

Abstract
In this study we explored the mechanisms of constitutive activation of c‐Met in lung adenocarcinoma cell lines. First, we examined levels of c‐Met and phospho‐c‐Met (Y1234/Y1235) in a panel of lung adenocarcinoma cell lines by Western blot analysis. c‐Met expression was found in 12 of 14 cell lines and an overall correlation between the expressions of c‐Met and phospho‐c‐Met was noted. c‐Met was constitutively activated particularly at high levels in five cell lines (PC3, LC‐2/ad, L27, H1648, and H2009). c‐Met amplification was identified in L27 and H1648 by single nucleotide polymorphism array analysis, but no mutations were identified in the Sema domain or in any part of the cytoplasmic domain of c‐Met. Experiments with neutralizing anti‐hepatocyte growth factor (HGF) antibody, scatter assay using Madin–Darby canine kidney cells, and Western blotting on conditioned media of the cell lines revealed that the constitutive phosphorylation of c‐Met was largely ligand‐independent. The inhibition of cell–matrix adhesion induced the dephosphorylation of c‐Met in the five cell lines tested. This was accompanied by downregulation of c‐Met in three of the five cell lines. In contrast, the inhibition of cell–cell adhesion by neutralizing E‐cadherin antibody had a minimal effect on the expression and phosphorylation of c‐Met. These results reveal three features of the constitutive activation of c‐Met in our panel of lung adenocarcinoma cell lines: (i) it correlates with c‐Met overexpression, either with or without gene amplification; (ii) it is largely ligand‐independent; and (iii) it depends on cell–matrix adhesion. (Cancer Sci 2008; 99: 14–22)
Lung cancer is the leading cause of cancer death in many developed countries, including the USA and Japan.( 1 ) Lung adenocarcinoma, one of the major histologic subtypes, is rising in incidence.( 2 ) Patients with lung carcinoma have a poor prognosis: those with stage I disease have a 5‐year survival of only 70%.( 3 ) Molecular‐targeted therapy provides a new therapeutic method in this setting and is now under intense investigation.( 4 ) Epidermal growth factor receptor (EGFR) inhibitors have proven to be effective in a subset of lung adenocarcinomas with the EGFR mutation.( 5 ) The response rate, however, is only 10% for tumors without the EGFR mutation. There is clearly a pressing need for other effective molecular‐targeted therapies. c‐Met might offer promise as an attractive new target among the other tyrosine kinases.( 6 )
Numerous studies have documented the gene mutation, overexpression, and amplification of c‐Met in various cancers.( 6 , 7 , 8 ) The c‐Met gene is highly expressed in non‐small‐cell lung cancer( 9 , 10 , 11 ) and c‐Met overexpression is associated with an advanced cancer stage and shorter patient survival.( 9 , 11 ) Our group recently investigated the expression and activation of c‐Met in surgically resected lung adenocarcinomas using antiphospho‐c‐Met antibody.( 12 , 13 ) As expected, our results indicated correlations of phospho‐c‐Met with high levels of hepatocyte growth factor (HGF) and high levels of c‐Met expression. Intriguingly, however, we also found positive phospho‐c‐Met expression in cases with little to no expression of HGF. In spite of the implication that c‐Met activation is both ligand‐dependent and ligand‐independent, it was difficult for us to dissect the mechanism of c‐Met activation using tissue sections alone.
Several mechanisms of c‐Met activation have been identified. Initial studies have shown that HGF is generally secreted from stromal cells and acts in a paracrine manner on c‐Met, which is widely expressed in epithelial tissues.( 6 , 7 , 8 ) However, the malignant tumor cells might also acquire the ability to secrete HGF, which, once secreted, acts on cancer cells in an autocrine manner.( 14 , 15 , 16 ) In some cancer cells, c‐Met might be constitutively activated in a ligand‐independent manner by gene mutation or overexpression with or without gene amplification.( 6 , 7 , 8 ) Intriguingly, previous studies have also suggested that c‐Met activation might take place through cell–matrix adhesion in the absence of HGF in some tumor cells.( 17 , 18 , 19 )
To begin this study, we examined the status of c‐Met activation in a panel of lung adenocarcinoma cell lines using an antiphospho‐c‐Met (Y1234/Y1235) antibody. This antibody has been established to recognize the c‐Met molecule phosphorylated at Y1234 and Y1235, tyrosine residues crucially involved in c‐Met activation.( 20 ) Having seen the constitutive activation of c‐Met in a majority of the cell lines, we went on to investigate the mechanisms behind the constitutive activation of c‐Met.
Materials and Methods
Cell lines and medium. Lung adenocarcinoma cell lines were obtained from several sources: H23, H522, H1395, H1648, H1975, and H2009 were from American Type Culture Collection; A549, ABC‐1, PC3, and VMRC‐LCD from the Japanese Cancer Research Resources Bank (Osaka, Japan); HLC‐1, LC‐2/ad, and RERF‐LC‐KJ from the RIKEN Cell Bank (Tsukuba, Japan). Lung adenocarcinoma cell line L27 (a kind gift from Dr S. Hirohashi, National Cancer Center Institute, Tokyo, Japan) has been described.( 21 ) In addition, H460 (large cell carcinoma) and H1299 (non‐small‐cell carcinoma), obtained from American Type Culture Collection, were included in the analysis. Lung fetal fibroblasts (HFLIII) were from RIKEN Cell Bank. All cell lines were maintained in the culture media recommended by the suppliers (RPMI‐1640 for cancer cell lines and Dulbecco's modified Eagle's medium for HFLIII) supplemented with 10% fetal calf serum, glutamine, and antibiotics in a humidified atmosphere with 5% CO2 and 95% air.
Reagents. Recombinant human HGF was purchased from R&D Systems (Minneapolis, MN). Lactacystin was obtained from Sigma‐Aldrich (St Louis, MO). Stock solution of lactacystin was prepared in water and added to the medium at a final concentration of 3 µmol/L.
