

Abstract
Heterozygous germline mutations of the tumor‐suppressor gene MEN1 are responsible for multiple endocrine neoplasia type 1 (MEN1), a dominantly inherited familial cancer syndrome characterized by pituitary, parathyroid, and enteropancreatic tumors. Various mutations have been identified throughout the entire gene region in patients with MEN1 and related disorders. Neither mutation hot spot nor phenotype–genotype correlation has been established in MEN1 although some missense mutations may be specifically associated with a phenotype of familial isolated hyperparathyroidism. The gene product menin has been implicated in multiple roles, including gene transcription, maintenance of genomic integrity, and control of cell division and differentiation. These multiple functions are likely to be conferred by association with multiple protein factors. Occurrence of MEN1‐causing missense mutations throughout menin also suggests the requirement of multiple binding factors for its full tumor‐suppressive activity. The effect of menin depletion is highly tissue specific, but its underlying mechanism remains to be elucidated. A DNA test for MEN1 germline mutations is a useful tool for diagnosis of MEN1 although it needs further improvements. (Cancer Sci 2009; 100: 209–215)
The tumor suppressor gene MEN1 was isolated by positional cloning in 1997 as the gene responsible for multiple endocrine neoplasia type 1 (MEN1),( 1 ) a dominantly inherited familial cancer syndrome characterized by the combined occurrence of pituitary, parathyroid, enteropancreatic, and other tumors (Fig. 1).( 2 , 3 ) Since then, approximately 500 different germline mutations that cause loss of MEN1 gene function have been identified in patients with MEN1 and related disorders.( 4 , 5 , 6 , 7 , 8 , 9 , 10 ) The germline mutations are heterozygous, and somatic loss of the normal MEN1 allele has been observed in the tumors arising in MEN1, in agreement with the Knudson's two‐hit model.( 11 ) Somatic inactivation of both MEN1 alleles has also been detected in some sporadic endocrine tumors, indicating involvement of this gene in the development of sporadic tumors. So far, MEN1 gene mutations have not been implicated in any other human diseases.
Figure 1.
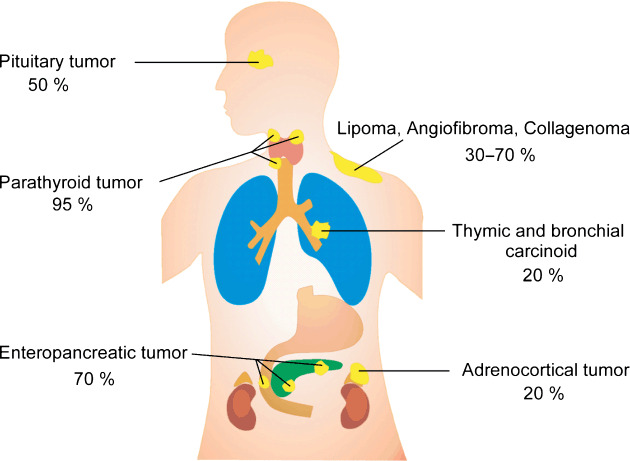
Estimated penetrance of tumors arising in MEN1.
The product of the MEN1 gene, menin, is a 610‐amino acid protein that exhibits no apparent sequence similarity to any other known protein.( 1 ) Thus, its biochemical function can not be deduced from its structure. In the last decade, a number of studies have demonstrated physical and functional associations between menin and diverse proteins of known function, shedding light on its molecular function. These menin‐interacting proteins include nuclear proteins such as transcription factors, histone deacetylases, and histone methyltransferases, suggesting that menin is involved in gene transcription. Several lines of evidence also suggest that menin is involved in the maintenance of genomic integrity. Although menin is localized mainly to cell nuclei, possible extranuclear functions have not been excluded because various cytoplasmic proteins bind to menin.
Although the molecular function of menin is still poorly understood, identification of the MEN1 gene has enabled a direct DNA test for predisposition to MEN1. In the present review, recent findings on the MEN1 gene are summarized and its mutations are discussed from basic and clinical points of view.
Structure and expression of the MEN1 gene
The human MEN1 gene comprises 10 exons distributed over 7 kb in the chromosome region 11q13 and encodes mRNA of approximately 2.8 kb (Fig. 2).( 1 ) Exons 2 through 10 encode the 610‐amino acid protein menin. Several benign polymorphisms within the exons and introns of the MEN1 gene have been documented.
Figure 2.
