

Abstract
Micro RNAs (miRNAs) are non‐coding small RNAs and constitute a novel class of negative gene regulators that are found in both plants and animals. Several miRNAs play crucial roles in cancer cell growth. To identify miRNAs specifically deregulated in anaplastic thyroid cancer (ATC) cells, we performed a comprehensive analysis of miRNA expressions in ARO cells and primary thyrocytes using miRNA microarrays. MiRNAs in a miR‐17‐92 cluster were overexpressed in ARO cells. We confirmed the overexpression of those miRNAs by Northern blot analysis in ARO and FRO cells. In 3 of 6 clinical ATC samples, miR‐17‐3p and miR‐17‐5p were robustly overexpressed in cancer lesions compared to adjacent normal tissue. To investigate the functional role of these miRNAs in ATC cells, ARO and FRO cells were transfected with miRNA inhibitors, antisense oligonucleotides containing locked nucleic acids. Suppression of miR‐17‐3p caused complete growth arrest, presumably due to caspase activation resulting in apoptosis. MiR‐17‐5p or miR‐19a inhibitor also induced strong growth reduction, but only miR‐17‐5p inhibitor led to cellular senescence. On the other hand, miR‐18a inhibitor only moderately attenuated the cell growth. Thus, we have clarified functional differences among the members of the cluster in ATC cells. In conclusion, these findings suggest that the miR‐17‐92 cluster plays an important role in certain types of ATCs and could be a novel target for ATC treatment. (Cancer Sci 2008; 99: 1147–1154)
Micro RNAs (miRNAs) are non‐coding, single‐stranded small RNAs and constitute a novel class of gene regulators that are found in both plants and animals.( 1 ) Mature miRNAs, ranging from 18 to 25 nucleotides in length, processed by two‐step cleavage involving Drosha and Dicer are thought to negatively regulate messenger RNA (mRNA). The mature miRNA binds to target mRNA and induces its cleavage or translational repression depending on the degree of complementarity.( 2 ) Although hundreds of miRNAs have been already cloned, only a small number of them have been characterized.
Recently, several miRNAs have been reported to be involved in cell proliferation or apoptosis in various types of cancers.( 3 , 4 ) MiR‐15a and miR‐16 induce apoptosis by targeting BCL2, and these miRNAs are frequently deleted or underexpressed in chronic lymphocytic leukemia.( 5 ) Let‐7 expression is reduced in lung cancer with poor prognosis,( 6 ) and inversely correlates with expression of RAS protein, suggesting a possible mechanism for cancer cell proliferation.( 7 ) Compared to these underexpressed miRNAs, miR‐21 has an antiapoptotic function and is overexpressed in glioblastoma. Knockdown of miR‐21 in glioblastoma cells induced caspase activation, resulting in apoptotic cell death.( 8 ) Thus, miRNAs can act as both tumor suppressor and oncogene.
The miR‐17‐92 cluster, composed of seven miRNAs (miR‐17‐5p, miR‐17‐3p, miR‐18a, miR‐19a, miR‐20a, miR‐19b, and miR‐92–1) and located in intron 3 of the C13orf25 gene, is overexpressed in lung cancer and B‐cell lymphoma.( 9 , 10 ) Enforced expression of truncated clusters comprising miR‐17‐5p~19b (miR‐17‐19b), the vertebrate‐specific portion of the miR‐17‐92 cluster, accelerated tumor development in a mouse B‐cell lymphoma model, suggesting oncogenic function of miR‐17‐19b. On the other hand, O'Donnell et al. have reported that expression of oncogenic E2F1 is negatively regulated by miR‐17‐5p and miR‐20a, members of the cluster, implying that they act as a tumor suppressors.( 11 ) Thus, the function of the cluster is still controversial.
In thyroid cancer, overexpression of several miRNAs has been reported. He et al. have reported that three miRNAs (miR‐221, miR‐222, and miR‐146) are overexpressed in papillary thyroid carcinomas (PTC) and regulate KIT expression.( 12 ) Another group has also shown that miR‐221, miR‐222 and miR‐181b are overexpressed in PTC, and inhibition of miR‐221 by antisense oligonucleotides led to attenuation of cell growth.( 13 ) In follicular thyroid cancers (FTC), miR‐197 and miR‐346 are significantly overexpressed.( 14 ) In vitro overexpression of either miRNA induced cell proliferation, whereas inhibition led to growth arrest. Very recently, Visone et al. have reported that significant decrease in miR‐30d, miR‐125b, miR‐26a, and miR‐30a‐5p was detected in human anaplastic thyroid cancers (ACT).( 15 )
ATC are highly aggressive and fatal tumors with less than 8 months of mean survival after diagnosis.( 16 ) Various treatment patterns including radiation and chemotherapy have been tried in ATC, but they are mostly unsuccessful.( 17 ) Therefore, the identification of miRNAs involved in proliferation or apoptosis in ATC cells has important therapeutic implications and may lead to establishment of a novel therapy for ATC. In the present study, we show that the miR‐17‐92 cluster, which is overexpressed in ARO and FRO cells, has a crucial role in cell growth and survival. These findings suggest that the miR‐17‐92 cluster might be a novel target for ATC treatment.
Materials and Methods
Cell culture. We used ATC cell lines ARO, FRO,( 18 ) and KTC‐2 (derived from anaplastic transformed PTC);( 19 ) PTC cell lines NPA and TPC‐1;( 20 ) FTC cell line WRO;( 21 ) and PT. All cells used in this study were of human origin. ARO, FRO, NPA, and WRO were kindly provided by Dr G. Juillard (University of California, Los Angeles, CA, USA). TPC‐1 and KTC‐2 were kindly provided by Dr Sato (Cancer Institute, Kanazawa University, Kanazawa, Japan) and Dr Kurebayashi (Kawasaki Medical School, Kurashiki, Japan), respectively. All cells (except PT) were maintained in RPMI‐1640 medium supplemented with 5% fetal bovine serum (FBS) and penicillin/streptomycin at 37°C in a humidified atmosphere with 5% CO2. PT were isolated from thyroid tissues obtained during subtotal thyroidectomy in patients with Graves’ disease and cultured as described previously.( 22 ) All experiments were performed after obtaining hospital ethical committee approval. Informed consent was obtained from each individual.