Antibodies. Rabbit polyclonal anti‐c‐Met and rabbit polyclonal antiphospho‐c‐Met (Y1234/Y1235) were purchased from IBL (Gunma, Japan) and Cell Signaling Technology (Danvers, MA), respectively. Rabbit polyclonal anti‐HGF α‐chain antibody was from IBL. Goat polyclonal anti‐β‐actin antibody was from Santa Cruz Biotechnology (Santa Cruz, CA). Goat polyclonal anti‐HGF neutralizing antibody was from R&D Systems. Mouse monoclonal anti‐E‐cadherin neutralizing antibody (HECD‐1) was from Takara Bio (Shiga, Japan).( 22 ) Anti‐rabbit immunoglobulin (Ig)G peroxidase conjugate was from Amersham (Arlington Hights, IL) and antigoat IgG peroxidase conjugate was from Sigma‐Aldrich.
DNA sequencing and single nucleotide polymorphism array analyses. The cDNA from cell lines were prepared by standard procedures. The coding regions of the Sema domain and the entire cytoplasmic domain in the c‐Met cDNAs were sequenced using standard polymerase chain reaction and sequencing techniques. The polymerase chain reaction primers used for the c‐Met mutational analysis were designed using the software Primer3 (http://frodo.wi.mit.edu/cgi‐bin/primer3/primer3_www.cgi), and their sequences are available upon request. Single nucleotide polymorphism array (human mapping 50K XbaI array; Affymetrix, Santa Clara, CA) analysis was carried out using the Genome Imbalance Map algorithm as described previously.( 23 )
Oligonucleotide microarray analysis. A comprehensive gene expression analysis was carried out using an oligonucleotide microarray as described previously (GeneChip Human Genome U133A array; Affymetrix).( 24 , 25 ) This array contains probe sets interrogating approximately 14 000 clusters from the UniGene database (Build 133). Analysis was carried out essentially according to the instructions from the Affymetrix GeneChip Expression Analysis Technical Manual.
Western blot analysis. Cells were lysed in a lysis buffer consisting of 20 mmol/L Tris‐HCl (pH 7.4), 150 mM NaCl, 50 mM NaF, and 1 mM Na3VO4 with a cocktail of proteinase inhibitors. After sonication, lysates were boiled at 98°C for 5 min and cleared by centrifugation. Protein concentrations were determined by the DC Protein Assay kit (Bio‐Rad). For Western blotting, equal amounts of protein samples were size‐separated on 8% polyacrylamide gels and electroblotted onto polyvinylidene difluoride membranes Nonspecific binding was blocked by immersion of the membranes for 1 h in 2% bovine serum albumin in Tris‐buffered saline at room temperature. The membranes were washed with Tris‐buffered saline buffer containing 0.1% Tween 20, incubated for 1 h at room temperature with primary antibodies (anti‐c‐Met antibody, 1:500 dilution; antiphospho‐c‐Met antibody, 1:500 dilution; anti‐HGF α‐chain antibody, 1:50 dilution; anti‐β‐actin antibody, 1:500 dilution), washed again, and incubated for 1 h with secondary antibody (antirabbit IgG peroxidase conjugate, 1:1000 dilution; antigoat IgG peroxidase conjugate, 1:1000). The antigen was detected using ECL Western Blotting Detection Reagents (Amersham) following the manufacturer's instructions.
Western blot analysis of conditioned media of carcinoma cell lines. Five lung adenocarcinoma cell lines (L27, LC‐2/ad, PC3, H1648, and H2009) and fibroblasts derived from fetal lung (HFLIII, as a positive control) were grown on 35 mm tissue culture dishes in their respective media without fetal bovine serum for 24 h. The media were then collected and centrifuged for removal of floating debris of carcinoma cells. Four milliliters of media was concentrated to 120–140 µL, using Amicon Ultra centrifugal filter devices (Millipore), and subjected to Western blot analysis.
Scatter assay. Six lung adenocarcinoma cell lines (A549, L27, LC‐2/ad, PC3, H1648, and H2009) were grown on 35 mm tissue culture dishes in their respective media without fetal bovine serum for 24 h. When cultures reached 80–90% confluence, the media were collected and centrifuged for removal of debris of carcinoma cells. The collected media were added directly to Madin–Darby canine kidney (MDCK) cells that had been seeded the previous day. After 24 h, the morphologic change of MDCK cells was observed under an inverted phase contrast microscope.
Inhibition of cell–matrix adhesion of carcinoma cell lines. Five lung adenocarcinoma cell lines (L27, LC‐2/ad, PC3, H1648, and H2009) were grown on 35 mm tissue culture dishes in their respective media for 48 h. After trypsinization, cells were plated and grown in low cell binding dishes coated with 2‐methacryloyloxyethyl phosphorylcholine (Nunc) in their respective media with 10% fetal bovine serum. Trypan Blue exclusion test showed that ≥50% of cells remained viable after culturing the five cell lines in low cell binding dishes.
Results
Western blot analysis of c‐Met and phospho‐c‐Met expressions. Before analyzing our panel of lung adenocarcinoma cell lines, we first checked the specificity of antiphospho‐c‐Met (Y1234/1235) antibody. Cells from the lung adenocarcinoma cell line A549 were cultured without serum for 24 h, then HGF was added to the medium at a final concentration of 50 ng/mL. Cell lysates were prepared 10 min after the addition of HGF and subjected to Western blot analysis. As shown in Fig. 1, a band for phospho‐c‐Met was clearly detectable in cells stimulated with HGF, but not in cells without HGF stimulation. Equal loading and transfer of the samples were confirmed by reprobing with anti‐c‐Met and anti‐β‐actin antibodies (Fig. 1, lower panels). After validating the antibody, we examined the expressions of c‐Met and phospho‐c‐Met in our panel of 16 cell lines that consisted of 14 adenocarcinoma, one large cell carcinoma (H460), and one non‐small‐cell lung carcinoma (H1299) cell lines. These cell lines were cultured without serum for 24 h, cell lysates were then prepared, and subjected to Western blotting analysis. The results are shown in Fig. 2. c‐Met was expressed in 12 of 14 lung adenocarcinoma cell lines and in H1299 and H460 cells. The expression was strong in six lung adenocarcinoma cell lines (PC3, L27, H1975, LC‐2/ad, H2009, and H1648). Phospho‐c‐Met was expressed in nine adenocarcinoma cell lines, and the expression was strong in five of them (PC3, L27, LC‐2/ad, H2009, and H1648). Phospho‐c‐Met was undetectable in H1299 and H460 cells. A comparison between c‐Met and phospho‐c‐Met revealed a good overall correlation between their expression levels.
