

Abstract
Retinoblastoma protein (RB) acts as a tumor suppressor in many tissue types, by promoting cell arrest via E2F‐mediated transcriptional repression. In addition to the aberrant forms of the RB gene found in different types of cancers, many viral oncoproteins including the simian virus 40 large T antigen target RB. However, cellular factors that inhibit RB function remain to be elucidated. Here, we report that RB interacts with the high mobility group protein A1 (HMGA1), a‐non‐histone architectural chromatin factor that is frequently overexpressed in cancer cells. HMGA1 binds the small pocket domain of RB, and competes with HDAC1. Subsequently, overexpression of HMGA1 abolishes the inhibitory effect of RB on E2F‐activated transcription from the cyclin E promoter. Under serum starvation, T98G cells had been previously shown to be arrested in the G0 phase in an RB‐mediated manner. The G0 phase was characterized by growth arrest and low levels of transcription, together with the hypophosphorylation of RB and the downregulation of HMGA1. In contrast, such serum‐depleted G0 arrest was abrogated in T98G cells overexpressing HMGA1. The overexpressed HMGA1 was found to form complexes with cellular RB, suggesting that downregulation of HMGA1 is required for G0 arrest. There were no phenotypic changes in HMGA1‐expressing T98G cells in the presence of serum, but the persistent expression of HMGA1 under serum starvation caused various nuclear abnormalities, which were similarly induced in T antigen‐expressing T98G cells. Our present findings indicate that overexpression of HMGA1 disturbs RB‐mediated cell arrest, suggesting a negative control of RB by HMGA1. (Cancer Sci 2007; 98: 1893–1901)
The retinoblastoma protein (RB) is known to be a key regulator of cell proliferation and arrest, and is therefore implicated in tumor suppression and cell differentiation.( 1 , 2 , 3 , 4 ) The principal role of RB is in the control of the cell cycle by repressing the E2F family of transcription factors, which regulate the expression of a number of genes involved in DNA synthesis and cell cycle progression.( 5 , 6 ) RB suppresses the E2F‐mediated transcription of genes in the G1 to the S phase by at least two mechanisms. First, RB binds to the transcriptional activation domain of E2F, and blocks its ability to stimulate gene expression.( 6 ) Second, the formation of the RB‐E2F complex results in active repression by recruiting appropriate co‐repressors that remodel chromatin to be transcriptionally inactive in the promoter region of E2F‐targeted genes. These co‐repressors include histone deacetylases (HDACs),( 7 , 8 , 9 , 10 ) histone methyltransferases,( 11 , 12 ) and DNA methyltransferases.( 13 , 14 ) RB also influences the accessibility of chromatin through the recruitment of ATP‐dependent chromatin remodeling factors such as Brahma (BRM) and BRM‐related gene (BRG).( 15 , 16 , 17 , 18 ) Thus, RB plays an important role in epigenetic gene regulation.
The ability of RB to repress E2F‐mediated transcription highly depends on the phosphorylation of RB by the cyclin‐dependent kinases (cdk),( 2 , 19 ) indicating that RB itself is governed by cell cycle machineries. In G0 and early G1 phases, RB is primarily unphosphorylated or hypophosphorylated, and becomes phosphorylated in the late G1 to S phase. Phosphorylation gradually increases throughout the S and G2/M phases. The initial phosphorylation occurs in the carboxyl terminal portion of RB by cyclin D/cdk4 and cdk6, and displaces HDACs from the pocket region of RB,( 19 ) leading to the blockade of the repressive role of RB. Subsequently, multiple phosphorylations in the pocket region of RB by cyclin E/cdk2 disrupt binding to E2F,( 19 ) resulting in the dissociation of RB from E2F. On the other hand, the G0 phase is a quiescent state where cells are resting or terminally differentiated as being ‘out of the cycle’.( 20 ) It was reported that the acute inactivation of RB alone was sufficient for G0‐arrested cells to re‐enter the cell cycle, suggesting that RB is required for maintaining G0 arrest. Similarly, cyclin C/cdk3 is involved in regulating the G0 to G1 transition (G0 exit) through specific phosphorylation of RB.( 20 ) However, the precise control of RB in the G0 phase remains to be elucidated.
The RB gene has been shown to be inactivated not only in retinoblastomas but also in a variety of cancer types.( 4 ) Importantly, several factors involved in the regulation of RB function, including cyclin D, cdk4 or the p16 cdk inhibitor, are frequently altered in many cancers,( 4 ) indicating that the deregulation of the RB pathway commonly contributes to tumor development. Recently, the mitotic checkpoint protein Mad2 was reported as an E2F‐regulated gene. Inactivation of the RB pathway causes inappropriate overexpression of Mad2 by E2F, and promotes aneuploidy in tumorigenesis,( 21 , 22 ) suggesting that RB is involved in the regulation of mitotic events. Three members of the RB family (pocket proteins) including RB, p107, and p130, have unique and overlapping functions.( 23 ) Compared with p107 and p130, the loss of RB function is closely linked to cancer. Viral oncoproteins such as simian virus 40 (SV40) large T antigen, adenovirus E1A, and human papilloma virus E7, target the pocket region of the RB family proteins and thereby disrupt their function.( 24 , 25 ) Like viral oncoproteins, mammalian E1A‐like inhibitor of differentiation 1 (EID‐1) was found to bind RB for inactivation.( 26 ) EID‐1 is highly and ubiquitously expressed in embryogenesis, and has not been associated with cancer. Since both phosphorylated and hypophosphorylated RB usually exist throughout the cell cycle, the function of RB seems not to be determined by this modification alone. Therefore, endogenous factors that inhibit RB may be present in growing cells.
During our investigations, we found that RB interacted with the high mobility group protein A1 (HMGA1), which is a non‐histone architectural chromatin protein, and participates in various cell regulation mechanisms.( 27 ) HMGA1 is characterized by the presence of three DNA‐binding motifs, called the adenine–thymine (AT) hook that preferentially binds the stretches of AT‐rich DNA sequences.( 27 , 28 ) In addition, this protein is reported to interact with several transcriptional factors and chromatin factors.( 29 , 30 , 31 ) HMGA1 is highly expressed during embryonic development, and then its expression is downregulated in differentiated adult cells.( 32 ) It is important to note that HMGA1 is frequently overexpressed in many types of cancers.( 33 ) The Myc family of transcription factors particularly appears to stimulate the expression of HMGA1.( 34 ) Furthermore, HMGA1 is rapidly induced by exposure to growth factors such as EGF.( 35 ) In fact, the expression levels of HMGA1 correlate with growth and metastatic activities of malignant cells.( 27 ) Transgenic mice overexpressing HMGA1 in all tissues develop lymphomas, and similarly to RB heterozygous mice, they have pituitary adenomas and thyroid tumors.( 36 ) These lines of evidence suggest that HMGA1 is a potential oncoprotein, and functionally cross‐talks with the RB pathway. Moreover, HMGA2, which is closely related to HMGA1, was recently reported to induce pituitary tumorigenesis by enhancing E2F1 activity.( 37 ) However, the functional relationship of overexpressed HMGA1 with RB remains unclear.