MiRNA microarray. Small RNAs (~200 nt) were extracted from ARO cells and PT using a mirVana miRNA Isolation Kit (Ambion, Austin, TX, USA). Five micrograms of the small RNAs were subjected to Custom microRNA Array Analysis Service (http://www.hssnet.co.jp/e/2/2_4_5_1.html) (Hokkaido System Science, Sapporo, Japan). The array contained 313 oligonucleotide probes for mature miRNA.
Northern blot analysis. Total RNA was extracted from cells using ISOGEN reagent (Nippon Gene, Tokyo, Japan). Ten micrograms of total RNA were separated on 15% denaturing polyacrylamide gel and electrotransferred onto Nylon Membrane Positively Charged (Roche Diagnostics, Basel, Switzerland). Oligonucleotides complementary to mature miRNAs were labeled with digoxigenin by terminal transferase‐mediated 3′ end‐labeling and used as probes. The sequences of oligonucleotides were as follows: miR‐17‐5p, 5′‐actacctgcactgtaagcactttg‐3′; miR‐17‐3p, 5′‐acaagtgccttcactgcagt‐3′; miR‐18a, 5′‐tatctgcactagatgcacctta3′; miR‐19a, 5′‐tcagttttgcatagatttgcaca‐3′; miR‐19b, 5′‐tcagttttgcatggatttgcaca‐3′; miR‐20a, 5′‐ctacctgcactataagcacttta‐3′; miR‐92–1, 5′‐caggccgggacaagtgcaata‐3′; miR‐21, 5′‐tcaacatcagtctgataagcta‐3′; let‐7, 5′‐gaggtagtaggttgtatagtt‐3′; miR‐106a, 5′‐gctacctgcactgtaagcactttt‐3′; miR‐106b, 5′‐atctgcactgtcagcacttta‐3′; 5S‐rRNA, 5′‐ttagcttccgagatcagacga‐3′. The membrane was then hybridized with hybridization mixture (0.25 M Na2HPO4[pH 7.2], 1 mM ethylenediamine tetraacetic acid (EDTA), 1% bovine serum albumin, 7% sodium dodecyl sulfate (SDS), 15% formamide, and the labeled probes) overnight at 43 or 45°C. After hybridization, the membrane was washed with wash mixture (20 mM Na2HPO4[pH 7.2], 1 mM EDTA, 1% SDS) followed by the washing buffer (0.1 M maleic acid, 0.15 M NaCl, 0.3% Tween‐20). After blocking with 1% Blocking Reagent (Roche Diagnostics), the hybridized membrane was incubated with alkaline phosphatase‐conjugated anti‐DIG antibody (Roche Diagnostics). The membrane was then washed with the washing buffer. After equilibration with the detection buffer (0.1 M Tris‐HCl [pH 9.5], 0.1 M NaCl), the membrane was incubated with the chemiluminescent substrate CDP Star (Roche Diagnostics). Detection was performed using a LAS3000 imaging system (Fujifilm, Tokyo, Japan). After detection, the membrane was stripped and used for re‐hybridization a few times.
Real‐time reverse transcription–polymerase chain reaction (RT‐PCR). The quantitative real‐time RT‐PCR for miRNA was performed using the TaqMan MicroRNA Assay System (Applied Biosystems, Foster City, CA, USA). Briefly, 10 ng of total RNA was reverse transcribed using a looped RT primer which is specific for each miRNA. The following amplification was performed using a corresponding TaqMan MicroRNA Assay Mix (Applied Biosystems) in a Thermal Cycler Dice Real‐time System (Takara Bio, Ohtsu, Japan). The cycle threshold value, which was determined using second derivative, was used to calculate the normalized expression of the indicated miRNAs using Q‐Gene software.( 23 )
RNA isolation from human ATC samples. Human ATC tissues and adjacent normal thyroid tissues were dissected from six formalin‐fixed paraffin‐embedded (FFPE) surgically resected samples. Total RNA was extracted using RecoverAll Total Nucleic Acid Isolation Kit for FFPE (Ambion) according to the manufacturer's protocol. Briefly, tissues were collected in microcentrifuge tubes from sections thicker than 20 µm and deparaffinized with 100% xylene followed by washing twice with 100% ethanol. Each sample was then digested with protease at 50°C for 3 h. For RNA isolation, sample solution was passed through the filter cartridge provided by manufacturer, and then DNase treatment was performed on the filter at room temperature for 30 min. After washing several times with washing solution, total RNA was eluted with heated nuclease free water.
Oligonucleotide transfection for suppression of endogenous miRNA. Locked nucleic acid (LNA) and deoxyribo nucleic acid (DNA) hybrid (LNA/DNA) antisense oligonucleotides (miR inhibitors) were chemically synthesized by a Greiner Bio‐one (Frickenhausen, Germany). The miR inhibitors contained LNA at eight consecutive centrally located bases (indicated by capital letters) as described previously.( 12 ) The sequences of the inhibitors were as follows: LNA‐17‐5p, 5′‐actacctgCACTGTAAgcactttg‐3′; LNA‐17‐3p, 5′‐acaagtGCCTTCACtgcagt; LNA‐18a, 5′‐tatctgcACTAGATGcacctta‐3′; LNA‐19a, 5′‐tcagtttTGCATAGAtttgcaca‐3′; LNA‐21, 5′‐tcaacatCAGTCTCTGAtaagcta‐3′. Transfection was done using the Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA).
Growth curves. Cells (ARO, 3 × 104; FRO, 1 × 104 cells) were plated in each well of 24‐well plates. Twelve hours later, the cells were transfected with miR inhibitors (first transfection). The second transfection was done at 48 h after first transfection. At the indicated times, the cells were detached by trypsinization, and the cell number was counted using a hemocytometer.
Imaging of caspase activation. Cells were incubated with 5 µM DRAQ5 (Biostatus, Leicestershire, UK) for 10 min followed by 25 µM D2R (Rhodamine 110 bi‐L‐aspartic acid amide) (Invitrogen Molecular Probes) for 15 min at 37°C. Cell images were obtained using a LSM510 META confocal microscope (Carl Zeiss, Oberkochen, Germany).