Figure 1.
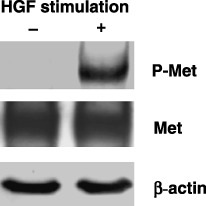
Validation of the specificity of antiphospho‐c‐Met (Y1234/1235) antibody. A549 lung adenocarcinoma cells were cultured without serum for 24 h then treated with hepatocyte growth factor (HGF) at a final concentration of 50 ng/mL in the medium. Phospho‐c‐Met was detectable in cells stimulated by HGF but not in cells without HGF stimulation. The lower panel shows a reprobing with anti‐c‐Met and anti‐β‐actin antibodies.
Figure 2.
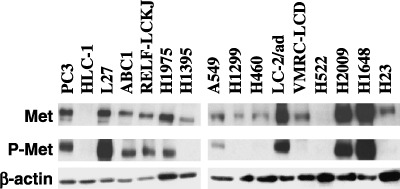
Expressions of c‐Met and phospho‐c‐Met in a panel of 14 lung adenocarcinoma cell lines and two lung carcinoma cell lines (H1299 and H460). Cell were cultured without serum for 24 h, lysed with RIPA buffer, and subjected to Western blot analysis. A good overall correlation was observed between the expression levels of c‐Met and phospho‐c‐Met.
DNA sequencing and single nucleotide polymorphism array analyses. Next we sequenced the coding regions of the Sema domain and the entire cytoplasmic domain of the c‐Met cDNA, the two domains in which almost all of the known c‐Met mutations have been identified in earlier studies.( 7 , 26 , 27 ) Our sequencing revealed no mutations in either domain. Next, we examined copy numbers of the c‐Met gene by single nucleotide polymorphism array analyses. The results are shown in Fig. 3. Gene amplifications were identified in L27 and H1648, both of which expressed c‐Met and phospho‐c‐Met at high levels (Fig. 2).
Figure 3.
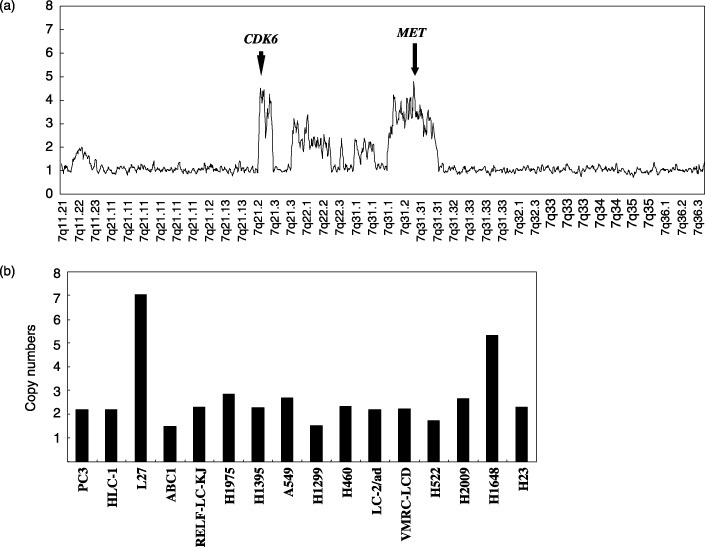
Amplifications of the c‐Met gene in lung adenocarcinoma cell lines. (a) Gene dosage analysis of L27 on chromosome 7q. The arrow indicates amplification of c‐Met on 7q31.2. The arrowhead indicates amplification of Cdk6 on 7q21.2. Here a moving point average of five consecutive probes is used for clarity. (b) Copy number changes of the c‐Met gene are shown for the lung adenocarcinoma cell lines. Amplification was identified in L27 and H1648. The data indicates average values for the six single nucleotide polymorphisms within the Met locus.
Western blot analysis of conditioned media of carcinoma cell lines. HGF and c‐Met have been found to colocalize in lung carcinoma cells( 15 ) and an autocrine HGF‐c‐Met loop has been identified in several cultured carcinoma cells.( 14 , 16 ) We thus began our Western blot experiments by exploring whether HGF was detectable in the concentrated media of lung adenocarcinoma cells. Cells were cultured without serum for 24 h, and the conditioned mediums of five lung adenocarcinoma cells (L27, LC‐2/ad, H1648, H2009, and PC3) and of human lung fibroblast (HFLIII) were concentrated to 29–33‐fold using an Amicon Ultra centrifugal filter device (Millipore) then subjected to Western blot analysis. When the active form of HGF is separated using sodium dodecyl sulfate–polyacrylamide gel electrophoresis in the presence of a reducing agent, it migrates as two distinct bands, the 64 kDa α‐chain and 32 kDa β‐chain. In contrast, the latent form of HGF migrates as a single band of 96 kDa. Thus, the latent and active forms of HGF can be distinguished by Western blotting. The results are shown in Fig. 4. The HFLIII fibroblast (positive control) showed both 64 kDa and 96 kDa bands, indicating that this cell secreted both latent and active forms of HGF. The H1648 cell showed only the 64 kDa band, indicating that this cell secreted only the active HGF. No HGF was detected in any of the other cell lines tested.
Figure 4.

Western blot analysis of conditioned mediums of carcinoma cell lines. After culturing cells without serum for 24 h, the conditioned mediums of five lung adenocarcinoma cells (L27, LC‐2/ad, H1648, H2009, and PC3) and of human lung fibroblast (HFLIII) were concentrated and subjected to Western blot analysis. Both active (64 kDa) and latent (96 kDa) bands were seen in the HFLIII fibroblasts (positive control). Hepatocyte growth factor (HGF), in contrast, was undetectable in all of the cancer cell lines except H1648.