In the present study, we report that HMGA1 bound to the pocket domain of RB, resulting in the competition with HDAC1 but not E2F1 for RB binding. Subsequently, the overexpression of HMGA1 abolished the inhibitory effect of RB on E2F‐activated transcription from the cyclin E promoter. Under serum starvation, RB‐mediated G0 phase in T98G cells was characterized by growth arrest and low levels of general transcription, together with hypophosphorylation of RB and downregulation of HMGA1. In contrast, serum‐starved G0 arrest was abrogated by exogenous expression of HMGA1 in cells. Exogenous HMGA1 was found to form complexes with cellular RB, suggesting that downregulation of HMGA1 is required for RB‐mediated G0 arrest. Together, our results indicate that HMGA1 is a cellular inhibitor of RB, and overexpression of HMGA1 disturbs RB‐mediated cell arrest. These data shed light on a trans‐acting control of RB function by HMGA proteins.
Materials and Methods
Yeast two‐hybrid screening. Yeast strain AH109 carrying the pAS2‐1‐pocket region of mouse RB (amino acids 355–779) was transformed with the mouse E11.5 and E17 whole embryo cDNA libraries constructed in pACT2 (Clontech). Plasmids harboring cDNA were recovered from both histidine‐ and adenine‐positive colonies, and were used for DNA sequencing analysis.
Plasmids. The human cDNA sequence for HMGA1 was cloned into pcDNA3 (pcDNA3‐FLAG‐HMGA1), and into pCAG‐EGFP‐IRES‐Puro (pCAG‐EGFP‐HMGA1). pSG5L‐HA‐RB was as previously described.( 38 )
Cell culture. T98G glioblastoma, Saos‐2, and MCF‐7 cells were cultured in a 1:1 mixture of Dulbecco's modified Eagle's minimum essential medium and Ham's F‐12 nutrient medium (Sigma) supplied with 10% (v/v) heat‐inactivated fetal bovine serum (FBS). For synchronization studies, cells were incubated in medium containing no FBS for 72 h, then were stimulated with medium containing 10% FBS. Both total RNA and genomic DNA were isolated using standard methods to determine RNA/DNA ratios.
Transfection and cell treatment. T98G and Saos‐2 cells were transfected with plasmid DNAs using a liposome‐mediated gene transfer method. For the luciferase assay, Saos‐2 cells (1 × 105 cells) were transfected with RB and HMGA1 expression vectors (1 µg each) using FuGene6 (Roche Applied Science) in 6‐well plates and, after 48 h, the cells were used for the luciferase assay.
Protein expression. The pGEX‐4T vector expressing glutathione S‐transferase (GST)‐fused human RB (amino acids 301–928) was provided by Dr Y. Taya. cDNAs for human HMGA1 were cloned into pET28a (Novagen). The expressions of these proteins and His‐fused MBD1(TRD) were verified as described previously.( 39 )
Antibodies. The antibodies used were anti‐RB from BD Pharmingen; anti‐HMGA1, anti‐E2F1, anti‐HDAC1, and anti‐GFP from Santa Cruz; anti‐FLAG (M5) and anti‐GFAP from Sigma; anti‐His tag from Qiagen; anti‐GST from DAKO; and antiβ‐tubulin from Amersham.
Immunoprecipitation. For immunoprecipitation of RB complexes, T98G cells were lyzed with a buffer (50 mM Tris (pH 8.0), 300 mM NaCl, 0.4% NP40, 10 mM MgCl2) supplemented with a cocktail of protease and phosphatase inhibitors. Extracts were cleared by centrifugation, and then diluted with a buffer containing 50 mM Tris (pH 8.0), 0.4% NP40, and 2.5 mM CaCl2. Extracts (250 µL) were precleared by incubation with 20 µL of protein A/G‐agarose beads (Amersham Pharmacia and Oncogene) for 30 min at 4°C. After incubation with the anti‐RB antibodies for 1 h at 4°C, 10 µL of protein A/G beads were added to the extracts for 1 h at 4°C. Following extensive washes, bound proteins were analyzed by Western blot analysis.
Chemical cross‐linking immunoprecipitation was carried out to examine cellular complexes of HMGA1 and RB. T98G cells were treated with dimethyl 3,3′‐dithiobispropionimidate‐2HCl (DTBP) (5 mM) (Pierce) in phosphate‐buffered saline (PBS), rinsed with an ice‐cold buffer (100 mM Tris‐HCl (pH 8.0), 150 mM NaCl), lyzed with a buffer containing 10 mM Tris (pH 7.5), 500 mM NaCl, 1% NP‐40, 5 mM EDTA, 5% glycerol, 0.1% SDS, 1% sodium deoxycholate, and supplemented with a cocktail of protease and phosphatase inhibitors. After being sonicated, the supernatants (250 µL) were incubated for 1 h at 4°C with specific antibodies or control immunoglobulin G (IgG), followed by an another hour‐incubation after the addition of 20 µL of protein A/G agarose beads. The bound proteins were analyzed by Western blot analysis.
In vitro binding and GST pull‐down assay. Bacterially expressed GST and GST‐fused RB (1 µg) were immobilized on glutathione‐agarose beads, and incubated with His‐tagged HMGA1 or MBD1(TRD) (1 µg) in a buffer containing 0.05% Triton X‐100, 50 mM HEPES (pH 7.4), 100 mM or 200 mM NaCl, 5% glycerol, 10 µM ZnCl, 1 mM dithiothreitol, and protease inhibitors for 1 h at 4°C. The input indicated 10% of the His‐tagged proteins. His‐HMGA1 was used for a competition assay using the RB immunoprecipitates.
Immunofluorescent analysis. After two washes with PBS, T98G cells were fixed with 4% paraformaldehyde in PBS, for 15 min at room temperature; and then treated with 0.2% Triton X‐100 for 5 min at 4°C. After washing the cells with PBS containing 0.5% bovine serum albumin (BSA), they were incubated with specific antibodies in PBS containing 0.2% BSA, for 1 h at room temperature. Samples were analyzed with an Olympus IX71 microscope using the Lumina Vision software (version 2.2).
Luciferase assay. Forty‐eight hours after the transfection with luciferase reporter pCycE‐luc, together with pSG5L‐HA‐RB and pcDNA3‐FLAG‐HMGA1, Saos‐2 cells were lyzed in a buffer provided by the manufacturer (Promega). pRL‐TK and insertless pcDNA3 were used as controls. Values reported are means and standard deviations of the results from three independent experiments.
Small interfering RNA‐mediated knockdown. Small interfering RNA (siRNA) duplexes were designed for targeting mRNAs encoding human HMGA1 (Japan Bio Services), listed in Table S1. The selected siRNA sequences were submitted to human genome and EST databases to ensure the target specificities. The siRNAs were transfected into the cells by using lipofectamine RNAiMAX (Invitrogen).
Quantitative real‐time reverse transcription‐polymerase chain reaction. Five micrograms of the total RNAs was reverse‐transcribed using Superscript III (Invitrogen) and random hexamers (Operon). For quantification, real‐time polymerase chain reaction (PCR) analysis was carried out using Power SYBR Green PCR Master Mix on an ABI Prism 7500 Sequence Detector (Applied Biosystems). PCR amplification was repeated at least three times. The relative fold induction was quantified by the comparative threshold cycle method, and β‐actin was used as an endogenous normalization control. Primer sets for HMGA1 and the E2F target genes are listed in Table S2.