Senescence‐associated β‐galactosidase (SA‐β‐gal) staining. Cells were fixed with 2% formaldehyde and 0.2% glutalaldehyde and assayed for SA‐β‐gal activity using X‐gal (5‐bromo‐4‐chloro‐3‐indolyl β‐D‐galactosidase) at pH 6.0, as described previously.( 24 , 25 ) SA‐β‐gal‐positive cells were detected by bright‐field microscopy. The percentages of positive cells were determined by scoring approximately 200–400 cells/field for each sample.
Western blot analysis. Cells were lyzed in a buffer containing 20 mM Tris‐HCl (pH 7.5), 1 mM EDTA, 150 mM NaCl, 0.5% Triton‐X, 2 mM phenylmethylsulfonyl fluoride, 50 mM sodium fluoride, 10 mM sodium pyrophosphate, 1 mM sodium orthovanadate, 5% glycerol, and Complete protease inhibitor cocktail (Roche Diagnositics). An equal amount of protein was separated by SDS‐polyarylamide gel electrophoresis and transferred onto nitrocellulose membrane Bio Trace (Pall Corporation, Pensacola, FL, USA). The following primary antibodies were used: anti‐PARP (Cell Signaling Technology, Beverly, MA, USA), anticaspase 9 (Cell Signaling Technology), anticaspase 3 (Cell Signaling Technology), anti‐RB 4H1 (Cell Signaling Technology), anti‐PTEN A2B1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), and anti‐β‐actin C4 (Santa Cruz Biotechnology). The antigen‐antibody complexes were visualized using horseradish peroxidase–conjugated antimouse or antirabbit IgG antibody (Cell Signaling Technology) and Chemi‐Lumi One system (Nacalai Tesque, Kyoto, Japan). The image was obtained using a LAS3000 imaging system (Fujifilm).
DNA agarose gel electrophoresis. Genomic DNA was extracted from the pellets used for protein extraction. The pellets were incubated with 500 µL TEN buffer containing 40 mM Tris‐HCl (pH 8.0), 1 mM EDTA, 150 mM NaCl, and proteinase K at 55°C overnight. Then, DNA was extracted using the phenol/chloroform/isoamylalcohol method and ethanol precipitation. A total of 400 ng of DNA was electrophoretically separated on 1.5% agarose gel. Ethidium bromide was used for staining and the image was obtained using a UV transilluminator.
Statistical analysis. Differences between groups were examined for statistical significance using anova followed by Fisher's protected least significant difference. P‐value not exceeding 0.05 was considered statistically significant.
Results
Distinct miRNA expression pattern in ATC cells. To identify miRNAs specifically deregulated in ATC cells, we performed a comprehensive analysis of miRNA expression in ARO cells and PT using miRNA microarrays. Seven percent of miRNAs were overexpressed (>2.0‐fold) and 7% were underexpressed (<0.5‐fold) in ARO cells compared to PT. Fold changes of representative miRNAs expression are listed in Table 1. All members of miR‐17‐92 cluster except miR‐17‐3p were overexpressed in ARO cells. With respect to cancer‐related miRNAs, miR‐16 was overexpressed, and let‐7 and miR‐21 were expressed at an almost normal level.
Table 1.
Expression pattern of miRNAs in ARO compared with PT
| miRNA | Fold change | miRNA | Fold change |
|---|---|---|---|
| hsa‐mir‐192 | 16.53 | hsa‐let‐7 | 1.02 |
| hsa‐mir‐196a | 11.95 | hsa‐mir‐17‐3p | 0.98 |
| hsa‐mir‐194 | 9.14 | hsa‐mir‐21 | 0.88 |
| hsa‐mir‐429 | 6.69 | hsa‐mir‐145 | 0.70 |
| has‐mir‐200b | 6.28 | hsa‐mir‐143 | 0.60 |
| hsa‐mir‐7 | 6.18 | hsa‐mir‐221 | 0.51 |
| hsa‐mir‐10a | 5.70 | hsa‐mir‐210 | 0.40 |
| hsa‐mir‐16 | 5.27 | hsa‐mir‐222 | 0.40 |
| hsa‐mir‐20a | 3.37 | hsa‐mir‐23a | 0.34 |
| hsa‐mir‐106b | 3.26 | hsa‐mir‐27a | 0.30 |
| hsa‐mir‐17‐5p | 2.25 | hsa‐mir‐24 | 0.30 |
| hsa‐mir‐106a | 2.20 | hsa‐mir‐199a | 0.25 |
| hsa‐mir‐19b | 2.12 | hsa‐mir‐148a | 0.19 |
| hsa‐mir‐18a | 1.46 | hsa‐mir‐100 | 0.19 |
| hsa‐mir‐92 | 1.38 | hsa‐mir‐138 | 0.08 |
| hsa‐mir‐19a | 1.29 | hsa‐mir‐125b | 0.05 |
Overexpression of miRNAs in miR‐17‐92 cluster in thyroid cancer cell lines. Next, we performed Northern blot analysis to confirm the microarray data and also to check the expressions of several miRNAs in various thyroid cancer cell lines. All members of the miR‐17‐92 cluster (miR‐17‐5p, miR‐17‐3p, miR‐18a, miR‐19a, miR‐20a, miR‐19b, and miR‐92‐1) were robustly overexpressed in ATC cell lines, ARO, and FRO (Fig. 1a). We also examined some other cancer‐related miRNAs. As shown in Figure 1b, let‐7 and miR‐21 were expressed at normal to slightly lower level. In ARO cells, the results of Northern blot analysis were mostly consistent with the microarray data.
Figure 1.