Blocking experiment with anti‐HGF neutralizing antibody. The previous experiment showed that HGF was undetectable in the conditioned media of most of the cell lines tested, indicating that the constitutive activation of c‐Met was ligand‐independent. It remains plausible, however, that HGF was present at levels which were too low to be detected by the Western blots but high enough to induce c‐Met phosphorylation. To pursue this possibility, we carried out a blocking experiment using anti‐HGF neutralizing antibody. In brief, after culturing cells without serum for 24 h, we added anti‐HGF neutralizing antibody at a final concentration of 1 µg/mL to the culture mediums of the five lung adenocarcinoma cells (L27, LC‐2/ad, H1648, H2009, and PC3) that had been confirmed to strongly express phospho‐c‐Met, then examined the effects of c‐Met phosphorylation at 30 min, 2 h, 8 h, and 24 h. As shown in Fig. 5a, the levels of c‐Met phosphorylation remained unchanged after the addition of the neutralizing antibody. In a separate experiment, a pre‐incubation of HGF with this anti‐HGF neutralizing antibody completely blocked the ability of HGF to induce c‐Met phosphorylation in A549 cells (Fig. 5b). This confirmed that the lack of effect on c‐Met phosphorylation could not have been due to a low potency of the anti‐HGF neutralizing antibody.
Figure 5.
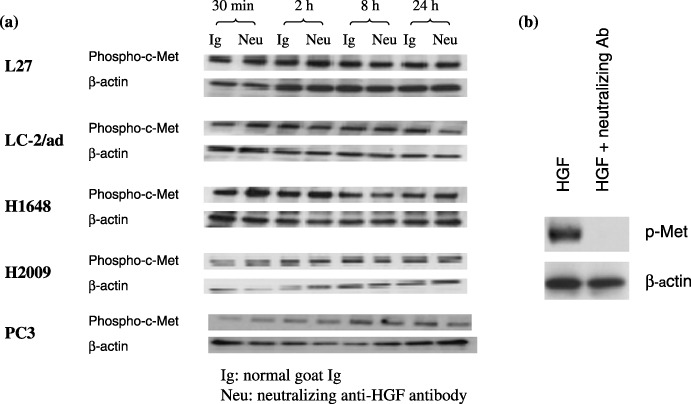
Blocking experiment with anti‐hepatocyte growth factor (HGF) neutralizing antibody. (a) After culturing cells without serum for 24 h, the culture mediums of five lung adenocarcinoma cells (L27, LC‐2/ad, H1648, H2009, and PC3) were treated with anti‐HGF neutralizing antibody then observed at 30 min, 2 h, 8 h, and 24 h to assess the effect on c‐Met phosphorylation. The levels of c‐Met phosphorylation remained unchanged after the addition of the neutralizing antibody. (b) Preincubation of HGF with this anti‐HGF neutralizing antibody completely blocked the ability of HGF to induce c‐Met phosphorylation in the A549 cells.
Scatter assay. MDCK cells scatter when exposed to recombinant HGF, thus providing a useful biologic assay for HGF.( 28 ) In this experiment, after culturing cells without serum for 24 h, we added the conditioned media of five lung adenocarcinoma cell lines (L27, LC‐2/ad, H1648, H2009, and PC3) and fibroblasts (HFLIII cell, positive control) directly to MDCK cells then observed the morphological changes of the MDCK cells (Fig. 6). The HFLIII and HI648 cell mediums induced moderate and mild scattering of the MDCK cells, whereas the mediums of L27, LC‐2/ad, H2009, and PC3 cells induced no MDCK cell scattering whatsoever. These results were in complete agreement with the results of the Western blot analysis of the conditioned mediums (Fig. 4), which showed secretion of HGF only in H1648 cells.
Figure 6.
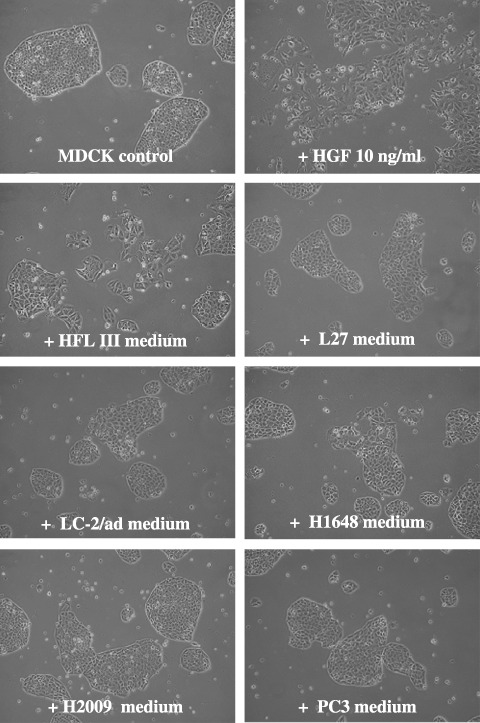
Scatter assay using Madin–Darby canine kidney (MDCK) cells. Conditioned mediums of five lung adenocarcinoma cells (L27, LC‐2/ad, H1648, H2009, and PC3) and fibroblasts (HFLIII cells, positive control) were added directly to MDCK cells, then the morphologic changes of the MDCK cells were observed at 24 h. The HFLIII cell medium elicited a moderate scattering of the MDCK cells. In contrast, the L27, LC‐2/ad, H2009, and PC3 cell mediums elicited no MDCK cell scattering whatsoever. The H1648 cell medium brought about a mild scattering of MDCK cells.
Oligonucleotide array analysis. To further confirm that there is little or no HGF expression in the cell lines with constitutive activation of c‐Met, we examined the gene expression profile data obtained by Affymetrix oligonucleotide array analysis. As shown in Fig. 7, levels of HGF mRNA were low in the five lung adenocarcinoma cells, PC3, L27, LC‐2/ad, H2009, and H1648, that had been confirmed to strongly express phospho‐c‐Met. Intriguingly, relatively high levels of HGF mRNA expression were found in HLC‐1 and H522 cells; these were the two exceptional cell lines in which the c‐Met protein was undetectable by Western blot analysis (Fig. 2).