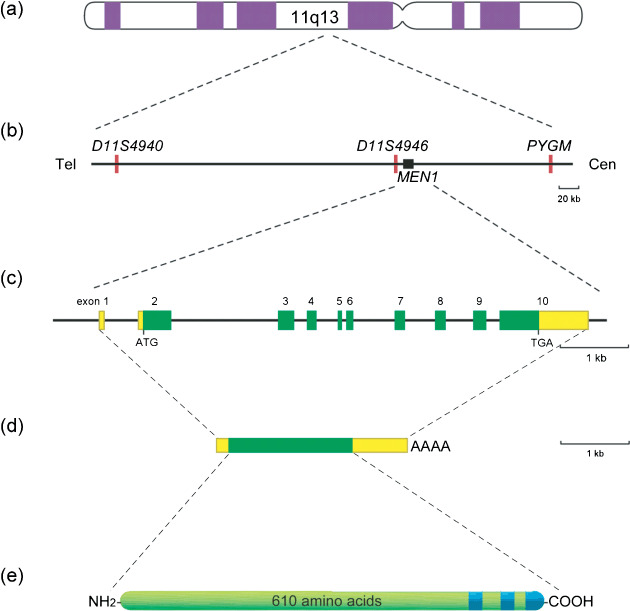
Chromosomal localization and structural organization of the human MEN1 gene. (a) Chromosome 11. (b) The approximately 300‐kb genomic region containing the MEN1 gene (closed box) and microsatellites (red bar) used as polymorphic DNA markers. Cen, centromere; Tel, telomere. (c) MEN1 gene. Green and yellow boxes indicate protein‐coding and non‐coding regions of exons, respectively. (d) Menin mRNA. Green and yellow boxes indicate translated and untranslated regions of the mRNA, respectively. (e) Menin. Blue regions indicate nuclear localization signals.
The primary structure of menin is not similar to previously identified proteins, providing few clues to its molecular function. Menin mRNA is expressed ubiquitously in the tissues and cell lines examined, although its expression levels are variable among different tissues.( 12 , 13 , 14 , 15 ) Menin orthologues have been identified in vertebrates and also invertebrates including fruit fly and snail, but not in the yeast Saccharomyces cerevisiae or nematode Caenorhabditis elegans.( 16 , 17 , 18 )
Germline mutations of the MEN1 gene
Prevalence of mutation in MEN1 and related disorders. Heterozygous germline mutations of the MEN1 gene have been identified in approximately 90% of familial MEN1, in which multiple patients are found in a family, and in a smaller fraction of sporadic MEN1 patients.( 4 , 19 ) These incomplete detection rates undoubtedly result from imperfect analyzing methods as well as genetic heterogeneity of diseases clinically diagnosed as MEN1.
A variant of MEN1 characterized by frequent occurrence of pituitary tumors and infrequent primary hyperparathyroidism (in contrast to classical MEN1 in which almost all patients exhibit hyperparathyroidism) has been considered as a disease entity distinct from classical MEN1. Accordingly, germline MEN1 mutations were rarely identified in this variant form. Recently, the CDKN1B gene, encoding cyclin‐dependent kinase inhibitor 1B (also known as p27Kip1 ), has been shown to be responsible for a part of this MEN1 variant.( 20 )
Germline MEN1 mutations are found in some patients with clinical presentations suggestive of MEN1 but who are not diagnosed with MEN1 according to the criteria. These include familial isolated hyperparathyroidism (FIHP) and apparently sporadic MEN1‐related tumors. FIHP is defined as hereditary primary hyperparathyroidism without the association of other disease or tumors, and is a mixture of genetically heterogeneous diseases.( 21 ) To date, approximately 40 FIHP families have been reported to have a germline MEN1 mutation.( 4 , 10 ) A few patients with sporadic parathyroid tumors, sporadic adrenocortical tumor, sporadic insulinoma, and sporadic carcinoid tumor have also been shown to carry germline MEN1 mutations.( 4 , 19 ) These mutation‐positive MEN1‐related disorders often turn out to be classical MEN1.
In contrast, familial isolated pituitary tumor, in which only pituitary tumors develop in multiple members of a family, are not associated with MEN1 gene mutations, indicating that this syndrome is a disease entity distinct from MEN1. Recently, the AIP gene, encoding the aryl hydrocarbon receptor‐interacting protein, has been shown to be responsible for a fraction of pituitary adenoma predisposition.( 22 )
Type and distribution of MEN1 germline mutations. Various MEN1 gene mutations have been identified in the germline of patients with MEN1 and related disorders (Fig. 3a). Frameshift, nonsense, and splice site mutations account for approximately 50, 20, and 10% of the mutations, respectively.( 4 ) These mutations cause premature termination of protein translation and often mRNA instability. Missense mutations and in‐frame deletions or insertions that do not cause truncation of menin account for 20–30% of the mutations. Large deletions encompassing the entire or partial gene regions have occasionally been documented. As a whole, disease‐causing mutations are scattered throughout the entire protein‐coding exons and their splicing junctions, and no obvious mutation hot spot has been identified.
Figure 3.
