7 and Apaf‐1−/– mouse embryonic fibroblast (MEF) cells; conversely, HSP70 upregulation protects cells from caspase‐independent apoptosis,( 37 , 39 , 40 ) indicating the essential role of HSP70 in regulation of the caspase‐independent apoptosis pathway. Herein, we addressed the possibility that BMA prevents the interaction between HSP70 and AIF/EndoG, thereby accelerating cytosolic AIF/EndoG translocation to the nucleus. Further investigation of the mode of action of BMA is currently in progress.
In conclusion, our study suggests that inhibition of V‐ATPase may be crucial for Bcl‐2/Bcl‐xL dysfunction and for AIF/EndoG translocation to the nucleus, leading to activation of the caspase‐independent apoptotic pathway. Taken together, our findings demonstrate the potential use of V‐ATPase inhibitors as anticancer drugs for Bcl‐2/Bcl‐xL‐overexpressing malignancies.
Abbreviations
| AIF | apoptosis‐inducing factor |
| Apaf | apoptosis protease‐activating factor |
| BMA | bafilomycin A1 |
| CMB | concanamycin B |
| DE | destruxin E |
| EndoG | endonuclease G |
| HSP | heat shock protein |
| MMP | mitochondrial membrane potential |
| Ms‐1/Bcl‐2 | human SCLC Ms‐1 cell line transfected with Bcl‐2 |
| Ms‐1/Bcl‐xL | human SCLC Ms‐1 cell line transfected with Bcl‐xL |
| Ms‐1/neo | human SCLC Ms‐1 cell line transfected with vector control |
| PARP | poly(ADP‐ribose) polymerase |
| SCLC | small cell lung carcinoma |
| TXL | taxol |
| V‐ATPase | vacuolar H+‐ATPase |
Acknowledgments
We thank Dr H. Osada and Dr S. Simizu (RIKEN) for kindly providing us with human bcl‐2 and bcl‐xL constructs. This work was supported by a grant from the Ministry of Education, Culture, Sports, Science, and Technology.
References
- 1. Gross A, McDonnell JM, Korsmeyer SJ. BCL‐2 family members and the mitochondria in apoptosis. Genes Dev 1999; 13: 1899–911. [DOI] [PubMed] [Google Scholar]
- 2. Boise LH, Gonzalez‐Garcia M, Postema CE et al . Bcl‐x, a bcl‐2‐related gene that functions as a dominant regulator of apoptotic cell death. Cell 1993; 74: 597–608. [DOI] [PubMed] [Google Scholar]
- 3. Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl‐2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 1993; 74: 609–19. [DOI] [PubMed] [Google Scholar]
- 4. Gross A, Jockel J, Wei MC, Korsmeyer SJ. Enforced dimerization of BAX results in its translocation, mitochondrial dysfunction and apoptosis. EMBO J 1998; 17: 3878–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kim R. Unknotting the roles of Bcl‐2 and Bcl‐xL in cell death. Biochem Biophys Res Commun 2005; 333: 336–43. [DOI] [PubMed] [Google Scholar]
- 6. Susin SA, Lorenzo HK, Zamzami N et al . Molecular characterization of mitochondrial apoptosis‐inducing factor. Nature 1999; 397: 441–6. [DOI] [PubMed] [Google Scholar]
- 7. Li LY, Luo X, Wang X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 2001; 412: 95–9. [DOI] [PubMed] [Google Scholar]
- 8. Widlak P, Li LY, Wang X, Garrard WT. Action of recombinant human apoptotic endonuclease G on naked DNA and chromatin substrates: cooperation with exonuclease and DNase I. J Biol Chem 2001; 276: 48 404–9. [DOI] [PubMed] [Google Scholar]