Figure 7.
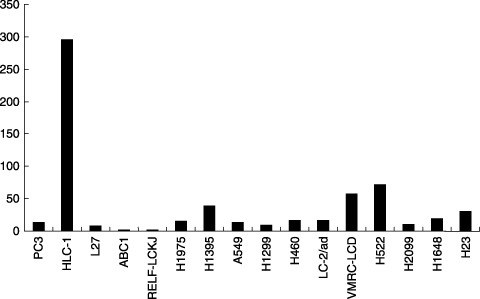
Hepatocyte growth factor (HGF) mRNA levels in a panel of lung carcinoma cell lines. Gene expression profile data were obtained by Affymetrix oligonucleotide array analysis. Levels of HGF mRNA were low in the five lung adenocarcinoma cells, PC3, L27, LC‐2/ad, H2009, and H1648, that had been confirmed to strongly express phospho‐c‐Met.
Effect of inhibition of cell–cell adhesion. The inhibition of cell–cell adhesion has been found to influence the phosphorylation levels of EGFR in several culture systems.( 29 , 30 ) With this in mind, we tested whether the inhibition of cell–cell adhesion with anti‐E‐cadherin neutralizing antibody (HECD‐1) affects the phosphorylation levels of c‐Met. To begin, we added anti‐E‐cadherin neutralizing antibody to several cell lines at a final concentration of 50 µg/mL and observed the effect of the antibody on cell–cell adhesion. As it turned out, the cell lines known to express phospho‐c‐Met at high levels, L27 and LC‐2/ad, showed a clear dissociation of cell–cell adhesion in response to anti‐E‐cadherin neutralizing antibody (Fig. 8a). Then we checked the effect of anti‐E‐cadherin neutralizing antibody on c‐Met phosphorylation in the presence of 10% serum at 3 h and 6 h. As shown in Fig. 8b, the addition of anti‐E‐cadherin neutralizing antibody had minimal effect on phosphorylation levels of c‐Met.
Figure 8.
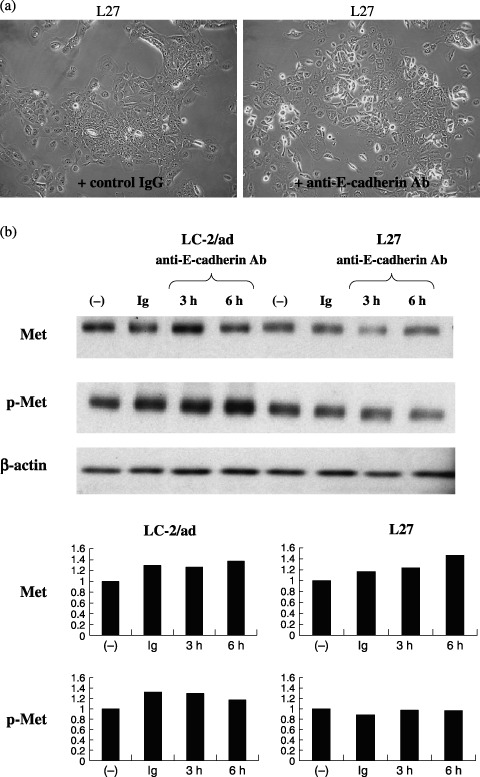
Effect of inhibition of cell–cell adhesion. (a) L27 and LC‐2/ad showed clear dissociation of cell–cell adhesion in response to anti‐E‐cadherin neutralizing antibody. (b) The addition of anti‐E‐cadherin neutralizing antibody had minimal effect on phosphorylation levels of c‐Met. The bar graph indicates the densitometric values relative to that of control culture after correction for β‐actin.
Effect of inhibition of cell–matrix adhesion. Previous studies have revealed that the inhibition of cell–matrix adhesion attenuates the phosphorylation of c‐Met.( 17 , 18 ) The five lung adenocarcinoma cell lines (H1648, H2009, PC3, L27, and LC‐2/ad) found to express high levels of c‐Met and phospho‐c‐Met were grown in standard cell culture dishes and in low cell binding dishes coated with 2‐methacryloyloxyethyl phosphorylcholine (Nunc) for 48 h in the presence of 10% serum. As shown in Fig. 9, the inhibition of cell–matrix adhesion dramatically reduced the levels of phospho‐c‐Met and total c‐Met protein in three cell lines (H2009, PC3, and L27). In H1648 and LC‐2/ad, phospho‐c‐Met levels were reduced, but the effect on total c‐Met levels was not evident, suggesting that the effect of cell detachment on total c‐Met levels might occur in a cell‐specific manner.
Figure 9.
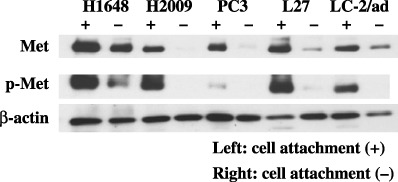
Effect of inhibition of cell–matrix adhesion. (a) The five lung adenocarcinoma cells (H1648, H2009, PC3, L27, and LC‐2/ad) confirmed to strongly express c‐Met and phospho‐c‐Met were grown in standard cell culture dishes and in low cell binding dishes coated with 2‐methacryloyloxyethyl phosphorylcholine for 48 h. The inhibition of the cell–matrix adhesion led to dramatic decreases in the levels of both phospho‐c‐Met protein and total c‐Met protein in three cell lines (H2009, PC3, and L27). In H1648 and LC‐2/ad, phospho‐c‐Met levels were reduced, but the effect on total c‐Met levels was not evident.
Effect of lactacystin on detachment‐induced c‐Met downregulation. The downregulation of c‐Met and phospho‐c‐Met following detachment from the substratum might have been attributable to either increased protein degradation or reduced transcription. To test the first possibility, we examined whether lactacystin, a specific proteosome inhibitor, inhibits the downregulation of c‐Met by cell detachment.