Results
RB interacts with HMGA1. To identify the factors that interact with RB, we carried out a yeast two‐hybrid screening using the region including the small pocket A and B of mouse RB (amino acids 355–779) as a bait (Fig. 1a). By screening approximately 3 × 106 independent transformants from 11.5 and 17‐day‐old mouse embryo cDNA libraries, we isolated a total of 52 cDNA clones encoding HMGA1 as well as previously known RB‐binding proteins including E2F1,( 40 ) HDAC1, and HDAC2.( 7 , 8 , 9 , 10 ) The present screening did not isolate HMGA2. To confirm the interaction between RB and HMGA1, we prepared His‐tagged human HMGA1, and subjected it to an in vitro pull‐down analysis (Fig. 1b). Briefly, GST and the GST‐fused pocket region of human RB (amino acids 301–928) were immobilized on glutathione‐agarose beads, and incubated with His‐HMGA1 in a binding buffer containing 100 mM or 200 mM NaCl. GST‐fused RB bound to His‐HMGA1, but not to methylated DNA‐binding protein MBD1 (TRD) fused to His‐tag as control.
Figure 1.
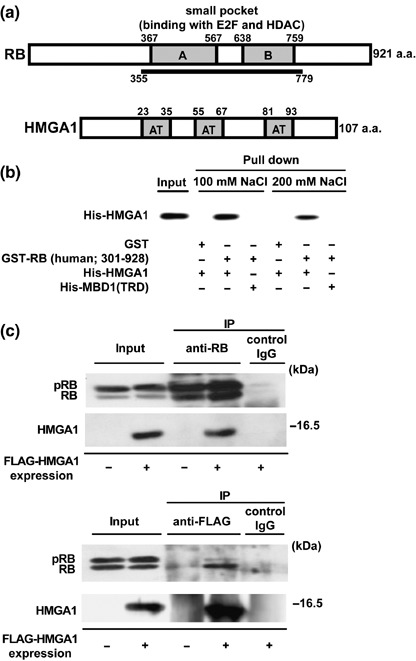
Interaction of retinoblastoma protein (RB) with high mobility group protein A1 (HMGA1). (a) Structures of RB and HMGA1. RB contains a small pocket region containing A and B. It binds the transcriptional factor E2F and histone deacetylase (HDAC). The pocket region of mouse RB (amino acids 355–779) was used as bait in a yeast two‐hybrid screen, resulting in the identification of HMGA1 isoform a (amino acids 1–107). HMGA1 (isoforms a and b) contains three copies of the AT hook motif (AT). (b) Direct binding of HMGA1 to RB. Recombinant GST and GST‐fused human RB (amino acids 301–928) were incubated with (His)6‐fused HMGA1 or the carboxyl terminal region of the methylated DNA binding protein MBD1, in a buffer containing 100 mM or 200 mM NaCl. Western blot analysis was carried out with anti‐His antibodies. The input shows 10% of each protein. (c) Complex formation between RB and HMGA1 in vivo. FLAG‐tagged HMGA1 was transiently expressed in T98G cells that express wild‐type RB, and immunoprecipitated by anti‐RB and anti‐FLAG antibodies. Western blot analysis of immunoprecipitates was carried out with anti‐RB and anti‐FLAG antibodies. The input shows 10% of each lysate. The phosphorylated and unphosphorylated/hypophosphorylated RB are shown as pRB and RB, respectively.
To check complex formation of RB and HMGA1 in vivo, we carried out an immunoprecipitation analysis (Fig. 1c). Human T98G cells were used in this study, and are known to express wild‐type RB and mutant‐type p53, and to arrest in the G0 phase in an RB‐mediated manner under serum starvation.( 20 , 41 ) Since currently available antibodies against HMGA1 were not suitable for the immunoprecipitation experiments, FLAG‐tagged HMGA1 was transiently expressed in T98G cells and immunoprecipitated by anti‐RB and anti‐FLAG antibodies. Western blot analysis revealed that HMGA1 was present in the immunoprecipitates with endogenous RB, but not in control immunoprecipitates. RB was detected in the immunoprecipitates with HMGA1. Two major forms for phosphorylated and hypophosphorylated RB were detected as upper and lower bands in input lanes, respectively. Immunoprecipitation by anti‐RB antibodies efficiently concentrated both forms of RB, while hypophosphorylated RB (functionally active) predominantly bound to HMGA1. Similar results were detected in MCF7 cells that also expressed wild‐type RB (data not shown). These results suggest that HMGA1 interacts with RB in vivo.
HMGA1 overcomes the repressive effect of RB on E2F‐mediated transcription. RB inhibits E2F‐activated transcription of cell growth‐regulated genes by recruiting co‐repressor proteins.( 7 , 8 , 9 , 10 ) For example, as shown in Fig. 2(b), E2F binds its binding motif in the promoter region of the cyclin E (CycE) gene, and is targeted by the RB‐HDAC complex for transcriptional repression. Interestingly, biochemical and structural studies revealed that HDAC1 binds to the pocket B of RB via the ‘L×C×E’ motif of this protein, while the opposite side of the pockets A and B of RB are recognized by E2F.( 42 ) Thus, RB‐binding proteins seem to interact with RB in different manners. To test the effect of HMGA1 on RB function, we purified endogenous RB‐containing complexes in T98G cells, by immunoprecipitation using anti‐RB antibodies (Fig. 2a). The precipitated complexes included E2F1, HDAC1, and RB, which agreed with previous reports.( 7 , 8 , 9 , 10 ) The RB‐containing complexes immobilized on glutathione‐agarose beads were then used for a competitive binding assay by adding recombinant His‐HMGA1. HDAC1 bound to RB was displaced by HMGA1 in a dose (HMGA1)‐dependent manner. In contrast, HMGA1 did not affect the binding of E2F1 to RB. The results suggest that high levels of HMGA1 disrupt the RB‐HDAC1 repressive complex, as was the case with HMGA2.( 37 ) In addition, several chromatin‐associated factors including HDAC2, SUV39, BRM, and BRG, have been reported to bind RB mechanistically alike HDAC1( 42 ) (See Discussion).
Figure 2.