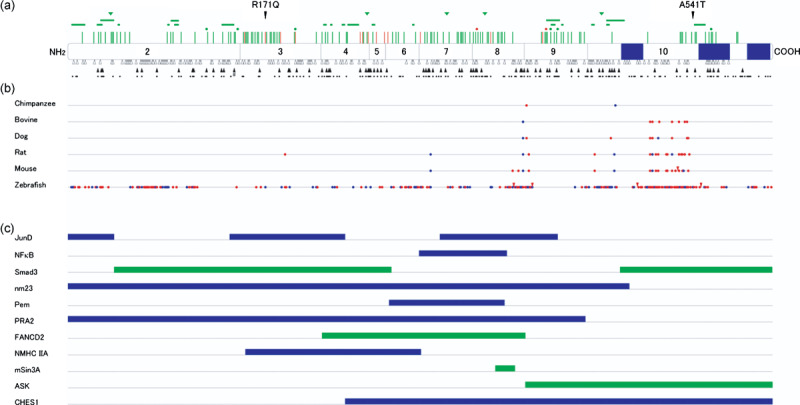
Mutations, evolutionary diversion, and binding domains of mein. (a) Germline MEN1 mutations identified in multiple endocrine neoplasia type 1 (MEN1) and related disorders. Locations of missense mutations (perpendicular lines), in‐frame deletions (dot, one amino acid; horizontal bar, two or more amino acids), and in‐frame insertions (triangles) are shown above the diagram of menin, with corresponding exons numbered. Green symbols indicate mutations causing MEN1, and orange symbols indicate those causing familial isolated hyperparathyroidism or apparently sporadic parathyroid tumor. Frameshift mutations (open triangles) and nonsense mutations (closed triangles) are shown below menin, with potential nonsense mutation sites indicated by dots. Splicing mutations and large deletions are not depicted. Blue boxes indicate nuclear localization signals. The normal polymorphisms R171Q and A541T are also indicated. #Nonsense mutation involving two base pair substitutions. (b) Homology between menin amino acid sequences among different vertebrate species. Sequences were retrieved from the internet website: http://www.ncbi.nlm.nih.gov/HomoloGene/. Blue and red dots indicate conservative and non‐conservative amino acid replacement compared with human menin, respectively. Red triangles indicate insertions of amino acid residues. (c) Menin regions implicated in its binding to interacting proteins. Blue bars represent the regions required for binding to proteins demonstrated to bind directly to menin. Green bars indicate the regions required for association with proteins not shown to bind directly to menin.
However, discrete distribution patterns are observed for different mutation types. The most noticeable is the uneven distribution of missense mutations and small in‐frame deletions and additions, which are more frequently identified in evolutionarily conserved regions of menin (Fig. 3b). These mutations do not cause gross alteration of menin's primary structure, but may replace or delete crucial amino acid residues or disrupt functionally important amino acid stretches. Nonsense mutations also exhibit a distinct uneven distribution pattern, but they frequently occur even in the evolutionarily non‐conserved regions. This uneven distribution of nonsense mutations is likely to be explained by the similarly uneven distribution of potential sites for nonsense mutations where stop codons can be generated by single nucleotide alterations (Fig. 3a). Frameshift mutations, caused by nucleotide insertions or deletions, are more uniformly distributed but still display some warm spots of mutation. These mutations are found frequently at a repetitive nucleotide sequence, suggesting that they occur by a replication‐slippage mechanism.
Phenotype–genotype correlation. Several mutations have been encountered repeatedly in apparently unrelated pedigrees and even in different ethnic groups( 4 ) but no characteristic phenotypes for particular mutations have been documented. A subtype of familial MEN1 that is characterized by an unusually high incidence of prolactinoma and a low frequency of enteropancreatic endocrine tumors has been noticed originally in the Burin peninsula of Newfoundland and hence referred to as MEN1Burin or the prolactinoma variant of MEN1.( 23 ) Several unrelated families affected with this subtype of MEN1 have been shown to harbor germline MEN1 mutations, including Y312X and R460X, which do not appear to be basically different from other nonsense mutations causative of classical MEN1. Thus, it is unlikely that particular MEN1 mutations are responsible for this subtype, and the molecular basis of the high penetrance of prolactinoma in certain MEN1 families is an enigma.
Subsets of FIHP and apparently sporadic parathyroid tumor have been shown to be caused by germline MEN1 mutations. Because hyperparathyroidism is the most frequent and early expression of MEN1, families or individuals showing these phenotypes may display the full manifestations of MEN1 subsequently, and those mutations may not be specific to the particular phenotype. However, approximately half of the mutations identified in FIHP are missense mutations or in‐frame deletions in contrast to only 30% prevalence of these mutations in typical MEN1.( 4 ) Some mutation carriers in these FIHP pedigrees are devoid of any signs of primary hyperparathyroidism, even at a relatively advanced age.( 24 ) These findings suggest that some mutations found in FIHP and apparently sporadic parathyroid tumor may specifically cause these MEN1‐related, milder disorders (Table 1).
Table 1.
Germline missense and in‐frame deletions identified only in patients with familial isolated hyperparathyroidism (FIHP) or sporadic parathyroid tumor
| Phenotype | Germline mutation | Exon |
|---|---|---|
| FIHP | D153V | 3 |
| V184E | 3 | |
| T197I | 3 | |
| E255K | 4 | |
| Q260P | 4 | |
| L267P | 5 | |
| P277H | 6 | |
| G305D | 7 | |
| Y353del | 8 | |
| A411P | 9 | |
| L414del | 9 | |
| Sporadic parathyroid tumor | R171W | 3 |
| S253W | 4 | |
| E274A | 5 | |
| E366D | 8 |
Molecular function of menin
Menin has amino acid sequences necessary for nuclear localization at its carboxy‐terminal region (2, 3), and is localized mainly to the nucleus.( 25 ) Menin has been shown to interact with a wide variety of proteins, including those related to transcriptional regulation, cell division, and DNA repair (Table 2). Although several studies have found menin in cytoplasmic and membrane fractions also,( 26 , 27 ) menin is thought to play a major role as a nuclear protein.
Table 2.