First, we tested whether lactacystin inhibits the downregulation of c‐Met following the addition of HGF; ligand stimulation is known to induce the downregulation of receptors by receptor internalization and the accompanying proteosome‐mediated degradation.( 31 ) Cells from the lung adenocarcinoma cell line A549 were cultured without serum for 24 h, then HGF was added to the medium at a final concentration of 50 ng/mL in the presence or absence of lactacystin. Our preliminary experiment had shown that in the A549 cell line, HGF‐induced downregulation of c‐Met was most apparent at 6 h after addition of HGF. Therefore, cell lysates were prepared 6 h after the addition of HGF and subjected to Western blot analysis. As shown in Fig. 10a, lactacystin inhibited the downregulation of c‐Met induced by the addition of HGF.
Figure 10.
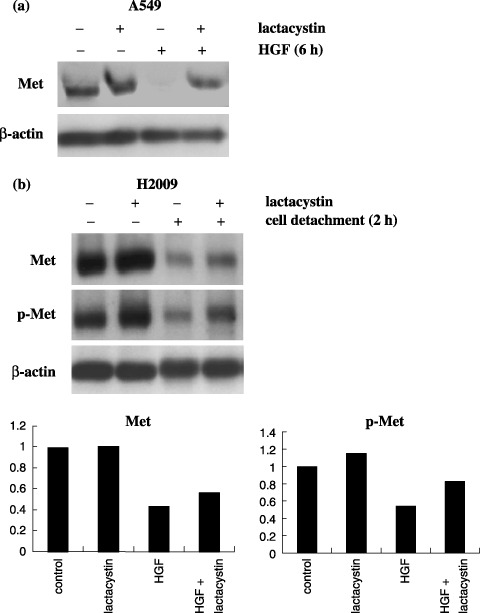
Effect of lactacystin on detachment‐induced c‐Met downregulation. (a) In A549 cells, lactacystin inhibited the downregulation of c‐Met that occurred 6 h after the addition of hepatocyte growth factor (HGF). (b) In H2009 cells, lactacystin had a modest inhibitory effect on the downregulation of phospho‐c‐Met and the total c‐Met that occurred 2 h after cell detachment. The bar graph indicates the densitometric values relative to that of the control culture after correction for β‐actin.
After confirming the inhibitory effect of lactacystin on ligand‐induced c‐Met downregulation, we further addressed the issue whether lactacystin inhibits the downregulation of c‐Met by cell detachment. For this experiment, of the five cell lines with constitutive c‐Met activation, we chose H2009 cells. This is because our preliminary experiment had shown that detachment‐induced c‐Met downregulation occurred most rapidly in H2009 cells (less than 2 h); we reasoned that use of H2009 cells would allow us to minimize the effect of altered transcription by cell detachment. The results are shown in Fig. 10b. Although there appeared to be some inhibitory effect of lactacystin, this turned out to be a modest effect after correction for β‐actin.
Discussion
Our first task in this study was to examine the levels of c‐Met expression and the patterns of c‐Met activation by Western blot analysis in a panel of 16 cell lines that consisted with 14 lung adenocarcinoma, one large cell (H460), and one non‐small‐cell lung carcinoma (H1299) cell lines. c‐Met was expressed in 12 of 14 lung adenocarcinoma cell lines and in H460 and H1299 cells. The expression was strong in six of them (PC3, L27, H1975, LC‐2/ad, H2009, and H1648). c‐Met activation, as manifested by phosphorylation of Y1234/1235, was observed in nine of the lung adenocarcinoma cell lines. Phospho‐c‐Met expression was high in the five lung adenocarcinoma cell lines (PC3, L27, LC‐2/ad, H2009, and H1648) that had been confirmed to overexpress c‐Met. Phospho‐c‐Met expression was undetectable in H460 and H1299 cells. Thus, c‐Met activation might take place constitutively in the cells that overexpress c‐Met.
The constitutive activation of c‐Met in cancer cells might be induced through: (i) c‐Met mutation; (ii) c‐Met overexpression with or without gene amplification; and (iii) autocrine activation by secreted HGF. Other published reports have proposed that the activation of tyrosine kinase receptors such as c‐Met might be regulated by cell–cell and/or cell–matrix adhesion.( 17 , 18 , 19 , 29 , 30 ) Our study now provides a comprehensive data set regarding the activation mechanisms of c‐Met in a panel of lung adenocarcinoma cell lines. Below we discuss these issues as they relate to the results of the present study.
Earlier papers have reported that HGF and c‐Met colocalize in several cancer cell lines( 15 ) and that the secreted HGF activates c‐Met in an autocrine manner.( 14 , 15 , 16 ) For further insight, we examined whether the constitutive activation of c‐Met is mediated by cancer cell‐derived HGF in an autocrine manner. Thorough attempts to detect HGF and HGF activity using three separate approaches (Western blot analysis, bioassay using MDCK cells, and a blocking experiment using a neutralizing antibody against HGF) confirmed the presence of HGF in only one of the five cell lines and failed to detect HGF activity in the remaining four cell lines. On this basis, we can reasonably conclude that the constitutive activation of c‐Met in lung adenocarcinoma cell lines takes place largely in a ligand‐independent manner. This conclusion in no way dismisses the importance of the ligand‐dependent activation of c‐Met in cancers; however, it would be equally important that cancer cells do acquire the mechanism for ligand‐independent activation of c‐Met.
c‐Met is overexpressed and amplified in adenocarcinomas of the stomach and colon.( 6 , 7 , 8 ) Missense mutations in the kinase domain of c‐Met have been reported in various cancers, including renal cell carcinoma and squamous cell carcinoma of the head and neck region.( 6 , 7 , 8 ) In lung cancer, missense mutations have been found in the juxtamembrane and extracellular (Sema) domains of small‐cell carcinomas and adenocarcinomas.( 7 , 26 ) In contrast, Shibata et al. identified amplification of a locus containing the c‐Met gene in four of 55 cases of primary lung adenocarcinomas, but they found no mutation of c‐Met in any cases examined.( 32 ) In this study, we found no mutation in the coding region of the Sema domain or in any part of the cytoplasmic domain of c‐Met cDNA. Instead, we found amplifications of the c‐Met gene in two of the five cell lines that had been confirmed to overexpress c‐Met. This suggests that c‐Met overexpression takes place through gene amplification in some cases and through other mechanisms in other cases.