- 9. Yang J, Liu X, Bhalla K et al . Prevention of apoptosis by Bcl‐2: release of cytochrome c from mitochondria blocked. Science 1997; 275: 1129–32. [DOI] [PubMed] [Google Scholar]
- 10. Tsujimoto Y, Cossman J, Jaffe E, Croce CM. Involvement of the bcl‐2 gene in human follicular lymphoma. Science 1985; 228: 1440–3. [DOI] [PubMed] [Google Scholar]
- 11. Wang S, Yang D, Lippman ME. Targeting Bcl‐2 and Bcl‐XL with nonpeptidic small‐molecule antagonists. Semin Oncol 2003; 30: 133–42. [DOI] [PubMed] [Google Scholar]
- 12. Wang JL, Liu D, Zhang ZJ et al . Structure‐based discovery of an organic compound that binds Bcl‐2 protein and induces apoptosis of tumor cells. Proc Natl Acad Sci USA 2000; 97: 7124–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Oltersdorf T, Elmore SW, Shoemaker AR et al . An inhibitor of Bcl‐2 family proteins induces regression of solid tumours. Nature 2005; 435: 677–81. [DOI] [PubMed] [Google Scholar]
- 14. Kitada S, Leone M, Sareth S, Zhai D, Reed JC, Pellecchia M. Discovery, characterization, and structure‐activity relationships studies of proapoptotic polyphenols targeting B‐cell lymphocyte/leukemia‐2 proteins. J Med Chem 2003; 46: 4259–64. [DOI] [PubMed] [Google Scholar]
- 15. Reed JC. Double identity for proteins of the Bcl‐2 family. Nature 1997; 387: 773–6. [DOI] [PubMed] [Google Scholar]
- 16. Kawatani M, Imoto M. Deletion of the BH1 domain of Bcl‐2 accelerates apoptosis by acting in a dominant negative fashion. J Biol Chem 2003; 278: 19 732–42. [DOI] [PubMed] [Google Scholar]
- 17. Futamura Y, Sawa R, Umezawa Y et al . Discovery of incednine as a potent modulator of the anti‐apoptotic function of Bcl‐xL from microbial origin. J Am Chem Soc 2008; 130: 1822–3. [DOI] [PubMed] [Google Scholar]
- 18. Imoto M, Umezawa K, Takahashi Y et al . Isolation and structure determination of inostamycin, a novel inhibitor of phosphatidylinositol turnover. J Nat Prod 1990; 53: 825–9. [Google Scholar]
- 19. Yoshimoto Y, Imoto M. Induction of EGF‐dependent apoptosis by vacuolar‐type H(+)‐ATPase inhibitors in A431 cells overexpressing the EGF receptor. Exp Cell Res 2002; 279: 118–27. [DOI] [PubMed] [Google Scholar]
- 20. Tzung SP, Kim KM, Basanez G et al . Antimycin A mimics a cell‐death‐inducing Bcl‐2 homology domain 3. Nat Cell Biol 2001; 3: 183–91. [DOI] [PubMed] [Google Scholar]
- 21. Takemoto Y, Watanabe H, Uchida K et al . Chemistry and biology of moverastins, inhibitors of cancer cell migration, produced by Aspergillus. Chem Biol 2005; 12: 1337–47. [DOI] [PubMed] [Google Scholar]
- 22. Tsuchiya A, Tashiro E, Yoshida M, Imoto M. Involvement of nuclear accumulation of heat shock protein 27 in leptomycin B‐induced apoptosis in HeLa cells. J Antibiotics 2005; 58: 810–16. [DOI] [PubMed] [Google Scholar]
- 23. Païs M, Das BC, Ferron P. Depsipeptides from Metarhizium anisopliae . Phytochemistry 1981; 20: 715–23. [Google Scholar]
- 24. Nelson N, Taiz L. The evolution of H+‐ATPases. Trends Biochem Sci 1989; 14: 113–16. [DOI] [PubMed] [Google Scholar]
- 25. Kinashi H, Someno K, Sakaguchi K. Isolation and characterization of concanamycins A, B and C. The J Antibiotics 1984; 37: 1333–43. [DOI] [PubMed] [Google Scholar]