Molecular function of menin deduced from its binding partners
| Binding partners | Suggested menin function | References |
|---|---|---|
| JunD † | Transcriptional repression | 28, 29, 30 |
| Nuclear factor‐κB † | Transcriptional repression | 31 |
| Pem † | Transcription, spermatogenesis | 34 |
| Smad1, Smad3, Smad5, Runx2 | Transcriptional repression, transforming growth factor‐β signal transduction, cell division, osteoblastic differentiation | 32, 33 |
| mSin3A, histone deacetylase | Transcriptional repression through histone deacetylation, DNA repair | 30, 39 |
| Mixed‐lineage leukemia oncoprotein–histone methlytransferase complex, lens epithelium‐derived growth factor | Transcriptional activation through histone methylation, hematopoietic cell differentiation | 35, 36, 37, 38, 39, 40, 41 |
| Estrogen receptor α † | Transcriptional activation | 42 |
| Fanconi anemia complementation group D2 | DNA repair | 45 |
| Check point suppressor 1 † | Transcriptional repression, DNA damage response | 48 |
| 32‐kDa subunit of replication protein A † | Cell division, DNA repair | 44 |
| Activator of S‐phase kinase | Cell division | 52 |
| Glial fibrillary acidic protein, vimentin | Cell division | 27 |
| Non‐muscle myosin type II‐A heavy chain † | Cell division | 51 |
| nm23β † | GTPase | 49, 50 |
Molecules demonstrated to bind directly to menin. The binding of the molecules without this symbol may be indirect.
Roles in gene transcription. The transcription factor JunD was the first to be shown to bind menin.( 28 ) Menin represses JunD‐activated transcription through a histone deacetylase (HDAC)‐dependent mechanism( 29 ) in which mSin3A, a general corepressor, recruits HDAC to menin.( 30 ) Menin has also been shown to interact directly with nuclear factor (NF)‐κB family members and repress NF‐κB‐mediated transcription.( 31 ) These findings suggest a potential mechanism of tumor suppression by menin, in which menin downregulates several target genes required for cell growth.
On the other hand, menin enhances Smad3‐ and transforming growth factor (TGF)‐β‐induced transcription.( 32 ) Because disruption of TGF‐β signaling has been observed in cancers, it is plausible that menin plays a role in the TGF‐β signaling pathways for cell growth inhibition. Menin has been implicated in osteoblastic differentiation in association with Smad family proteins and Runx2.( 33 ) Direct binding of menin and Pem, a homeobox‐containing DNA binding protein, has also been demonstrated, suggesting a role in spermatogenesis.( 34 )
Menin has been shown to associate with RNA polymerase II and SET1‐like histone methyltransferase (HMT) complexes that contain mixed‐lineage leukemia oncoprotein (MLL) or MLL2, a close homolog of MLL.( 35 , 36 ) Menin binds to gene loci of p27Kip1 , p18Ink4c , and several Hox family genes, and regulates hematopoiesis and myeloid transformation.( 37 , 38 , 39 , 40 ) Interestingly, p27Kip1 has been identified as a gene responsible for the MEN1 variant mentioned above.( 20 ) Most recently, menin has been demonstrated to serve as a molecular adapter for tethering lens epithelium‐derived growth factor with HMT complexes, and is involved in the control of hematopoietic cell growth and differentiation.( 41 ) Menin also interacts directly with estrogen receptor α in a hormone‐dependent manner.( 42 ) Activation of the estrogen‐responsive gene encoding trefoil factor 1 (TFF1) results in promoter recruitment of menin and histone modification. These findings indicate that menin is involved in the regulation of gene transcription through histone modification in association with multiple transcription factors and cofactors (Fig. 4).
Figure 4.
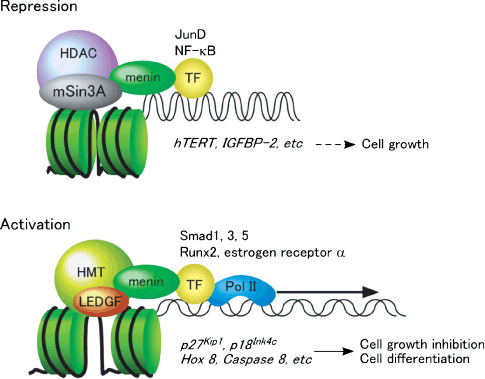
Postulated roles for menin in transcriptional repression and activation. In this model, menin represses pro‐cell growth genes by recruiting a general corepressor mSin3A and histone deacetylase (HDAC) to specific transcription factor (TF) binding sites, thus promoting histone deacetylation (upper). On the other hand, menin activates transcription of genes involved in cell growth inhibition and cell differentiation by recruiting lens epithelium‐derived growth factor (LEDGF) and histone methyltransferase (HMT) to specific TF binding sites, thus promoting histone methylation (lower).