Cell adhesion is classified into two types, cell–substratum adhesion and cell–cell adhesion. Cell–cell adhesion is mediated by adhesion molecules such as cadherins.( 33 ) Cell–substratum adhesion is mediated by adhesion molecules such as integrins, that is, receptors for the extracellular matrix component.( 34 ) Previous studies indicate that Met and EGFR activations are strongly influenced by cell–matrix( 17 , 18 , 19 ) and cell–cell adhesion.( 29 ) This prompted us to examine whether cell adhesion influences constitutive c‐Met activation in lung adenocarcinoma cells. The blocking of cell–cell adhesion with a neutralizing antibody to E‐cadherin (HECD‐1)( 22 ) left the expression levels of c‐Met and phospho‐c‐Met unchanged, confirming that the cell–cell adhesion was not required for the maintenance of constitutive activation of c‐Met. In contrast, the cell detachment induced marked reductions of phospho‐c‐Met levels in the five cell lines tested, which was accompanied with downregulation of total c‐Met levels in three of the five cell lines. The reduction was modestly attenuated by the addition of lactacystin, a proteosome inhibitor. Thus, integrin‐mediated signals somehow play a role in maintaining high levels of c‐Met activation in lung adenocarcinoma cells. However, it was of note that this effect of cell detachment appears to occur in a cell‐specific manner, as reduction of total c‐Met levels was not evident in H1648 or LC‐2/ad cells. Indeed, previous studies had shown that in the cell lines tested, cell detachment induced downregulation of phospho‐c‐Met without affecting total c‐Met levels.( 17 , 18 , 19 ) Thus, it would be of interest to investigate the molecular basis for this cell‐specific effect of detachment.
It is well known that ligand‐induced receptor activation is followed by receptor internalization and inactivation. In contrast, attachment‐induced receptor activation occurs in a sustained manner.( 17 ) Thus, there appears to be a fundamental difference in the two modes of receptor activation. The underlying cause for this difference is unknown, however, and we believe that this is an interesting issue that warrants further investigations. Moreover, interference with the integrin‐mediated signal might provide a new approach to inhibit the constitutive activation of c‐Met in cancer cells.
According to published reports, cancer cells that harbor gene amplification and/or point mutation of the c‐Met gene might be very sensitive to c‐Met inhibitors such as SU11274 or PHA‐665752.( 35 , 36 ) In lung adenocarcinomas, downregulation of c‐Met by small interfering RNA resulted in inhibition of phospho‐c‐Met, phospho‐Akt, and reduced cell viability.( 26 ) Recently, Engelman et al. reported that c‐Met amplification led to resistance to gefitinib in lung adenocarcinoma cell lines and tissues. Moreover, inhibition of c‐Met signaling restored sensitivity to gefitinib.( 37 ) Further investigations are warranted to confirm these findings and to determine whether inhibition of c‐Met signaling provides a new therapeutic modality in the treatment of patients with lung adenocarcinomas.
Acknowledgments
We thank Etsuko Ohara for her excellent technical assistance. This study was supported in part by the Foundation for Development of Community, the Vehicle Racing Commemorative Foundation, the Ministry of Health, Labor and Welfare, and a Grant‐in‐Aid for Scientific Research on Priority Areas from the Ministry of Education, Culture, Sports, Science and Technology.
References
- 1. Jemal A, Siegel R, Ward E et al . Cancer statistics, 2006. CA Cancer J Clin 2006; 56: 106–30. [DOI] [PubMed] [Google Scholar]
- 2. Janssen‐Heijnen ML, Coebergh JW. Trends in incidence and prognosis of the histological subtypes of lung cancer in North America, Australia, New Zealand and Europe. Lung Cancer 2001; 31: 123–37. [DOI] [PubMed] [Google Scholar]
- 3. Naruke T, Tsuchiya R, Kondo H, Asamura H, Nakayama H. Implications of staging in lung cancer. Chest 1997; 112: 242S–8S. [DOI] [PubMed] [Google Scholar]
- 4. Maulik G, Kijima T, Salgia R. Role of receptor tyrosine kinases in lung cancer. Meth Mol Med 2003; 74: 113–25. [DOI] [PubMed] [Google Scholar]
- 5. Riely GJ, Politi KA, Miller VA, Pao W. Update on epidermal growth factor receptor mutations in non‐small cell lung cancer. Clin Cancer Res 2006; 12: 7232–41. [DOI] [PubMed] [Google Scholar]
- 6. Christensen JG, Burrows J, Salgia R. c‐Met as a target for human cancer and characterization of inhibitors for therapeutic intervention. Cancer Lett 2005; 225: 1–26. [DOI] [PubMed] [Google Scholar]
- 7. Ma PC, Maulik G, Christensen J, Salgia R. c‐Met: structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev 2003; 22: 309–25. [DOI] [PubMed] [Google Scholar]
- 8. Jiang WG, Martin TA, Parr C, Davies G, Matsumoto K, Nakamura T. Hepatocyte growth factor, its receptor, and their potential value in cancer therapies. Crit Rev Oncol Hematol 2005; 53: 35–69. [DOI] [PubMed] [Google Scholar]
- 9. Takanami I, Tanana F, Hashizume T et al . Hepatocyte growth factor and c‐Met/hepatocyte growth factor receptor in pulmonary adenocarcinomas: an evaluation of their expression as prognostic markers. Oncology 1996; 53: 392–7. [DOI] [PubMed] [Google Scholar]
- 10. Tsao MS, Liu N, Chen JR et al . Differential expression of Met/hepatocyte growth factor receptor in subtypes of non‐small cell lung cancers. Lung Cancer 1998; 20: 1–16. [DOI] [PubMed] [Google Scholar]