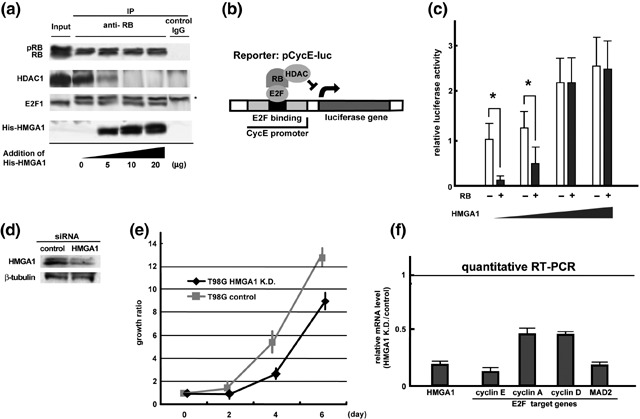
High mobility group protein A1 (HMGA1) inhibits retinoblastoma protein (RB)‐mediated repression of E2F‐target genes. (a) The effect of HMGA1 on E2F‐RB‐HDAC1 complexes. Endogenous RB‐containing complexes in T98G cells were immunoprecipitated by anti‐RB antibodies, and immobilized on glutathione‐agarose beads in a competitive binding assay using recombinant His‐HMGA1. The asterisk shows the band of the immunoglobulin. (b) Luciferase reporter assays. Hemagglutinin (HA)‐tagged RB and FLAG‐HMGA1 were expressed in human Saos‐2 cells that do not express RB genes. The effects of these combinations were examined using a Photinus pyralis luciferase reporter containing an E2F‐binding motif of the human cyclin E (CycE) gene promoter. (c) The effect of HMGA1 on RB‐mediated repression of the cyclin E promoter. The luciferase activities from insertless HA‐mock were normalized to 1. The asterisk indicates statistical difference using a student t‐test. (d) Specific siRNA‐mediated knockdown of HMGA1. β‐tubulin is shown as a control. (e) The effect of HMGA1 knockdown on cell proliferation. The cell numbers were determined on 0, 2, 4, and 6 days after the knockdown. Results were obtained from more than two independent experiments. Error bars indicate standard deviation. (f) The effect of HMGA1 knockdown on the expression of E2F target genes. A quantitative reverse transcription‐polymerase chain reaction (RT‐PCR) analysis showed that cyclin E1, cyclin A1, cyclin D1 and MAD2 were downregulated by depletion of HMGA1. Using triplicate experiments, the mRNA levels in control cells are normalized to 1.
We tested whether HMGA1 influenced RB‐mediated transcriptional repression, using luciferase reporter experiments (Fig. 2c). HA‐tagged RB and FLAG‐HMGA1 were expressed in human Saos‐2 cells that were null for RB genes. Western blot analyzes revealed that HA‐RB and FLAG‐HMGA1 were appropriately expressed in this assay (data not shown). The effects of the combined use of HA‐RB and FLAG‐HMGA1 were examined using a Photinus pyralis luciferase reporter containing an E2F‐binding motif of the human cyclin E gene promoter (Fig. 2b). In the absence of HMGA1, RB decreased cyclin E promoter activities by approximately five‐fold (Fig. 2c, left bars). HMGA1 alone tended to increase the luciferase activities in a dose‐dependent manner (Fig. 2c, white bars), suggesting a direct effect of HMGA1 on promoter activities. RB‐mediated repression of cyclin E promoter activities was antagonized by co‐expression of HMGA1 (Fig. 2c, right bars).
To check the effect of HMGA1 on cell proliferation and endogenous E2F target genes, we knocked‐down HMGA1 using synthesized siRNAs. Western blot analysis confirmed that HMGA1 was depleted by the specific knockdown in T98G cells (Fig. 2d). Using more than two independent assays, the depletion of HMGA1 significantly reduced the growth rate of the cells in comparison with the control (Fig. 2e). To investigate the effect of HMGA1 knockdown on the E2F activities, we checked the expression of E2F target genes (cyclin E1, cyclin A1, cyclin D1 and MAD2) using a quantitative reverse transcription (RT)‐PCR analysis (Fig. 2f). By triplicate experiments, the mRNA levels in the control cells were normalized to 1.0. The HMGA1 knockdown markedly reduced the expression of these factors. These data suggest that the overexpression of HMGA1 inhibits the repressive effect of RB on E2F‐mediated transcription, probably by disrupting the RB‐HDAC complexes, and that HMGA1 promotes cell growth and induction of the E2F target genes.
Hypophosphorylation of RB and downregulation of HMGA1 in cellular G0 phase. It is of great importance that RB represses E2F‐responsive genes to induce cell cycle arrest for tumor suppression and cell differentiation.( 3 , 4 ) The G0 phase is a quiescent arrest state for mammalian cells where the cells are not growing.( 20 , 43 ) To investigate the cellular G0 entry and exit, T98G cells were precultured in the presence of 10% FBS for 48 h, and then serum‐depleted for 72 h. They were subsequently cultured by adding serum (Fig. 3a). Cells mostly arrested at 12–24 h after serum starvation, and started to regrow 12–24 h after serum addition. The quiescent state in the G0 phase is well‐characterized by low levels of general transcription activities.( 44 ) Therefore, we isolated total RNAs and genomic DNAs at each cell cycle stage, and measured RNA/DNA ratios (Fig. 3b). Twelve hours after serum‐free culture, the RNA/DNA ratio markedly decreased to less than two‐fold (G0 entry), and continued to decrease by 72 h (G0 phase). The addition of serum in the culture medium at 72 h restored the RNA/DNA ratio at 96 h (G0 exit).
Figure 3.

Hypophosphorylated retinoblastoma protein (RB) and downregulation of high mobility group protein A1 (HMGA1) in serum‐starved G0 phase. (a) Culture of T98G cells. T98G cells were precultured in the presence of 10% fetal bovine serum (FBS) for 48 h, serum‐depleted for 72 h, and subsequently grown by adding 10% serum. White and black arrows show serum‐free and serum addition, respectively. The timings of G0 entry and exit are indicated. (b) Low levels of general transcription activities in the quiescent G0 phase. Total RNAs and genomic DNAs were isolated at each stage from the serum‐starved cells to measure RNA/DNA ratios. This ratio in growing conditions (at 0 h) was normalized to 1. The asterisk indicates statistical differences using a student t‐test. (c) Expression statuses of RB and HMGA1 in G0 arrest. Western blot analysis was carried out using anti‐RB and anti‐HMGA1 antibodies. The phosphorylated and unphosphorylated/hypophosphorylated RB are shown as pRB and RB, respectively. (d) Downregulation of HMGA1 in the G0 phase. Immunostaining analysis using anti‐HMGA1 and antiglial fibrillary acidic protein (GFAP) antibodies was carried out in T98G cells.
To examine the involvement of RB and HMGA1 in the quiescent state, Western blot analysis was carried out using anti‐RB and anti‐HMGA1 antibodies (Fig. 3c). There were both phosphorylated and hypophosphorylated forms of RB under growth‐stimulated conditions (0 h). RB was dephosphorylated at 24–72 h after serum depletion, and then phosphorylated after serum addition at 72 h. In addition, HMGA1 level remarkably decreased under serum‐free conditions. Thus, hypophosphorylated RB coexisted with downregulated HMGA1 in the G0 phase of serum‐starved T98G cells. Immunostaining analysis using anti‐HMGA1 and antiglial fibrillary acidic protein (GFAP) antibodies further emphasized the downregulation of HMGA1, together with somewhat fibroblast‐like morphologies of T98G cells, 72 h after serum depletion (Fig. 3d). These results suggest that both hypophosphorylation of RB and downregulation of HMGA1 are characteristics of serum‐starved G0 phase in T98G cells.