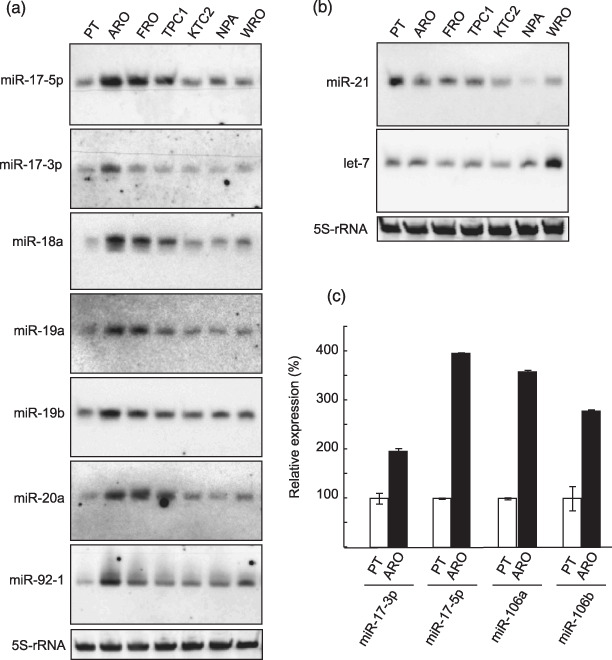
Expression of miRNAs in thyroid cancer cell lines. (a) Northern blot analysis for members of the miR‐17‐92 cluster. (b) Northern blot analysis for other cancer‐related miRNAs. (a,b) 5S ribosomal RNA (5S‐rRNA) was used as a loading control. Similar results were obtained in at least two independent experiments. (c) Real‐time reverse transciption–polymerase chain reaction (RT‐PCR) for miRNA with highly similar sequence. The expression level of indicated miRNA was measured as described in ‘Materials and Methods’. U6 small nuclear RNA was used as an internal control. Each bar indicates the mean and standard error of the data collected in triplicate.
MiR‐106a and miR‐106b have high homologous sequence to miR‐17‐5p (Fig. 3c), raising the possibility that signals detected with the miR‐17‐5p probe on Northern blot analysis were caused by cross‐hybridization to miR‐106a and miR‐106b. We thus utilized the TaqMan real‐time RT‐PCR assay, which is basically capable of discriminating single mismatched nucleotides.( 26 ) As shown in Figure 1c, not only miR‐17‐5p, but also miR‐106a and miR‐106b, were overexpressed in ARO cells. We also observed overexpression of miR‐17‐3p which has no homologous miRNA, leading to the conclusion that the miR‐17‐92 cluster was bona fide overexpressed in ARO cells (Fig. 1c).
Figure 3.
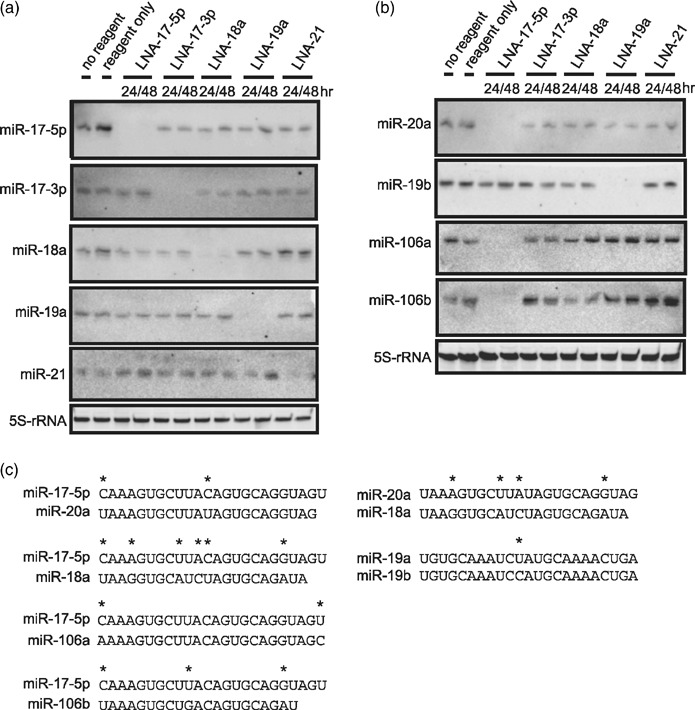
Suppression of miRNA by miR inhibitors. (a,b) ARO cells were transfected with indicated miR inhibitors. Total RNA was extracted at indicated time‐points and subjected to Northern blot analysis using indicated probes. 5S‐rRNA was used as a loading control. Similar results were obtained in at least two independent experiments. (c) Alignments of homologous miRNAs are shown. An asterisk indicates mismatched base.
Overexpression of miR‐17‐3p and miR‐17‐5p in human ATC samples. We next analyzed expression levels of miR‐17‐3p and miR‐17‐5p in clinical human ATC samples and paired adjacent normal tissue. TaqMan real‐time RT‐PCR assay was also used in this experiment. In 3 of 6 cases, those miRNAs were robustly overexpressed in ATC lesions compared to normal portions (Fig. 2). Two cases showed decreased expression in ATC lesions, and one case showed no change. This result suggests that the miR‐17‐92 cluster is overexpressed in some types of ATCs and may have a role in ATC pathogenesis.
Figure 2.
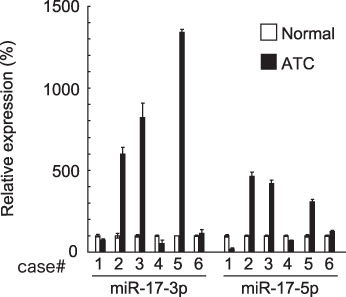
Expression of miR‐17‐3p and miR‐17‐5p in human ATC. Real‐time reverse transcription–polymerase chain reaction (RT‐PCR) was performed using RNA from six human ATC tissues (filled bars) and paired normal tissues (empty bars). The expression level of indicated miRNA was measured as described in ‘Materials and Methods’. U6 small nuclear RNA was used as an internal control. Each bar indicates the mean and standard error of the data collected in triplicate.