Roles in the maintenance of genomic integrity. As slightly elevated chromosomal aberration frequency has been noticed in blood cells from MEN1 patients( 43 ) it has long been suspected that menin may have a role in DNA repair or the maintenance of genomic integrity. Menin has been shown to bind to the 32‐kDa subunit of replication protein A, which is essential for DNA replication, recombination, and repair, and is also implicated in the regulation of apoptosis and gene expression.( 44 ) Menin has also been shown to be associated with Fanconi anemia complementation group D2 (FANCD2), which is involved in DNA repair and mutated in patients with Fanconi anemia, a hereditary cancer syndrome.( 45 ) Loss of menin expression in mouse embryonic fibroblasts has been shown to increase sensitivity to DNA damage.( 45 ) The general corepressor mSin3A, which has been shown to be associated with HDAC and menin, has also been implicated in the repair of DNA damage.( 46 )
In addition, the Drosophila null mutant for menin has been demonstrated to exhibit hypersensitivity to ionizing radiation and DNA crosslinking agents, and an elevated rate of mutation.( 47 ) Genetic screening for factors that modify the effect of menin in Drosophila identified CHES1, a forkhead transcription factor implicated in transcriptional repression and DNA damage‐induced G2 arrest.( 48 ) These findings strongly suggest a role for menin in the maintenance of genomic integrity.
Implications of non‐nuclear localization of menin. We demonstrated that rat nm23β, the counterpart of human nm23‐H1, which has long been known as a tumor‐metastasis suppressor, interacts directly with menin and enhances its GTP‐hydrolyzing activity.( 49 , 50 ) Although a nuclear role as a transcription factor has also been implicated for nm23, it is localized mainly in the cytoplasm. Other non‐nuclear proteins shown to bind to menin include glial fibrillary acidic protein (GFAP), vimentin, and non‐muscle myosin heavy chain type II‐A (NMHC IIA). Menin has been demonstrated to colocalize with GFAP and vimentin in the cytoplasm of glioma cells during S–G2 phase of the cell cycle,( 27 ) and with NMHC IIA in the cleavage furrow of dividing cells.( 51 ) Menin also interacts functionally with the activator of S‐phase kinase (ASK) and represses ASK‐induced cell proliferation, suggesting that menin exerts an inhibitory effect on cell division through the association with ASK.( 52 ) The association with non‐nuclear proteins such as nm23, GFAP, vimentin, and NMHC IIA may reflect the functional sequestration of menin at the S–G2 phase of cell division.
Implications of MEN1 mutations. Analysis of protein binding domains has revealed that distinct parts of menin are used for association with different partners and that all of the menin regions appear to be required for these direct or functional associations (Fig. 3c). This is consistent with the finding that missense mutations and small in‐frame deletions and additions, which do not truncate menin, are dispersed, although not uniformly, throughout the menin‐coding regions. It is noteworthy that each missense mutation disrupts associations with particular binding partners but not with others. These findings suggest that multiple binding partners are indispensable for the tumor‐suppressor function of menin, although all of these associations may not necessarily be essential. It is also possible, however, that mutant menin having only a single amino acid change may be degraded rapidly and lose its activity, even if its binding domains remain intact in its primary structure.( 53 )
Tissue‐specific function of menin. Although menin is expressed ubiquitously in various tissues, the effects of menin depletion are highly tissue specific, as seen in the human disease MEN1 and in the heterozygous MEN1 mutant mouse, which closely mimics the human disease.( 54 ) Moreover, total depletion of menin results in proliferation of pancreatic islet β cells but no obvious consequences in the liver.( 55 ) Menin has been shown to bind and regulate several genes, including p27Kip1 , p18Ink4c , Hox family genes,( 36 , 37 , 38 , 39 , 40 , 41 ) and hTERT gene encoding human telomerase reverse transcriptase,( 56 ) which appear to be relevant to tumor suppression or cell differentiation. However. recent application of the genome‐wide search for menin‐binding genes revealed a broader role for menin in gene transcription.( 57 ) It is likely that menin may differentially regulate transcription of a subset of the potential target genes in association with cell‐specific factors. The exact mechanisms for a tissue‐specific function of menin remain to be elucidated.
DNA test for MEN1
Because the optimal therapies for MEN1‐associated tumors, especially for multicentric parathyroid and pancreatic tumors, are different from those for solitary sporadic tumors, accurate differential diagnosis between these hereditary and non‐hereditary tumors is mandatory before planning treatment.( 3 ) The diagnostic DNA test for MEN1 will be beneficial to the patients of endocrine tumors suggestive of MEN1 and offspring of patients definitely diagnosed with MEN1.
The test for the probands usually requires mutation screening over the entire MEN1 gene region because there are no mutation hot spots in the MEN1 gene. The presence of a pathogenic mutation indicates that they must be treated as MEN1 patients. Absence of identifiable mutation, however, does not exclude the diagnosis of MEN1, because several types of mutations, including whole gene deletions,( 58 ) escape detection by the routine method, which usually analyzes nucleotide sequences of polymerase chain reaction‐amplified exons and their nearby sequences. Furthermore, even if a nucleotide change is found in the non‐coding regions, it is often difficult to distinguish a causative mutation from a benign polymorphism.( 59 ) Although association analysis between mutations and disease is useful in these cases, this requires the examination of multiple family members and, therefore, may not always be possible.
Thus, the DNA test for MEN1 is still not always easy or perfect. Nevertheless, identification of the MEN1 gene has greatly simplified the presymptomatic diagnosis of MEN1 in most cases. Even for proband patients who already have some MEN1‐related tumors, confirmation of the diagnosis by DNA test will be beneficial when a MEN1 phenocopy, such as a chance association of several endocrine tumors, is suspected. The benefits of the DNA test are more obvious when an apparently healthy relative of a MEN1 patient is examined for the causative mutation identified in the proband. MEN1 mutation carriers should be monitored for the development of tumors by yearly biochemical tests and less often by imaging examinations.( 3 ) On the other hand, mutation non‐carriers are released from the unnecessary and costly periodic check up and fear of the disease. Use of the DNA test, however, should be decided from not only a medical but also an ethical standpoint, especially when the test is applied to minors.