- 26. Yoshimori T, Yamamoto A, Moriyama Y, Futai M, Tashiro Y. Bafilomycin A1, a specific inhibitor of vacuolar‐type H+‐ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J Biol Chem 1991; 266: 17 707–12. [PubMed] [Google Scholar]
- 27. Kim R, Emi M, Tanabe K. Role of mitochondria as the gardens of cell death. Cancer Chemother Pharmacol 2006; 57: 545–53. [DOI] [PubMed] [Google Scholar]
- 28. Liu X, Zou H, Slaughter C, Wang X. DFF, a heterodimeric protein that functions downstream of caspase‐3 to trigger DNA fragmentation during apoptosis. Cell 1997; 89: 175–84. [DOI] [PubMed] [Google Scholar]
- 29. Sakahira H, Enari M, Nagata S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature 1998; 391: 96–9. [DOI] [PubMed] [Google Scholar]
- 30. Hirata R, Graham LA, Takatsuki A, Stevens TH, Anraku Y. VMA11 and VMA16 encode second and third proteolipid subunits of the Saccharomyces cerevisiae vacuolar membrane H+‐ATPase. J Biol Chem 1997; 272: 4795–803. [DOI] [PubMed] [Google Scholar]
- 31. Nishi T, Forgac M. The vacuolar H+‐ATPases – nature's most versatile proton pumps. Nat Rev Mol Cell Biol 2002; 3: 94–103. [DOI] [PubMed] [Google Scholar]
- 32. Sennoune SR, Luo D, Martinez‐Zaguilan R. Plasmalemmal vacuolar‐type H+‐ATPase in cancer biology. Cell Biochem Biophys 2004; 40: 185–206. [DOI] [PubMed] [Google Scholar]
- 33. De Milito A, Fais S. Tumor acidity, chemoresistance and proton pump inhibitors. Future Oncol 2005; 1: 779–86. [DOI] [PubMed] [Google Scholar]
- 34. Lu X, Qin W, Li J et al . The growth and metastasis of human hepatocellular carcinoma xenografts are inhibited by small interfering RNA targeting to the subunit ATP6L of proton pump. Cancer Res 2005; 65: 6843–9. [DOI] [PubMed] [Google Scholar]
- 35. Tafani M, Cohn JA, Karpinich NO, Rothman RJ, Russo MA, Farber JL. Regulation of intracellular pH mediates Bax activation in HeLa cells treated with staurosporine or tumor necrosis factor‐alpha. J Biol Chem 2002; 277: 49 569–76. [DOI] [PubMed] [Google Scholar]
- 36. Hu Y, Benedict MA, Wu D, Inohara N, Nunez G. Bcl‐XL interacts with Apaf‐1 and inhibits Apaf‐1‐dependent caspase‐9 activation. Proc Natl Acad Sci USA 1998; 95: 4386–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ravagnan L, Gurbuxani S, Susin SA et al . Heat‐shock protein 70 antagonizes apoptosis‐inducing factor. Nat Cell Biol 2001; 3: 839–43. [DOI] [PubMed] [Google Scholar]
- 38. Kalinowska M, Garncarz W, Pietrowska M, Garrard WT, Widlak P. Regulation of the human apoptotic DNase/RNase endonuclease G: involvement of Hsp70 and ATP. Apoptosis 2005; 10: 821–30. [DOI] [PubMed] [Google Scholar]
- 39. Nylandsted J, Rohde M, Brand K, Bastholm L, Elling F, Jaattela M. Selective depletion of heat shock protein 70 (Hsp70) activates a tumor‐specific death program that is independent of caspases and bypasses Bcl‐2. Proc Natl Acad Sci USA 2000; 97: 7871–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chinnathambi S, Tomanek‐Chalkley A, Bickenbach JR. HSP70 and EndoG modulate cell death by heat in human skin keratinocytes in vitro . Cells Tissues Organs 2008; 187: 131–40. [DOI] [PubMed] [Google Scholar]