Persistent expression of HMGA1 abrogates serum‐starved G0 arrest. To check whether the downregulation of HMGA1 is required for the quiescent state, we established green fluorescent protein (GFP)‐expressing T98G cells (T98G GFP), and GFP‐fused HMGA1‐expressing T98G cells (T98G GFP‐HMGA1) (Fig. 4a). In addition to the immunostaining data, Western blot analysis using anti‐GFP antibodies showed that either GFP or GFP‐HMGA1 was equally expressed in these cells (Fig. 4b). Using the procedure indicated in Fig. 3(a), T98G GFP and T98G GFP‐HMGA1 cells were analyzed under serum starvation. Western blot analysis was carried out to analyze the expressions of RB, HMGA1, and GFP‐HMGA1 in T98G GFP‐HMGA1 cells (Fig. 4c). GFP‐HMGA1 was stably expressed using the viral cytomegalovirus (CMV) promoter throughout the culture conditions. In contrast, endogenous HMGA1 levels decreased under serum starvation, as similarly shown in Fig. 3(c), suggesting that HMGA1 was originally regulated by mitogenic stimulation. On the other hand, dephosphorylation of RB slowed down in serum‐starved T98G GFP‐HMGA1 cells, since phosphorylated RB still remained 72 h after serum depletion. In addition, we confirmed that HMGA1 was upregulated by epidermal growth factor or 17β‐estradiol in MCF7 cells, and that it was downregulated by retinoic acid‐induced differentiation in mouse embryonic cells (Fig. S1). These data suggest that HMGA1 expression highly depends on proliferative and undifferentiated states. Furthermore, as previously reported( 45 ) MCF7 cells overexpressing GFP‐HMGA1 formed colonies in soft agar (Fig. S2), suggesting that HMGA1 is potentially oncogenic.
Figure 4.
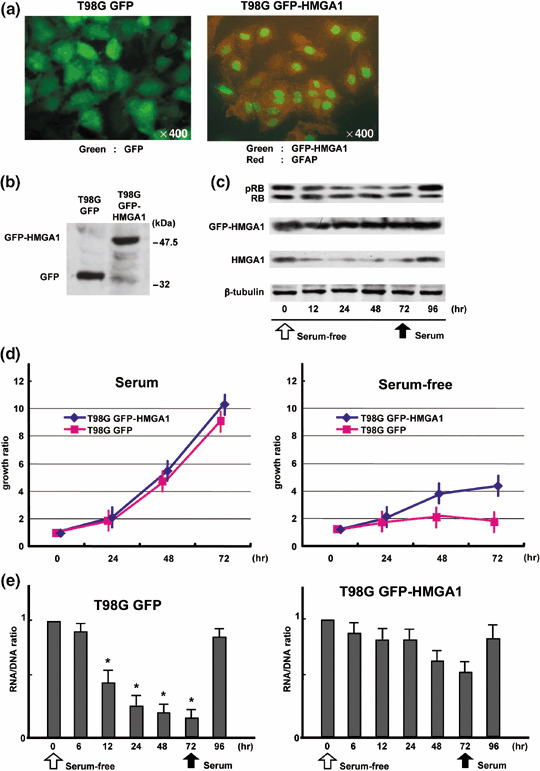
Persistent expression of high mobility group protein A1 (HMGA1) abrogates serum‐starved G0 arrest. (a) Establishment of green fluorescent protein (GFP)‐expressing T98G cells (T98G GFP) and GFP‐fused HMGA1‐expressing T98G cells (T98G GFP‐HMGA1). (b) Expression of GFP and GFP‐HMGA1 in T98G cells. Western blot analysis was carried out using anti‐GFP antibodies. (c) Expression statuses of retinoblastoma protein (RB), GFP‐HMGA1, and HMGA1 in G0 arrest. GFP‐HMGA1 was constantly expressed, while endogenous HMGA1 was downregulated under serum starvation. In comparison with Fig. 3(c), dephosphorylation of RB was slowed down in T98G GFP‐HMGA1 cells. (d) Growth rates of T98G GFP‐HMGA1 and T98G GFP cells in the presence and absence of serum. Both cell types comparatively proliferated under serum‐containing conditions. T98G GFP‐HMGA1 cells gradually proliferated under serum starvation, while T98G GFP cells were arrested 24 h after serum‐free conditions. (e) RNA/DNA ratio in T98G GFP‐HMGA1 cells during serum depletion. Analysis was carried out as shown in Fig. 3(b). The asterisk indicates statistical difference using a student t‐test.
We next examined the growth rates of T98G GFP‐HMGA1 and T98G GFP cells in the presence and absence of serum (Fig. 4d). Regardless of the expression of GFP‐HMGA1, both cells comparably proliferated under serum‐containing conditions. On the other hand, there were significant differences in growth rate between these cells under serum‐free conditions. T98G GFP‐HMGA1 cells continued to gradually grow even under serum starvation, while T98G GFP cells as well as the original T98G cells (data not shown) mostly arrested 24 h after incubation in serum‐free conditions. To examine whether T98G GFP‐HMGA1 cells entered the G0 phase under serum depletion, we analyzed the RNA/DNA ratio to assess general transcriptional activities (Fig. 4e). In T98G GFP cells, the RNA/DNA ratio markedly decreased 12–72 h after the start of serum starvation. In contrast, the significant decrease in RNA/DNA ratio was lost in T98G GFP‐HMGA1 cells. These data suggest that the persistent expression of HMGA1 abrogates RB‐mediated G0 arrest.
Inhibition of RB‐mediated G0 arrest by HMGA1 leads to chromosome instability. To demonstrate whether persistently expressed HMGA1 targets RB, we carried out an immunoprecipitation analysis in T98G GFP‐HMGA1 cells (Fig. 5a). Using T98G GFP‐HMGA1 cells, we examined the existence of GFP‐HMGA1 complexed with RB. As shown in Fig. 3(a), T98G GFP‐HMGA1 cells and T98G GFP cells were grown in serum‐containing medium, which was then replaced by serum‐free medium. Immunoprecipitation using anti‐GFP and anti‐RB antibodies showed that RB was present in the precipitates with GFP‐HMGA1, and that GFP‐HMGA1 coprecipitated with RB. Hypophosphorylated RB, rather than its phosphorylated form, tended to hbind GFP‐HMGA1. The precipitates with species‐matched control IgG gave no band for GFP‐HMGA1 or RB.
Figure 5.