Conclusion and perspectives
Much knowledge about mutations and functions of the MEN1 gene has been accumulated since its discovery in 1997. The mutation analyses revealed a distinct variant of MEN1, which was later found to be caused by a different gene, CDKN1B/p27Kip1 . The DNA test for MEN1 has proven to be useful for the management of patients with MEN1 and related disorders, although the test is not perfect and requires improvements. Functional analyses have revealed multiple roles for menin, including gene transcription, maintenance of genomic integrity, and control of cell division and differentiation, which are likely to be conferred by association with multiple menin‐binding proteins. However, the exact mechanisms underlying endocrine organ‐specific tumorigenesis in MEN1 remain to be explored. Elucidation of menin's molecular function will facilitate the understanding of cell growth‐controlling mechanisms common to various endocrine cells and discovery of new targets for endocrine tumor therapy.
Acknowledgments
This study was supported in part by a Grant‐in‐Aid for the Third Comprehensive 10‐Year Strategy for Cancer Control and for Cancer Research (17‐20) from the Ministry of Health, Labour, and Welfare of Japan.
References
- 1. Chandrasekharappa SC, Guru SC, Manickam P et al . Positional cloning of the gene for multiple endocrine neoplasia‐type 1. Science 1997; 276: 404–7. [DOI] [PubMed] [Google Scholar]
- 2. Gardner DG. Recent advances in multiple endocrine neoplasia syndromes. Adv Intern Med 1997; 42: 597–627. [PubMed] [Google Scholar]
- 3. Brandi ML, Gagel RF, Angeli A et al . Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 2001; 86: 5658–71. [DOI] [PubMed] [Google Scholar]
- 4. Lemos MC, Thakker RV. Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Hum Mutat 2008; 29: 22–32. [DOI] [PubMed] [Google Scholar]
- 5. Balogh K, Hunyady L, Patocs A et al . MEN1 gene mutations in Hungarian patients with multiple endocrine neoplasia type 1. Clin Endocrinol 2007; 67: 727–34. [DOI] [PubMed] [Google Scholar]
- 6. Toledo RA, Lourenco DM Jr, Coutinho FL et al . Novel MEN1 germline mutations in Brazilian families with multiple endocrine neoplasia type 1. Clin Endocrinol 2007; 67: 377–84. [DOI] [PubMed] [Google Scholar]
- 7. Tham E, Grandell U, Lindgren E, Toss G, Skogseid B, Nordenskjöld M. Clinical testing for mutations in the MEN1 gene in Sweden: a report on 200 unrelated cases. J Clin Endocrinol Metab 2007; 92: 3389–95. [DOI] [PubMed] [Google Scholar]
- 8. Vierimaa O, Ebeling TM, Kytölä S et al . Multiple endocrine neoplasia type 1 in Northern Finland: clinical features and genotype phenotype correlation. Eur J Endocrinol 2007; 157: 285–94. [DOI] [PubMed] [Google Scholar]
- 9. Jiang XH, Lu JL, Cui B et al . MEN1 mutation analysis in Chinese patients with multiple endocrine neoplasia type 1. Endocr Relat Cancer 2007; 14: 1073–9. [DOI] [PubMed] [Google Scholar]
- 10. Hannan FM, Nesbit MA, Christie PT et al . Familial isolated primary hyperparathyroidism caused by mutations of the MEN1 gene. Nat Clin Prac Endocrinol Metab 2008; 4: 53–8. [DOI] [PubMed] [Google Scholar]
- 11. Knudson AG. Hereditary cancer: two hits revisited. J Cancer Res Clin Oncol 1996; 122: 135–40. [DOI] [PubMed] [Google Scholar]
- 12. Stewart C, Parente F, Piehl F et al . Characterization of the mouse Men1 gene and its expression during development. Oncogene 1998; 17: 2485–93. [DOI] [PubMed] [Google Scholar]
- 13. Maruyama K, Tsukada T, Hosono T et al . Structure and distribution of rat menin mRNA. Mol Cell Endocrinol 1999; 156: 25–33. [DOI] [PubMed] [Google Scholar]
- 14. Ikeo Y, Sakurai A, Suzuki R et al . Proliferation‐associated expression of the MEN1 gene as revealed by in situ hybridization: possible role of the menin as a negative regulator of cell proliferation under DNA damage. Lab Invest 2000; 80: 797–804. [DOI] [PubMed] [Google Scholar]
- 15. Wautot V, Khodaei S, Frappart L et al . Expression analysis of endogenous menin, the product of the multiple endocrine neoplasia type 1 gene, in cell lines and human tissues. Int J Cancer 2000; 85: 877–81. [DOI] [PubMed] [Google Scholar]
- 16. Maruyama K, Tsukada T, Honda M et al . Complementary DNA structure and genomic organization of Drosophila menin. Mol Cell Endocrinol 2000; 168: 135–40. [DOI] [PubMed] [Google Scholar]