MiR inhibitors abolish endogenous miRNA expressions. The miR‐17‐92 cluster has been reported to be overexpressed in certain types of cancers; however, the function of each individual miRNA in the cluster is still controversial. Since miR‐17‐19b is vertebrate‐specific portion of the miR‐17‐92 cluster and has been reported to sufficiently act as an oncogene in a mouse B cell lymphoma model,( 9 ) we focused in greater detail on miRNAs in miR‐17‐19b. To explore biological significance of the members of miR‐17‐19b, ARO cells were transfected with the corresponding miR inhibitor for the each member. Transfection efficiency was initially assessed using fluorescein‐isothiocyanate–conjugated DNA oligonucleotides and fluorescence microscope, and it was almost 100% in our hands (data not shown). Since miRNA can bind to target mRNA with an imperfect match, we used cells treated with transfection reagent only as a control instead of using scrambled oligonucleotides to avoid any unpredictable effects. We then checked specificity of each miR inhibitor by Northern blot analysis. As shown in Figure 3a, target miRNAs became undetectable up to 48 h after transfection and the effect was sequence‐specific. However, at 72 h after transfection, the target miRNA expression was slightly restored (data not shown). In addition, miR‐20a, which has a similar sequence to miR‐17‐5p (2‐base difference; Fig. 3c), and miR‐19b, similar to miR‐19a (1‐base difference; Fig. 3c), were also undetectable in cells transfected with LNA‐17‐5p and LNA‐19a, respectively (Fig. 3b). Moreover, expression of miR‐106a and miR‐106b, which also have a homologous sequence to miR‐17‐5p (2‐base and 3‐base difference, respectively; Fig. 3c), were similarly affected by LNA‐17‐5p (Fig. 3b). Considering that probes for miR‐106a and miR‐106b probably detect all of miR‐17‐5p, miR‐20a, miR‐106a, and miR‐106b, LNA‐17‐5p seemed to block all of those miRNAs. On the other hand, the expression of miR‐18a, which also shares some homology with miR‐17‐5p (6‐base difference; Fig. 3c), was not altered by LNA‐17‐5p (Fig. 3a). Similarly, miR‐20a expression was not affected with LNA‐18a (4‐base difference; Fig. 3b,c).
Effects of inhibition of endogenous miRNA on ARO and FRO cell growth. We next investigated the effect of inhibition of the members of miR‐17‐19b on ARO and FRO cell growth. As shown in Figure 4a,b, LNA‐17‐3p totally suppressed the cell growth, and many detached cells were also observed (data not shown). LNA‐17‐5p and LNA‐19a also dramatically inhibited the growth (Fig. 4a,b). In contrast to LNA‐17‐3p, detached cells were barely detected with these miR inhibitors (data not shown). On the other hand, the growth reduction by LNA‐18a was moderate, and LNA‐21 did not affect at all (Fig. 4a,b). In ARO cells, LNA‐17‐5p, LNA‐17‐3p, LNA‐18a, and LNA‐19a suppressed the growth in a dose‐dependent fashion, and the inhibitory effect by LNA‐17‐3p at lower concentration (e.g. 10 pmol/well) was far greater than that of any other miR inhibitors (Fig. 4c).
Figure 4.
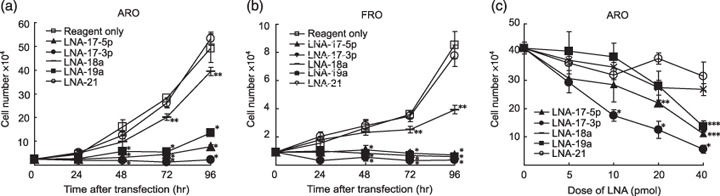
Effect of miR inhibitors on ATC cell growth. (a,b) Time course curves. The indicated cells were transfected with indicated miR inhibitors (40 pmol/well) as described in ‘Materials and Methods’. The number of cells was counted at indicated times after initial transfection. *P < 0.0001 versus reagent only, LNA‐18a and LNA‐21. **P < 0.0001 versus reagent only and LNA‐21. (c) Dose‐dependent curve. ARO cells were transfected with indicated dose of miR inhibitors (0–40 pmol/well) as described in ‘Materials and Methods’. The number of cells was counted at 72 h after transfection (24 h after second transfection). *P < 0.01 versus LNA‐18a, LNA‐19a, and LNA‐21. **P < 0.0001 versus LNA‐21a. ***P < 0.0001 versus LNA‐18a and LNA‐21. (a–c) Each point indicates the mean and SD of three wells. Similar results were obtained in three independent experiments.
LNA‐17‐3p induces apoptosis. Since cell detachments were exclusively observed in cells transfected with LNA‐17‐3p, we next explored the mechanism of growth inhibition by each miR inhibitor. We first investigated caspase activation using a D2R substrate after transfection with miR inhibitors. D2R is a cell‐permeable non‐fluorescent derivative of rhodamine 110. Upon enzymatic cleavage by caspases, this substrate is converted into fluorescent rhodamine 110, enabling us to measure the caspase activity in living cells. Cells were simultaneously stained with DRAQ‐5 to evaluate nuclear morphology. As shown in Figure 5a, intense activation of caspases was found only in the cells transfected with LNA‐17‐3p. Nuclear apoptotic changes, such as fragmentation or chromatin condensation, were also observed (Fig. 5a, arrowheads). Despite the significant growth reduction by LNA‐17‐5p or LNA‐19a (Fig. 4), there was no caspase activation in cells with those inhibitors (Fig. 5a). We then performed Western blot analysis to check the activation of caspase 3 and 9, key mediators of apoptosis. As shown in Figure 5b, both caspases were most clearly activated by LNA‐17‐3p. LNA‐17‐3p also caused cleavage of PARP (Fig. 5b) and DNA fragmentation (Fig. 5c), supporting the evidence of apoptosis induced by LNA‐17‐3p.
Figure 5.
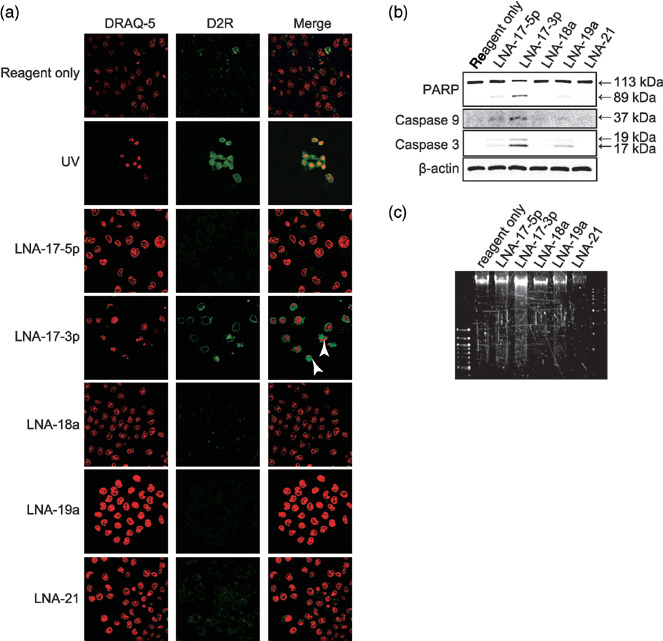
Effect of miR inhibitors on apoptosis in ARO cells. ARO cells were transfected with indicated miR inhibitors. (a) Images were obtained at 72 h after transfection (24 h after second transfection). D2R and DRAQ‐5 were excited with 488 and 633 nm lasers, respectively. Representative images are shown. Nuclear fragmentation is indicated by arrowhead. UV exposure was used as a positive control. (b) Cells were harvested at 72 h after transfection (24 h after second transfection), and Western blot analysis was performed using indicated primary antibodies. β‐actin was used as a loading control. (c) Agarose gel electrophoresis of DNA extracted from the cells used in (b). Similar results were observed in three independent experiments.