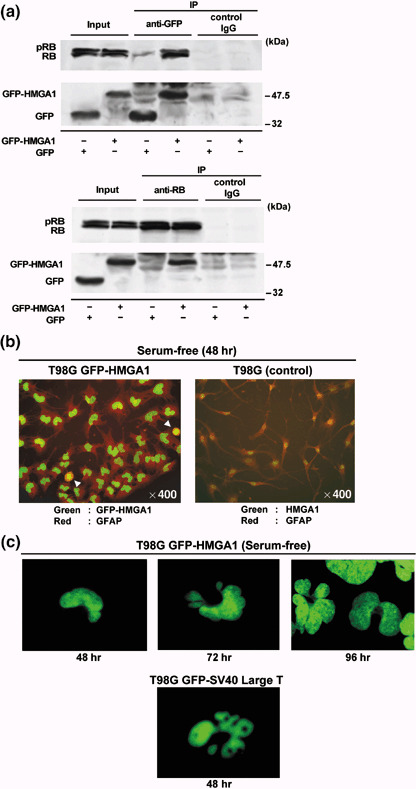
Inhibition of retinoblastoma protein (RB)‐mediated G0 arrest by high mobility group protein A1 (HMGA1) causes chromosome instability. (a) Green fluorescent protein (GFP)‐HMGA1 targets RB in serum‐starved T98G GFP‐HMGA1 cells. As shown in Fig. 3(a), T98G GFP‐HMGA1 cells and T98G GFP cells were placed in serum‐free conditions. Immunoprecipitation was carried out using anti‐GFP and anti‐RB antibodies. (b, c) Chromosome abnormalities in serum‐starved T98G GFP‐HMGA1 cells. Most of T98G GFP‐HMGA1 cells had a U‐shape or segmented nuclei 48 h after serum‐free culture (b). Mitotic cells were found with condensed chromosomes (indicated by arrowheads). The nuclei of the T98G GFP‐HMGA1 cells became multiple segmented and enlarged with micronuclei at 72 and 96 h (c). Simian virus 40 large T antigen (SV40 Large T) fused to GFP caused similar chromosomal abnormalities in T98G cells at 48 h after transfection, in the presence of serum.
During our close observations of T98G‐derived cells under serum starvation, we unexpectedly found that various nuclear structural abnormalities appeared in T98G GFP‐HMGA1 cells, but not in T98G or T98G GFP cells (Fig. 5b). As shown in Fig. 4(a), GFP‐HMGA1 constantly labeled the whole chromosomes in the cell cycle. Most of T98G GFP‐HMGA1 cells had a U‐shape or had segmented nuclei 48 h after serum‐free culture. In addition, mitotic GFP‐HMGA1 cells were detected with condensed chromosomes (Fig. 5b, arrowheads). The nuclei of the T98G GFP‐HMGA1 cells further became multiple segmented, and were enlarged with micronuclei at 72 and 96 h (Fig. 5c). These cells later seemed to stop undergoing nuclear changes, and survived instead of rapidly becoming apoptotic (data not shown). SV40 large T antigen (SV40 Large T) is known to inactivate RB, and frequently induces chromosomal instability in cell transformation.( 24 ) Therefore, GFP‐fused SV40 Large T was transiently expressed in T98G cells. Very similar chromosomal changes were detected in SV40 Large T‐expressing T98G cells 48 h after transfection in the presence of serum. Collectively, these results suggest that the overexpression of HMGA1 inhibits transcriptional control by RB‐E2F1 complexes, leading to the abrogation of serum‐starved G0 arrest and the chromosome instability.
Discussion
The present study showed that: (i) RB interacts with HMGA1 as a negative control; (ii) the overexpression of HMGA1 abrogates RB‐mediated G0 arrest; and (iii) disturbed G0 arrest by HMGA1 causes nuclear and chromosomal instabilities. Although the functional regulation of RB by intramolecular phosphorylation has been established,( 2 , 19 ) this study rather emphasized the significance of a trans‐acting (intermolecular) control of RB function. Biochemical experiments showed that HMGA1 bound to the pocket domain of RB, which is involved in interactions with E2F, chromatin modifying factors such as HDAC proteins, and viral oncoproteins such as SV40 large T antigen.( 8 , 24 ) Although HDAC1 and SV40 large T antigen contain the ‘L×C×E’ motif that specifically binds RB,( 8 , 42 ) E2F and HMGA proteins do not. However, a previous report,( 37 , 46 ) and the present study suggested that HMGA1 and HMGA2 competed with HDAC1 but not with E2F for RB binding, resulting in the displacement of HDAC1 from RB. The observations that the interaction of RB with ‘L×C×E’‐containing proteins was affected by HMGA proteins suggest that the ‘L×C×E’ motif and HMGA proteins target an identical area in the pocket domain of the RB molecule. In addition, such effect of HMGA proteins is similar to the initial phosphorylation of RB by cyclin D/cdk4 and cdk6,( 2 , 19 ) suggesting that RB is controlled by multiple overlapping mechanisms. Furthermore, the overexpression of HMGA1 abolished the RB/E2F‐dependentrepression of cyclin E promoter, possibly due to the loss of RB–HDAC association, leading to cell cycle progression. Considering its unique patterns of expression, HMGA1 is likely to act as a negative regulator of RB especially during embryonic development and in transformed cells.
HMGA1 is a chromatin architectural factor that plays a crucial role in cell regulation.( 27 ) The upregulation of HMGA1 is regarded as a molecular hallmark of malignant phenotypes, and is related to the degree of tumor aggressiveness. In sporadic human cancers, it is important to note that RB regulators (cyclin D, cdk4, and p16 cdk inhibitors) and RB itself are frequently altered. Since RB regulators affect the phosphorylation status of RB in the cell cycle, there may be a distinct mechanism that controls RB function for precise cell regulation. Transgenic mice overexpressing HMGA1 in all tissues develop lymphoma, pituitary adenomas and thyroid tumors,( 36 ) as previously reported in RB heterozygous mice. In addition, in HMGA2 transgenic mice, it was recently reported that HMGA2 interacted with RB, and induced E2F1 activity in mouse pituitary adenomas, and the loss of E2F1 function suppressed pituitary tumorigenesis.( 37 ) Moreover, mouse 3T3‐L1 fibroblasts were transformed by Hmga1 that was deleted for the carboxyl terminal sequences, and showed a higher E2F activity than the wild‐type cells, and a reduced G0/G1 fraction.( 46 ) These observations may be at least in part explained by the inhibition of RB by HMGA proteins, followed by the activation of E2F1.
RB phosphorylation by cyclin C/Cdk3 mediates the G0–G1 transition,( 20 ) suggesting that the inactivation of RB promotes G0 exit. In our study, HMGA1‐overexpressing T98G cells were not arrested in RB‐mediated G0 phase under serum‐free conditions. The cell cycle G0–G1 transition is controlled by the RB family proteins including RB, p107, and p130, which inhibit E2F family members.( 3 , 4 ) In the G0 phase, p130 seems to serve as a global E2F‐dependent repressor but does not regulate all E2F downstream genes.( 19 , 47 ) For example, cyclin E was reported to be overexpressed under serum starvation in RB‐deficient fibroblasts, suggesting that this gene could be selectively regulated by RB. The escape from the G0 state was observed in cells that abundantly expressed cyclin E.( 19 ) In addition, RB‐deficient fibroblasts under serum starvation exit the G0/G1 state immediately after serum stimulation. Fibroblasts lacking all three pocket proteins had a shorter cell cycle, and were not able to remain in the G0/G1 phase.( 48 ) Moreover, the overexpression of HMGA1 in MCF‐7 breast cancer cells increased the S phase population, together with a corresponding reduction of the G0/G1 population.( 45 ) Other reports showed that the expression of HMGA1 reduced the proportion of the G0/G1 population in several cell lines, and HMGA1‐transfected cells entered the S phase significantly earlier than untransfected cells, leading to an abnormally extended G2‐M phase.( 49 ) These lines of evidence indicate that the interactions between RB and HMGA proteins are biologically significant.