- 17. Rubin GM, Yandell MD, Wortman JR et al . Comparative genomics of the eukaryotes. Science 2000; 287: 2204–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Van Kesteren RE, Syed NI, Munno DW et al . Synapse formation between central neurons requires postsynaptic expression of the MEN1 tumor suppressor gene. J Neurosci 2001; 21: RC161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tsukada T, Ymaguchi K, Kameya T. The MEN1 gene and associated diseases: an update. Endocr Pathol 2001; 12: 259–73. [DOI] [PubMed] [Google Scholar]
- 20. Pellegata NS, Quintanilla‐Martinez L, Siggelkow H et al . Germ‐line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci USA 2006; 103: 15 558–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Simonds WF, James‐Newton LA, Agarwal SK et al . Familial isolated hyperparathyroidism: clinical and genetic characteristics of 36 kindreds. Medicine 2002; 81: 1–26. [DOI] [PubMed] [Google Scholar]
- 22. Vierimaa O, Georgitsi M, Lehtonen R et al . Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science 2006; 312: 1228–30. [DOI] [PubMed] [Google Scholar]
- 23. Hao W, Skarulis MC, Simonds WF et al . Multiple endocrine neoplasia type 1 variant with frequent prolactinoma and rare gastrinoma. J Clin Endocrinol Metab 2004; 89: 3776–84. [DOI] [PubMed] [Google Scholar]
- 24. Honda M, Tsukada T, Tanaka H et al . A novel mutation of the MEN1 gene in a Japanese kindred with familial isolated primary hyperparathyroidism. Eur J Endocrinol 2000; 142: 138–43. [DOI] [PubMed] [Google Scholar]
- 25. Guru SC, Goldsmith PK, Burns AL et al . Menin, the product of the MEN1 gene, is a nuclear protein. Proc Natl Acad Sci USA 1998; 95: 1630–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huang SC, Zhuang Z, Weil RJ et al . Nuclear/cytoplasmic localization of the multiple endocrine neoplasia type 1 gene product, menin. Lab Invest 1999; 79: 301–10. [PubMed] [Google Scholar]
- 27. Lopez‐Egido JR, Cunningham J, Berg M, Oberg K, Bongcam‐Rudloff E, Gobl AE. Menin's interaction with glial fibrillary acidic protein and vimentin suggests a role for the intermediate filament network in regulating menin activity. Exp Cell Res 2002; 278: 175–83. [DOI] [PubMed] [Google Scholar]
- 28. Agarwal SK, Guru SC, Heppner C et al . Menin interacts with the AP1 transcription factor JunD and represses JunD‐activated transcription. Cell 1999; 96: 143–52. [DOI] [PubMed] [Google Scholar]
- 29. Gobl AE, Berg M, Lopez‐Egido JR, Öberg K, Skogseid B, Westin G. Menin represses JunD‐activated transcription by a histone deacetylase‐dependent mechanism. Biochim Biophys Acta 1999; 1447: 51–6. [DOI] [PubMed] [Google Scholar]
- 30. Kim H, Lee J‐E, Cho E‐J, Liu JO, Youn H‐D. Menin, a tumor suppressor, represses JunD‐mediated transcriptional activity by association with an mSin3A‐histone deacetylase complex. Cancer Res 2003; 63: 6135–9. [PubMed] [Google Scholar]
- 31. Heppner C, Bilimoria KY, Agarwal SK et al . The tumor suppressor protein menin interacts with NF‐κB proteins and inhibits NF‐κB‐mediated transactivation. Oncogene 2001; 20: 4917–25. [DOI] [PubMed] [Google Scholar]
- 32. Kaji H, Canaff L, Lebrun JJ, Goltzman D, Hendy GN. Inactivation of menin, a Smad3‐interacting protein, blocks transforming growth factor type β signaling. Proc Natl Acad Sci USA 2001; 98: 3837–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sowa H, Kaji H, Hendy GN et al . Menin is required for bone morphogenetic protein 2‐ and transforming growth factor β‐regulated osteoblastic differentiation through interaction with Smads and Runx2. J Biol Chem 2004; 279: 40 267–75. [DOI] [PubMed] [Google Scholar]
- 34. Lemmens IH, Forsberg L, Pannett AAJ et al . Menin interacts directly with the homeobox‐containing protein Pem. Biochem Biophys Res Commun 2001; 286: 426–31. [DOI] [PubMed] [Google Scholar]
- 35. Hughes CM, Rozenblatt‐Rosen O, Milne TA et al . Menin associates with a trithorax family histone methyltransferase complex and with the Hoxc8 locus. Mol Cell 2004; 13: 587–97. [DOI] [PubMed] [Google Scholar]
- 36. Yokoyama A, Wang Z, Wysocka J et al . Leukemia proto‐oncoprotein MLL forms a SET1‐like histone methyltransferase complex with menin to regulate Hox gene expression. Mol Cell Biol 2004; 24: 5639–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Milne TA, Hughes CM, Lloyd R et al . Menin and MLL cooperatively regulate expression of cyclin‐dependent kinase inhibitors. Proc Natl Acad Sci USA 2005; 102: 749–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yokoyama A, Somervaille TCP, Smith KS, Rozenblatt‐Rosen O, Meyerson M, Cleary ML. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL‐associated leukemogenesis. Cell 2005; 123: 207–18. [DOI] [PubMed] [Google Scholar]