- 11. Masuya D, Huang C, Liu D et al . The tumour–stromal interaction between intratumoral c‐Met and stromal hepatocyte growth factor associated with tumour growth and prognosis in non‐small‐cell lung cancer patients. Br J Cancer 2004; 90: 1555–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Inoue T, Kataoka H, Goto K et al . Activation of c‐Met (hepatocyte growth factor receptor) in human gastric cancer tissue. Cancer Sci 2004; 95: 803–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nakamura Y, Niki T, Goto A et al . c‐Met activation in lung adenocarcinoma tissues: an immunohistochemical analysis. Cancer Sci 2007; 98: 1006–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yoshinaga Y, Fujita S, Gotoh M, Nakamura T, Kikuchi M, Hirohashi S. Human lung cancer cell line producing hepatocyte growth factor/scatter factor. Jpn J Cancer Res 1992; 83: 1257–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wong AS, Pelech SL, Woo MM et al . Coexpression of hepatocyte growth factor‐Met: an early step in ovarian carcinogenesis? Oncogene 2001; 20: 1318–28. [DOI] [PubMed] [Google Scholar]
- 16. Nakashiro K, Hara S, Shinohara Y et al . Phenotypic switch from paracrine to autocrine role of hepatocyte growth factor in an androgen‐independent human prostatic carcinoma cell line, CWR22R. Am J Pathol 2004; 165: 533–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang R, Kobayashi R, Bishop JM. Cellular adherence elicits ligand‐independent activation of the Met cell‐surface receptor. Proc Natl Acad Sci USA 1996; 93: 8425–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang R, Ferrell LD, Faouzi S, Maher JJ, Bishop JM. Activation of the Met receptor by cell attachment induces and sustains hepatocellular carcinomas in transgenic mice. J Cell Biol 2001; 153: 1023–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Qiao H, Hung W, Tremblay E et al . Constitutive activation of met kinase in non‐small‐cell lung carcinomas correlates with anchorage‐independent cell survival. J Cell Biochem 2002; 86: 665–77. [DOI] [PubMed] [Google Scholar]
- 20. Ferracini R, Longati P, Naldini L, Vigna E, Comoglio PM. Identification of the major autophosphorylation site of the Met/hepatocyte growth factor receptor tyrosine kinase. J Biol Chem 1991; 266: 19 558–64. [PubMed] [Google Scholar]
- 21. Niki T, Kohno T, Iba S et al . Frequent co‐localization of Cox‐2 and laminin‐5 gamma2 chain at the invasive front of early‐stage lung adenocarcinomas. Am J Pathol 2002; 160: 1129–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shimoyama Y, Hirohashi S, Hirano S et al . Cadherin cell‐adhesion molecules in human epithelial tissues and carcinomas. Cancer Res 1989; 49: 2128–33. [PubMed] [Google Scholar]
- 23. Ishikawa S, Komura D, Tsuji S et al . Allelic dosage analysis with genotyping microarrays. Biochem Biophys Res Commun 2005; 333: 1309–14. [DOI] [PubMed] [Google Scholar]
- 24. Matsubara D, Niki T, Ishikawa S et al . Differential expression of S100A2 and S100A4 in lung adenocarcinomas: clinicopathological significance, relationship to p53 and identification of their target genes. Cancer Sci 2005; 96: 844–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang T, Niki T, Goto A et al . Hypoxia increases the motility of lung adenocarcinoma cell line A549 via activation of the epidermal growth factor receptor pathway. Cancer Sci 2007; 98: 506–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ma PC, Kijima T, Maulik G et al . c‐Met mutational analysis in small cell lung cancer: novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer Res 2003; 63: 6272–81. [PubMed] [Google Scholar]
- 27. Ma PC, Jagadeeswaran R, Jagadeesh S et al . Functional expression and mutations of c‐Met and its therapeutic inhibition with SU11274 and small interfering RNA in non‐small cell lung cancer. Cancer Res 2005; 65: 1479–88. [DOI] [PubMed] [Google Scholar]
- 28. Kenworthy P, Dowrick P, Baillie‐Johnson H et al . The presence of scatter factor in patients with metastatic spread to the pleura. Br J Cancer 1992; 66: 243–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shen X, Kramer RH. Adhesion‐mediated squamous cell carcinoma survival through ligand‐independent activation of epidermal growth factor receptor. Am J Pathol 2004; 165: 1315–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Qian X, Karpova T, Sheppard AM, McNally J, Lowy DR. E‐cadherin‐mediated adhesion inhibits ligand‐dependent activation of diverse receptor tyrosine kinases. EMBO J 2004; 23: 1739–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jeffers M, Taylor GA, Weidner KM, Omura S, Vande Woude GF. Degradation of the Met tyrosine kinase receptor by the ubiquitin‐proteasome pathway. Mol Cell Biol 1997; 17: 799–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shibata T, Uryu S, Kokubu A et al . Genetic classification of lung adenocarcinoma based on array‐based comparative genomic hybridization analysis: its association with clinicopathologic features. Clin Cancer Res 2005; 11: 6177–85. [DOI] [PubMed] [Google Scholar]
- 33. Hirohashi S, Kanai Y. Cell adhesion system and human cancer morphogenesis. Cancer Sci 2003; 94: 575–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guo W, Giancotti FG. Integrin signalling during tumour progression. Nat Rev Mol Cell Biol 2004; 5: 816–26. [DOI] [PubMed] [Google Scholar]
- 35. Berthou S, Aebersold DM, Schmidt LS et al . The Met kinase inhibitor SU11274 exhibits a selective inhibition pattern toward different receptor mutated variants. Oncogene 2004; 23: 5387–93. [DOI] [PubMed] [Google Scholar]
- 36. Smolen GA, Sordella R, Muir B et al . Amplification of MET may identify a subset of cancers with extreme sensitivity to the selective tyrosine kinase inhibitor PHA‐665752. Proc Natl Acad Sci USA 2006; 103: 2316–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Engelman JA, Zejnullahu K, Mitsudomi T et al . MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007; 316: 1039–43. [DOI] [PubMed] [Google Scholar]