LNA‐17‐5p induces cell senescence. We then examined SA‐β‐gal activity in ARO cells transfected with miR inhibitors. Senescent and/or terminally differentiated cells are known to show enlarged granular phenotype and to possess SA‐β‐gal activity at pH 6.0.( 24 ) As shown in Figure 6a,b, the number of SA‐β‐gal‐positive and enlarged cells was significantly increased in cells treated with LNA‐17‐5p but not with LNA‐17‐3p or LNA‐19a. This finding suggests that one of the mechanisms of growth inhibition by LNA‐17‐5p is senescence‐like growth arrest.
Figure 6.
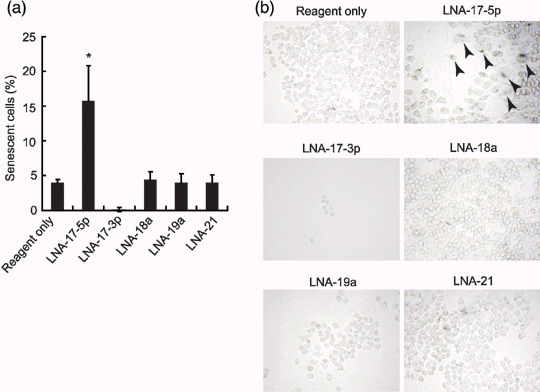
Effects of miR inhibitors on cellular senescence in ARO cells. (a) After senescence‐associated β‐galactosidase (SA‐β‐gal) staining, the number of senescent cells was counted at 72 h after transfection (24 h after second transfection). Each bar indicates the mean and SD of 10 fields. Similar results were obtained in at least two independent experiments. *P < 0.0001 versus others. (b) Representative images are shown. Enlarged and positively stained senescent cells are indicated by arrowheads. All images were obtained using a same magnification.
LNA‐17‐5p and LNA‐19a induce expression of retinoblastoma protein 1 (RB1) and PTEN, respectively. To explore further mechanisms of the growth arrest by LNA‐17‐5p and LNA‐19a, we also checked protein expression of tumor suppressors RB1 and PTEN after transfecion with those miR inhibitors. RB1 and PTEN have been predicted to be targets of miR‐17‐5p (miR‐20a, miR‐106a, and miR‐106b) and miR‐19a (miR‐19b), respectively.( 27 ) As shown in Figure 7, the expression of RB1 and PTEN was clearly increased in cells transfected with LNA‐17‐5p and LNA‐19a, respectively.
Figure 7.
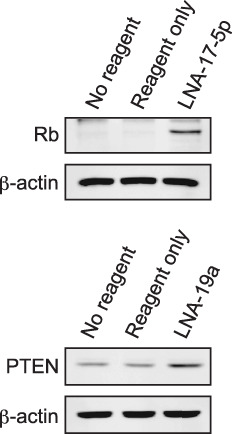
Effects of miR inhibitors on target protein expression. ARO cells were transfected with indicated miR inhibitors (400 pmol in 6‐cm culture dish). Cells were harvested 96 h after transfection, and Western blot anlaysis was performed using indicated primary antibodies. β‐actin was used as a loading control. Similar results were obtained at least two independent experiments.
Discussion
The miR‐17‐92 cluster located on the C13orf25 gene has been reported to act as an oncogene in cooperation with MYC in B‐cell lymphoma.( 9 ) On the other hand, O'Donnell et al. have reported that MYC directly binds to the cluster locus and regulates the transcription of miR‐17‐92 pri‐miRNA. MiR‐17‐5p and miR‐20a, members of the miR‐17‐92 cluster, negatively regulate the expression of E2F1 which promotes cell‐cycle progression. This finding suggests that these miRNAs have tumor suppressor activity.( 11 ) These two studies seem to contradict each other. However, E2F1 is reported to induce apoptosis when the level of this protein is excessive.( 28 ) Since MYC also directly induces E2F1 transcription, this mechanism may be present to achieve efficient cell proliferation by tightly regulating the E2F1 expression. Thus, the overall consequence of overexpressing the miR‐17‐92 cluster probably depends on cellular environment and level of target mRNA expression.
In the present work, we have shown the clear overexpression of miRNAs in the miR‐17‐92 cluster in ATC cell lines and some of the clinical ATC cases. We have also revealed their growth‐promoting effects using miR inhibitors. Contrary to our findings, Visone et al. have recently reported that no miR was overexpressed in 10 human ATC samples using microarray technique.( 15 ) This discrepancy could be due to their method of analysis in which mean of fold change was calculated and the cut‐off of two‐fold was applied. In addition, it is generally observed that differences in expression level measured by microarray are usually smaller (underestimated) than actual differences. Our results suggest that at least in some of ATC cases, the miR‐17‐92 cluster play a role. However, the mechanism of regulating the cluster in each case remains to be explored.
Inhibition of miR‐17‐3p caused apoptosis with caspase activation. Interestingly, the massive caspase activation was not observed in cells transfected with any other miR inhibitors. MiR‐17‐3p clearly has a distinct function from other members of the cluster. To our knowledge, however, there is no report regarding the target mRNA of miR‐17‐3p. Further studies are required to clarify the function of miR‐17‐3p in ATC.