The expression of HMGA1 produces various effects on cell regulation. HMGA1‐overexpressing cells proliferate rapidly while they undergo apoptosis or senescence probably due to a conflict with mitogenic pressure and the inability to proceed to the cell cycle.( 27 ) T98G cells, which overexpressed HMGA1 in our study, expressed wild‐type RB and were resistant to apoptosis possibly due to a mutation of p53. Overexpression of HMGA1 induces cellular senescence in normal cells.( 50 ) Thus, the effects of overexpression of HMGA1 may depend on cellular conditions. Our study further showed that there were no phenotypic changes in HMGA1‐expressing T98G cells in the presence of serum, but the persistent expression of HMGA1 under serum starvation caused nuclear and chromosomal abnormalities. There are two possible explanations for these observations. First, HMGA1 binds AT‐rich DNA sequences and change the genome‐wide chromatin status, since the position of nucleosomes consisting of core histones and the linker histone H1 can be affected by HMGA proteins,( 51 , 52 ) possibly in association with chromosome instability. Second, the activity of E2F to express the spindle checkpoint protein Mad2 increases as a result of RB inactivation, and produces a hyperactive spindle checkpoint, resulting in abnormal mitotic events and aneuploidy.( 21 , 53 ) Under serum‐free conditions, T98G cells were arrested in the G0/G1 phase, and did not undergo mitosis and cytokinesis. In contrast, HMGA1‐expressing T98G cells were not arrested in the G0/G1 phase and entered cell division. Considering the evident nuclear changes, the resulting chromosomal instability may include aneuploidy and endoreduplication. As the size of tumor tissue increases during tumorigenesis, the inside of the tumor suffers malnutritional conditions like serum starvation. Most tumor cells may become necrotic or apoptotic, while a subset of cells may survive and adapt to the environment as a malignant transformation. Our preliminary data indicated that the high expression of HMGA1 was observed in glioblastoma cells in hypovascular or necrotic tissues. It was interesting to see that HMGA1‐positive cells tended to have abnormal nuclear shapes and failed to undergo proper mitosis (Fig. S3). The present study has shed additional insights into the negative regulation of RB by HMGA1, and its relation to cellular regulation.
Acknowledgments
The authors thank Dr C. Takahashi (Kyoto University) for the pSG5L‐HA‐RB plasmid, Dr Y. Taya (National Cancer Institute) for GST‐fused human RB‐expressing pGEX‐4T, and the members of our laboratory for their helpful suggestions. This work was supported by a Grant‐in‐Aid for the Scientific Research on Priority Areas, a Grant‐in‐Aid for 21st Century COE Research from the Ministry of Education, Culture, Sports, Science and Technology (M. N. and S.W.), and a research grant from the Takeda Science Foundation (M. N.). Y.U. was a COE Junior Research Associate.
References
- 1. Friend SH, Bernards R, Rogelj S et al . A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature 1986; 323: 643–6. [DOI] [PubMed] [Google Scholar]
- 2. Chen PL, Scully P, Shew JY, Wang JY, Lee WH. Phosphorylation of the retinoblastoma gene product is modulated during the cell cycle and cellular differentiation. Cell 1989; 58: 1193–8. [DOI] [PubMed] [Google Scholar]
- 3. Lipinski MM, Jacks T. The retinoblastoma gene family in differentiation and development. Oncogene 1999; 18: 7873–82. [DOI] [PubMed] [Google Scholar]
- 4. Liu H, Dibling B, Spike B, Dirlam A, Macleod K. New roles for the RB tumor suppressor protein. Curr Opin Genet Dev 2004; 14: 55–64. [DOI] [PubMed] [Google Scholar]
- 5. Bosco GW, Orr‐Weaver TL. DNA replication control through interaction of E2F‐RB and the origin recognition complex. Nat Cell Biol 2001; 3: 289–95. [DOI] [PubMed] [Google Scholar]
- 6. Nevins JR. The Rb/E2F pathway and cancer. Hum Mol Genet 2001; 10: 699–703. [DOI] [PubMed] [Google Scholar]
- 7. Kennedy BK, Liu OW, Dick FA, Dyson N, Harlow E, Vidal M. Histone deacetylase‐dependent transcriptional repression by pRB in yeast occurs independently of interaction through the LXCXE binding cleft. Proc Natl Acad Sci USA 2001; 98: 8720–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Luo RX, Postigo AA, Dean DC. Rb interacts with histone deacetylase to repress transcription. Cell 1998; 92: 463–73. [DOI] [PubMed] [Google Scholar]
- 9. Brehm A, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature 1998; 391: 597–601. [DOI] [PubMed] [Google Scholar]
- 10. Magnaghi‐Jaulin L, Groisman R, Naguibneva I et al . Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature 1998; 391: 601–5. [DOI] [PubMed] [Google Scholar]
- 11. Nielsen SJ, Schneider R, Bauer UM et al . Rb targets histone H3 methylation and HP1 to promoters. Nature 2001; 412: 561–5. [DOI] [PubMed] [Google Scholar]
- 12. Vandel L, Nicolas E, Vaute O, Ferreira R, Ait‐Si‐Ali S, Trouche D. Transcriptional repression by the retinoblastoma protein through the recruitment of a histone methyltransferase. Mol Cell Biol 2001; 21: 6484–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pradhan S, Kim GD. The retinoblastoma gene product interacts with maintenance human DNA (cytosine‐5) methyltransferase and modulates its activity. EMBO J 2002; 21: 779–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Robertson KD, Ait‐Si‐Ali S, Yokochi T, Wade PA, Jones PL, Wolffe AP. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F‐responsive promoters. Nat Genet 2000; 25: 338–42. [DOI] [PubMed] [Google Scholar]
- 15. Dunaief JL, Strober BE, Guha S et al . The retinoblastoma protein and BRG1 form a complex and cooperate to induce cell cycle arrest. Cell 1994; 79: 119–30. [DOI] [PubMed] [Google Scholar]
- 16. Strober BE, Dunaief JL. Guha, Goff SP. Functional interactions between the hBRM/hBRG1 transcriptional activators and the pRB family of proteins. Mol Cell Biol 1996; 16: 1576–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Trouche D, Le Chalony C, Muchardt C, Yaniv M, Kouzarides T. RB and hbrm cooperate to repress the activation functions of E2F1. Proc Natl Acad Sci USA 1997; 94: 11 268–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wong AK, Shanahan F, Chen Y et al . BRG1, a component of the SWI‐SNF complex, is mutated in multiple human tumor cell lines. Cancer Res 2000; 60: 6171–7. [PubMed] [Google Scholar]
- 19. Harbour JW, Luo RX, Dei Santi A, Postigo AA, Dean DC. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell 1999; 98: 859–69. [DOI] [PubMed] [Google Scholar]
- 20. Ren S, Rollins BJ. Cyclin C/cdk3 promotes Rb‐dependent G0 exit. Cell 2004; 117: 239–51. [DOI] [PubMed] [Google Scholar]