- 39. Chen Y‐X, Yan J, Keeshan K et al . The tumor suppressor menin regulates hematopoiesis and myeloid transformation by influencing Hox gene expression. Proc Natl Acad Sci USA 2006; 103: 1018–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Caslini C, Yang Z, El‐Osta M, Milne TA, Slany RK, Hess JL. Interaction of MLL amino terminal sequences with menin is required for transformation. Cancer Res 2007; 67: 7275–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yokoyama A, Cleary ML. Menin critically links MLL proteins with LEDGF on cancer‐associated target genes. Cancer Cell 2008; 14: 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dreijerink KMA, Mulder KW, Winkler GS, Höppener JWM, Lips CJM, Timmers HTM. Menin links estrogen receptor activation to histone H3K4 trimethylation. Cancer Res 2006; 66: 4929–35. [DOI] [PubMed] [Google Scholar]
- 43. Gustavson KH, Jansson R, Öberg K. Chromosomal breakage in multiple endocrine adenomatosis (type I and II). Clin Genet 1983; 23: 143–9. [DOI] [PubMed] [Google Scholar]
- 44. Sukhodolets KE, Hickman AB, Agarwal SK et al . The 32‐kilodalton subunit of replication protein A interacts with menin, the product of the MEN1 tumor suppressor gene. Mol Cell Biol 2003; 23: 493–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jin S, Mao H, Schnepp RW et al . Menin associates with FANCD2, a protein involved in repair of DNA damage. Cancer Res 2003; 63: 4204–10. [PubMed] [Google Scholar]
- 46. Silverstein RA, Ekwall K. Sin3: a flexible regulator of global gene expression and genome stability. Curr Genet 2005; 47: 1–17. [DOI] [PubMed] [Google Scholar]
- 47. Busygina V, Suphapeetiporn K, Marek LR, Stowers RS, Xu T, Bale AE. Hypermutability in a Drosophila model for multiple endocrine neoplasia type 1. Hum Mol Genet 2004; 13: 2399–408. [DOI] [PubMed] [Google Scholar]
- 48. Busygina V, Kottemann MC, Scott KL, Plon SE, Bale AE. Multiple endocrine neoplasia type 1 interacts with forkhead transcription factor CHES1 in DNA damage response. Cancer Res 2006; 66: 8397–403. [DOI] [PubMed] [Google Scholar]
- 49. Ohkura N, Kishi M, Tsukada T, Yamaguchi K. Menin, a gene product responsible for multiple endocrine neoplasia type 1, interacts with the putative tumor metastasis suppressor nm23. Biochem Biophys Res Commun 2001; 282: 1206–10. [DOI] [PubMed] [Google Scholar]
- 50. Yaguchi H, Ohkura N, Tsukada T, Yamaguchi K. Menin, the multiple endocrine neoplasia type 1 gene product, exhibits GTP‐hydrolyzing activity in the presence of the tumor metastasis suppressor nm23. J Biol Chem 2002; 277: 38 197–204. [DOI] [PubMed] [Google Scholar]
- 51. Obungu VH, Burns AL, Agarwal SK, Chandrasekharappa SC, Adelstein RS, Marx SJ. Menin, a tumor suppressor, associates with nonmuscle myosin II‐A heavy chain. Oncogene 2003; 22: 6347–58. [DOI] [PubMed] [Google Scholar]
- 52. Schnepp RW, Hou Z, Wang H et al . Functional interaction between tumor suppressor menin and activator of S‐phase kinase. Cancer Res 2004; 64: 6791–6. [DOI] [PubMed] [Google Scholar]
- 53. Yaguchi H, Ohkura N, Takahashi M, Nagamura Y, Kitabayashi I, Tsukada T. Menin missense mutants associated with multiple endocrine neoplasia type 1 are rapidly degraded via the ubiquitin–proteasome pathway. Mol Cell Biol 2004; 24: 6569–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Crabtree JS, Scacheri PC, Ward JM et al . A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors. Proc Natl Acad Sci USA 2001; 98: 1118–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Scacheri PC, Crabtree JS, Kennedy AL et al . Homozygous loss of menin is well tolerated in liver, a tissue not affected in MEN1. Mamm Genome 2004; 15: 872–7. [DOI] [PubMed] [Google Scholar]
- 56. Lin S‐Y, Elledge SJ. Multiple tumor suppressor pathways negatively regulate telomerase. Cell 2003; 113: 881–9. [DOI] [PubMed] [Google Scholar]
- 57. Scacheri PC, Davis S, Odom DT et al . Genome‐wide analysis of menin binding provides insights into MEN1 tumorigenesis. PLoS Genet 2006; 2: 406–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kishi M, Tsukada T, Shimizu S et al . A large germline mutation of the MEN1 gene in a family with multiple endocrine neoplasia type 1. Jpn J Cancer Res 1998; 89: 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kishi M, Tsukada T, Shimizu S et al . A novel splicing mutation (894–9G→A) of the MEN1 gene responsible for multiple endocrine neoplasia type 1. Cancer Lett 1999; 142: 105–10. [DOI] [PubMed] [Google Scholar]