Although LNA‐17‐5p and LNA‐19a induced relatively strong growth reduction, only LNA‐17‐5p caused some degree of cellular senescence in ARO cells. Lazzerini Denchi et al. have proposed that the model in which sustained E2F activity induces cellular senescence, whereas temporal E2F activation evokes cell proliferation.( 29 ) Because MYC is overexpressed in ARO cells compared to PT,( 30 ) inhibition of miR‐17‐5p and miR‐20a by LNA‐17‐5p might cause prolonged E2F1 activation which results in cellular senescence. However, further investigation is required to clarify the detailed mechanism.
One of predicted targets for miR‐19a and miR‐19b is tumor suppressor PTEN.( 31 ) As expected, definite up‐regulation of PTEN was observed in ARO cells transfected with LNA‐19a, suggesting that one of targets of miR‐19a and miR‐19b is PTEN. Germline mutations of PTEN are found in patients with Cowden syndrome, which predisposes to breast and thyroid neoplasia. Recent evidence suggests that reduced expression of PTEN plays a crucial role in thyroid cancer.( 32 , 33 , 34 , 35 ) Moreover, Frisk et al. have reported that PTEN inactivation is involved in highly malignant or late‐stage thyroid cancer, especially the anaplastic subtype.( 36 ) Our results suggest that overexpression of miR‐19a and miR‐19b might be associated with translational suppression of PTEN and induce cell growth in ATC.
We also found overexpression of miR‐106a and miR‐106b, and LNA‐17‐5p inhibited the expression of these homologous miRNAs. Since one of the predicted targets of these miRNAs is also E2F1,( 31 ) their suppression might result in cellular senescence by the above‐mentioned mechanism. Moreover, we demonstrated that the tumor suppressor RB1 was markedly up‐regulated by LNA‐17‐5p. This is consistent with the report suggesting a post‐transcriptional regulation of RB1 by miR‐106a overexpression in colon cancer.( 37 ) Therefore, up‐regulation of RB1 by LNA‐17‐5p might result in a negative action in ATC proliferation.
When one or two of five miRNAs (miR‐17‐3p, miR‐17‐5p, miR‐19a, miR‐19b, and miR‐20a) were inhibited, the cell growth was significantly reduced in ATC cells. On the other hand, suppression of miR‐18a only moderately reduced cell growth. There is a possibility that this slight growth reduction was due to the weak binding of LNA‐18a to miR‐17‐5p or miR‐20a. Although the expression of miR‐17‐5p and miR‐20a was not changed after transfection with LNA‐18a in the Northern blot analysis performed under denaturing conditions, LNA‐18a may weakly bind to miR‐17‐5p or miR‐20a physiologically and inhibit their function in actual live cells.
Hayashita et al. have reported that the miR‐17‐92 cluster was overexpressed in the most aggressive form of lung cancer, small‐cell cancer and might play a role in its development.( 10 ) Very recently, Matsubara et al. have also demonstrated that inhibition of miR‐17‐5p and miR‐20a caused apoptosis in lung cancer cells, whereas miR‐18a and miR‐19a did not show significant change in cell growth.( 38 ) The function of each miRNA in the cluster seems to be dependent on type of cells. In ATC cells, down‐regulation of PTEN might be more important for cell growth as mentioned above.
In PTC, two groups have concordantly reported that miR‐221 and miR‐222 are overexpressed and modulate KIT expression.( 12 , 13 ) However, in our microarray data, the expression of those miRNAs in ARO cells was underexpressed. ARO cells harbor the BRAFV600E mutation and are accordingly thought to originate from PTC,( 39 ) suggesting that those miRNAs may play a role in PTC carcinogenesis, but are no longer critical after anaplastic transformation.
In conclusion, our results demonstrate that the miR‐17‐92 cluster plays an important role in cell growth in some ATC, and that miR inhibitors containing LNA efficiently block cell proliferation and even induce cell death. The miR inhibitors against miR‐17‐92 cluster could be a novel therapeutic approach to certain types of ATC.
Acknowledgments
This work was supported in part by a Grant‐in‐Aid for Scientific Research (No. 18790637 and No. 18591030) and the Global COE Program from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, the President's discretionary fund of Nagasaki University, and the Nagasaki Igakudousoukai Fund for medical research.
References
- 1. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004; 116: 281–97. [DOI] [PubMed] [Google Scholar]
- 2. Ambros V. MicroRNA pathways in flies and worms: growth, death, fat, stress, and timing. Cell 2003; 113: 673–6. [DOI] [PubMed] [Google Scholar]
- 3. Esquela‐Kerscher A, Slack FJ. Oncomirs – microRNAs with a role in cancer. Nat Rev Cancer 2006; 6: 259–69. [DOI] [PubMed] [Google Scholar]
- 4. Calin GA, Croce CM. MicroRNA‐cancer connection: the beginning of a new tale. Cancer Res 2006; 66: 7390–4. [DOI] [PubMed] [Google Scholar]
- 5. Cimmino A, Calin GA, Fabbri M et al . miR‐15 and miR‐16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA 2005; 102: 13 944–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Takamizawa J, Konishi H, Yanagisawa K et al . Reduced expression of the let‐7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res 2004; 64: 3753–6. [DOI] [PubMed] [Google Scholar]
- 7. Johnson SM, Grosshans H, Shingara J et al . RAS is regulated by the let‐7 microRNA family. Cell 2005; 120: 635–47. [DOI] [PubMed] [Google Scholar]
- 8. Chan JA, Krichevsky AM, Kosik KS. MicroRNA‐21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res 2005; 65: 6029–33. [DOI] [PubMed] [Google Scholar]
- 9. He L, Thomson JM, Hemann MT et al . A microRNA polycistron as a potential human oncogene. Nature 2005; 435: 828–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hayashita Y, Osada H, Tatematsu Y et al . A polycistronic microRNA cluster, miR‐17‐92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res 2005; 65: 9628–32. [DOI] [PubMed] [Google Scholar]