- 21. Van Deursen JM. Rb loss causes cancer by driving mitosis mad. Cancer Cell 2007; 11: 1–3. [DOI] [PubMed] [Google Scholar]
- 22. Sotillo R, Hernando E, Diaz‐Rodriguez E et al . Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell 2007; 11: 9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Paggi MG, Baldi A, Bonetto F, Giordano A. Retinoblastoma protein family in cell cycle and cancer: a review. J Cell Biochem 1996; 62: 418–30. [DOI] [PubMed] [Google Scholar]
- 24. Lee C, Cho Y. Interactions of SV40 large T antigen and other viral proteins with retinoblastoma tumour suppressor. Rev Med Virol 2002; 12: 81–92. [DOI] [PubMed] [Google Scholar]
- 25. Felsani A, Mileo AM, Paggi MG. Retinoblastoma family proteins as key targets of the small DNA virus oncoproteins. Oncogene 2006; 25: 5277–85. [DOI] [PubMed] [Google Scholar]
- 26. Miyake S, Sellers WR, Safran M et al . Cells degrade a novel inhibitor of differentiation with E1A‐like properties upon exiting the cell cycle. Mol Cell Biol 2000; 20: 8889–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Reeves R. Molecular biology of HMGA proteins: hubs of nuclear function. Gene 2001; 277: 63–81. [DOI] [PubMed] [Google Scholar]
- 28. Geierstanger BH, Volkman BF, Kremer W, Wemmer DE. Short peptide fragments derived from HMG‐I/Y proteins bind specifically to the minor groove of DNA. Biochemistry 1994; 33: 5347–55. [DOI] [PubMed] [Google Scholar]
- 29. Currie RA. Functional interaction between the DNA binding subunit trimerization domain of NF‐Y and the high mobility group protein HMG‐I (Y). J Biol Chem 1997; 272: 30 880–8. [DOI] [PubMed] [Google Scholar]
- 30. Foti D, Iuliano R, Chiefari E, Brunetti A. A nucleoprotein complex containing Sp1, C/EBP beta, and HMGI‐Y controls human insulin receptor gene transcription. Mol Cell Biol 2003; 23: 2720–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yie J, Merika M, Munshi N, Chen G, Thanos D. The role of HMG I (Y) in the assembly and function of the IFN‐beta enhanceosome. EMBO J 1999; 18: 3074–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chiappetta G, Avantaggiato V, Visconti R et al . High level expression of the HMGI (Y) gene during embryonic development. Oncogene 1996; 13: 2439–46. [PubMed] [Google Scholar]
- 33. Sgarra R, Rustighi A, Tessari MA et al . Nuclear phosphoproteins HMGA and their relationship with chromatin structure and cancer. FEBS Lett 2004; 574: 1–8. [DOI] [PubMed] [Google Scholar]
- 34. Wood LJ, Mukherjee M, Dolde CE et al . HMG‐I/Y, a new c‐Myc target gene and potential oncogene. Mol Cell Biol 2000; 20: 5490–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Holth LT, Thorlacius AE, Reeves R. Effects of epidermal growth factor and estrogen on the regulation of the HMG‐I/Y gene in human mammary epithelial cell lines. DNA Cell Biol 1997; 16: 1299–309. [DOI] [PubMed] [Google Scholar]
- 36. Fedele M, Pentimalli F, Baldassarre G et al . Transgenic mice overexpressing the wild‐type form of the HMGA1 gene develop mixed growth hormone/prolactin cell pituitary adenomas and natural killer cell lymphomas. Oncogene 2005; 24: 3427–35. [DOI] [PubMed] [Google Scholar]
- 37. Fedele M, Visone R, De Martino I et al . HMGA2 induces pituitary tumorigenesis by enhancing E2F1 activity. Cancer Cell 2006; 9: 459–71. [DOI] [PubMed] [Google Scholar]
- 38. Sellers WR, Novitch BG, Miyake S et al . Stable binding to E2F is not required for the retinoblastoma protein to activate transcription, promote differentiation, and suppress tumor cell growth. Genes Dev 1998; 12: 95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fujita N, Shimotake N, Ohki I et al . Mechanism of transcriptional regulation by methyl‐CpG binding protein MBD1. Mol Cell Biol 2000; 20: 5107–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chellappan SP, Hiebert S, Mudryj M, Horowitz JM, Nevins JR. The E2F transcription factor is a cellular target for the RB protein. Cell 1991; 65: 1053–61. [DOI] [PubMed] [Google Scholar]
- 41. Harada K, Kurisu K, Sadatomo T et al . Growth inhibition of human glioma cells by transfection‐induced P21 and its effects on telomerase activity. J Neurooncol 2000; 47: 39–46. [DOI] [PubMed] [Google Scholar]
- 42. Dahiya A, Gavin MR, Luo RX, Dean DC. Role of the LXCXE binding site in Rb function. Mol Cell Biol 2000; 20: 6799–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ohta T, Fukuda M, Wanebo HJ, Jogo K, Yamaguchi S. Behavior of the cell cycle‐associated proteins in an unusual G0‐arrestable cancer cell line. Exp Cell Res 1996; 225: 85–92. [DOI] [PubMed] [Google Scholar]
- 44. Ladd AC, Pyatt R, Gothot A et al . Orderly process of sequential cytokine stimulation is required for activation and maximal proliferation of primitive human bone marrow CD34+ hematopoietic progenitor cells residing in G0. Blood 1997; 90: 658–68. [PubMed] [Google Scholar]
- 45. Reeves R, Edberg DD, Li Y. Architectural transcription factor HMGI (Y) promotes tumor progression and mesenchymal transition of human epithelial cells. Mol Cell Biol 2001; 21: 575–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pierantoni GM, Battista S, Pentimalli F et al . A truncated HMGA1 gene induces proliferation of the 3T3‐L1 pre‐adipocytic cells: a model of human lipomas. Carcinogenesis 2003; 24: 1861–9. [DOI] [PubMed] [Google Scholar]
- 47. Canhoto AJ, Chestukhin A, Litovchick L, DeCaprio JA. Phosphorylation of the retinoblastoma‐related protein p130 in growth‐arrested cells. Oncogene 2000; 19: 5116–22. [DOI] [PubMed] [Google Scholar]
- 48. Sage J, Mulligan GJ, Attardi LD et al . Targeted disruption of the three Rb‐related genes leads to loss of G (1) control and immortalization. Genes Dev 2000; 14: 3037–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Baldassarre G, Battista S, Belletti B et al . Negative regulation of BRCA1 gene expression by HMGA1 proteins accounts for the reduced BRCA1 protein levels in sporadic breast carcinoma. Mol Cell Biol 2003; 23: 2225–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Narita M, Narita M, Krizhanovsky V et al . A novel role for high‐mobility group a proteins in cellular senescence and heterochromatin formation. Cell 2006; 126: 503–14. [DOI] [PubMed] [Google Scholar]
- 51. Reeves R, Nissen MS. Interaction of high mobility group‐I (Y) nonhistone proteins with nucleosome core particles. J Biol Chem 1993; 268: 21 137–46. [PubMed] [Google Scholar]
- 52. Zhao K, Kas E, Gonzalez E, Laemmli UK . SAR‐dependent mobilization of histone H1 by HMG‐I/Y in vitro: HMG‐I/Y is enriched in H1‐depleted chromatin. EMBO J 1993; 12: 3237–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hernando E, Nahle Z, Juan G et al . Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature 2004; 430: 797–802. [DOI] [PubMed] [Google Scholar]