- 11. O'Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c‐Myc‐regulated microRNAs modulate E2F1 expression. Nature 2005; 435: 839–43. [DOI] [PubMed] [Google Scholar]
- 12. He H, Jazdzewski K, Li W et al . The role of microRNA genes in papillary thyroid carcinoma. Proc Natl Acad Sci USA 2005; 102: 19 075–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pallante P, Visone R, Ferracin M et al . MicroRNA deregulation in human thyroid papillary carcinomas. Endocr Relat Cancer 2006; 13: 497–508. [DOI] [PubMed] [Google Scholar]
- 14. Weber F, Teresi RE, Broelsch CE, Frilling A, Eng C. A limited set of human MicroRNA is deregulated in follicular thyroid carcinoma. J Clin Endocrinol Metab 2006; 91: 3584–91. [DOI] [PubMed] [Google Scholar]
- 15. Visone R, Pallante P, Vecchione A et al . Specific microRNAs are downregulated in human thyroid anaplastic carcinomas. Oncogene 2007; 26: 7590–5. [DOI] [PubMed] [Google Scholar]
- 16. Ain KB. Anaplastic thyroid carcinoma: behavior, biology, and therapeutic approaches. Thyroid 1998; 8: 715–26. [DOI] [PubMed] [Google Scholar]
- 17. Vini L, Harmer C. Management of thyroid cancer. Lancet Oncol 2002; 3: 407–14. [DOI] [PubMed] [Google Scholar]
- 18. Fagin JA, Matsuo K, Karmakar A, Chen DL, Tang SH, Koeffler HP. High prevalence of mutations of the p53 gene in poorly differentiated human thyroid carcinomas. J Clin Invest 1993; 91: 179–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kurebayashi J, Otsuki T, Tanaka K, Yamamoto Y, Moriya T, Sonoo H. Medroxyprogesterone acetate decreases secretion of interleukin‐6 and parathyroid hormone‐related protein in a new anaplastic thyroid cancer cell line, KTC‐2. Thyroid 2003; 13: 249–58. [DOI] [PubMed] [Google Scholar]
- 20. Tanaka J, Ogura T, Sato H, Hatano M. Establishment and biological characterization of an in vitro human cytomegalovirus latency model. Virology 1987; 161: 62–72. [DOI] [PubMed] [Google Scholar]
- 21. Estour B, Van Herle AJ, Juillard GJ et al . Characterization of a human follicular thyroid carcinoma cell line (UCLA RO 82 W‐1). Virchows Arch B Cell Pathol Incl Mol Pathol 1989; 57: 167–74. [DOI] [PubMed] [Google Scholar]
- 22. Kawabe Y, Eguchi K, Shimomura C et al . Interleukin‐1 production and action in thyroid tissue. J Clin Endocrinol Metab 1989; 68: 1174–83. [DOI] [PubMed] [Google Scholar]
- 23. Muller PY, Janovjak H, Miserez AR, Dobbie Z. Processing of gene expression data generated by quantitative real‐time RT‐PCR. Biotechniques 2002; 32: 1372–4,6,8–9. [PubMed] [Google Scholar]
- 24. Dimri GP, Lee X, Basile G et al . A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA 1995; 92: 9363–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bulgin D, Podtcheko A, Takakura S et al . Selective pharmacologic inhibition of c‐Jun NH2‐terminal kinase radiosensitizes thyroid anaplastic cancer cell lines via induction of terminal growth arrest. Thyroid 2006; 16: 217–24. [DOI] [PubMed] [Google Scholar]
- 26. Chen C, Ridzon DA, Broomer AJ et al . Real‐time quantification of microRNAs by stem‐loop RT‐PCR. Nucleic Acids Res 2005; 33: e179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Griffiths‐Jones S, Grocock RJ, Van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res 2006; 34: D140–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Trimarchi JM, Lees JA. Sibling rivalry in the E2F family. Nat Rev Mol Cell Biol 2002; 3: 11–20. [DOI] [PubMed] [Google Scholar]
- 29. Lazzerini Denchi E, Attwooll C, Pasini D, Helin K. Deregulated E2F activity induces hyperplasia and senescence‐like features in the mouse pituitary gland. Mol Cell Biol 2005; 25: 2660–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ishigaki K, Namba H, Nakashima M et al . Aberrant localization of beta‐catenin correlates with overexpression of its target gene in human papillary thyroid cancer. J Clin Endocrinol Metab 2002; 87: 3433–40. [DOI] [PubMed] [Google Scholar]
- 31. Lewis BP, Shih IH, Jones‐Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell 2003; 115: 787–98. [DOI] [PubMed] [Google Scholar]
- 32. Weng LP, Brown JL, Eng C. PTEN coordinates G (1) arrest by down‐regulating cyclin D1 via its protein phosphatase activity and up‐regulating p27 via its lipid phosphatase activity in a breast cancer model. Hum Mol Genet 2001; 10: 599–604. [DOI] [PubMed] [Google Scholar]
- 33. Halachmi N, Halachmi S, Evron E et al . Somatic mutations of the PTEN tumor suppressor gene in sporadic follicular thyroid tumors. Genes Chromosomes Cancer 1998; 23: 239–43. [DOI] [PubMed] [Google Scholar]
- 34. Gimm O, Perren A, Weng LP et al . Differential nuclear and cytoplasmic expression of PTEN in normal thyroid tissue, and benign and malignant epithelial thyroid tumors. Am J Pathol 2000; 156: 1693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bruni P, Boccia A, Baldassarre G et al . PTEN expression is reduced in a subset of sporadic thyroid carcinomas: evidence that PTEN‐growth suppressing activity in thyroid cancer cells mediated by p27kip1. Oncogene 2000; 19: 3146–55. [DOI] [PubMed] [Google Scholar]
- 36. Frisk T, Foukakis T, Dwight T et al . Silencing of the PTEN tumor‐suppressor gene in anaplastic thyroid cancer. Genes Chromosomes Cancer 2002; 35: 74–80. [DOI] [PubMed] [Google Scholar]
- 37. Volinia S, Calin GA, Liu CG et al . A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci USA 2006; 103: 2257–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Matsubara H, Takeuchi T, Nishikawa E et al . Apoptosis induction by antisense oligonucleotides against miR‐17‐5p and miR‐20a in lung cancers overexpressing miR‐17–92. Oncogene 2007; 26: 6099–105. [DOI] [PubMed] [Google Scholar]
- 39. Namba H, Nakashima M, Hayashi T et al . Clinical implication of hot spot BRAF mutation, V599E, in papillary thyroid cancers. J Clin Endocrinol Metab 2003; 88: 4393–7. [DOI] [PubMed] [Google Scholar]