

Abstract
Cancer chemotherapy and radiotherapy kill cancer cells by inducing DNA damage, unless the lesions are repaired by intrinsic repair pathways. DNA double‐strand breaks (DSB) are the most deleterious type of damage caused by cancer therapy. Homologous recombination (HR) is one of the major repair pathways for DSB and is thus a potential target of cancer therapy. Cells with a defect in HR have been shown to be sensitive to a variety of DNA‐damaging agents, particularly interstrand crosslink (ICL)‐inducing agents such as mitomycin C and cisplatin. These findings have recently been applied to clinical studies of cancer therapy. ERCC1, a structure‐specific endonuclease involved in nucleotide excision repair (NER) and HR, confers resistance to cisplatin. Patients with ERCC1‐negative non‐small‐cell lung cancer were shown to benefit from adjuvant cisplatin‐based chemotherapy. Imatinib, an inhibitor of the c‐Abl kinase, has been investigated as a sensitizer in DNA‐damaging therapy, because c‐Abl activates Rad51, which plays a key role in HR. Furthermore, proteins involved in HR have been shown to repair DNA damage induced by a variety of other chemotherapeutic agents, including camptothecin and gemcitabine. These findings highlight the importance of HR machinery in cancer therapy. (Cancer Sci 2008; 99: 187–194)
DNA‐damaging chemotherapeutic drugs and ionizing radiation induce a variety of DNA lesions in cancer cells as well as in normal cells. Among these lesions, DNA double‐strand breaks (DSB) are the most serious, and they eventually lead to cell death unless properly repaired. DSB are repaired by non‐homologous end‐joining (NHEJ), homologous recombination (HR), and single‐strand annealing (SSA).( 1 ) NHEJ rejoins DSB by directly ligating the broken DNA ends, consequently generating small deletions or mutations. SSA repairs DSB by annealing complementary DNA on both sites of the broken DNA, resulting in the loss of a repeat and the DNA sequence between the repeats. HR repairs DSB by using the sister chromatid or homologous chromosome to ensure accurate repair for the maintenance of genome stability.
Non‐homologous end‐joining is a main pathway in the repair of DSB that are induced by ionizing radiation in mammals. DNA‐dependent protein kinase (DNA‐PK), consisting of the catalytic subunit DNA‐PKcs and the DNA binding complex Ku70/80, plays a key role in NHEJ. The DNA ligase IV‐XRCC4 complex re‐ligates the broken DNA ends. Artemis processes complex DNA ends prior to repair. Cells deficient in the NHEJ pathway show extreme sensitivity to ionizing radiation, suggesting that specific inhibitors of NHEJ may be used as radio‐sensitizers.
In contrast to NHEJ, the HR machinery consists of complex pathways.( 2 ) The MRN complex (Mre11‐Rad50‐NBS1) recognizes DSB and resects the 5′ ends at break sites, followed by an ordered assembly with replication protein A (RPA), Rad52, Rad51, and Rad54. Rad51, the central player at early stages of HR, forms a nucleoprotein filament on single‐strand DNA and catalyzes homologous DNA pairing and strand exchange. The complexity of the HR machinery in higher eukaryotes stems partly from the functional significance of Rad51 paralogs. Five members (Rad51B, Rad51C, Rad51D, XRCC2, and XRCC3), sharing 20–30% sequence identity with Rad51, form two protein complexes, Rad51B‐Rad51C‐Rad51D‐XRCC2 and Rad51C‐XRCC3. The proteins’ biochemical properties and biological functions reveal that they are involved in HR by assisting Rad51 function. In addition to its role in the early stages of HR, Rad51C and/or its associated proteins were shown to play a role at the late stages of HR by resolving Holliday junctions, which are four‐way branched intermediates critical for crossover that are formed after strand exchange mediated by the Rad51 assembly. Like Rad51 paralogs, Rad54 is a multifunctional protein that is involved in several steps of HR by assisting Rad51 function, promoting Holliday junction migration, and remodeling chromatin. In addition to these critical proteins, many accessory proteins are involved in HR. Thus, HR is a complex process required for the repair of DSB after exposure to DNA‐damaging agents.
Nucleotide excision repair and homologous recombination act together to repair interstrand crosslinks
Interstrand crosslinking (ICL) agents such as mitomycin C and cisplatin form DNA adducts, which are repaired by a combination of nucleotide excision repair (NER) and HR.( 3 ) Several NER proteins were identified from patients with xeroderma pigmentosum (XP), characterized by hypersensitivity to ultraviolet (UV) light and a high risk of skin cancer. Among these, XP complementation group F (XPF) forms a heterodimer with ERCC1, and the XPF/ERCC1 complex serves as a structure‐specific endonuclease to remove 3′ ends (Fig. 1). Cells with ERCC1 mutation show hypersensitivity to ICL agents. Accordingly, high levels of ERCC1 in cancers confer resistance to platinum‐based chemotherapy, whereas low levels of ERCC1 are well correlated with favorable responses of cancers to the therapy.( 4 , 5 , 6 ) These findings led to a clinical study that investigated the association between ERCC1 expression levels in cancer tissues and the response to cisplatin‐based adjuvant chemotherapy in surgically treated non‐small‐cell lung cancer (NSCLC) patients.( 7 ) Patients with ERCC1‐negative tumors were shown to benefit from cisplatin‐based adjuvant chemotherapy, whereas patients with ERCC1‐positive tumors did not. Furthermore, ERCC1 polymorphisms, codon 118C/T and C8092A, were shown to be associated with a response to platinum‐based chemotherapy.( 8 , 9 )
Figure 1.
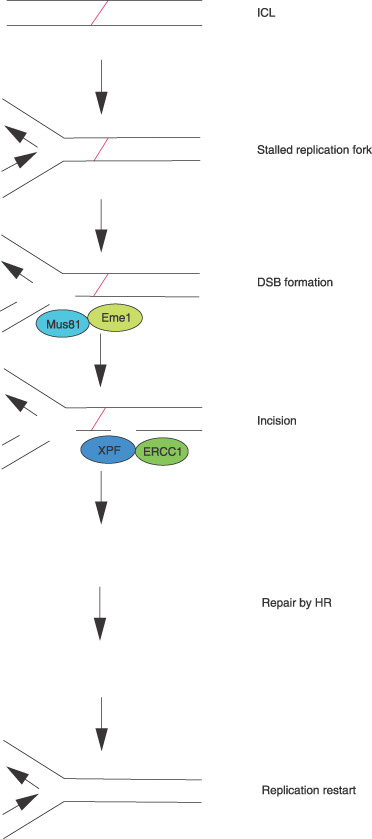
Proposed model of interstrand crosslink (ICL) repair in mammalian cells. A replication fork, if it encounters ICL, is stalled, and a double‐strand break (DSB) is generated by Mus81‐Eme1.( 31 ) Subsequently, a‐3′ end near the ICL is incised by XPF‐ERCC1, resulting in the release of the ICL from one of the strands. The DNA lesion may be bypassed by translesional synthesis and is eventually repaired by homologous recombination (HR) after the ICL is excised.( 3 )
The significance of low levels of ERCC1 in sensitization to ICL agents also contributes to the combination therapy of ICL agents with drugs that reduce ERCC1 levels. Fludarabine, a purine nucleotide analog, is used for the treatment of chronic lymphocytic leukemia (CLL) and has been shown to reduce ERCC1 levels.( 10 ) The reduction in ERCC1 levels was well correlated with the degree of synergy between fludarabine and the DNA minor groove ICL agent SJG‐136, indicating the clinical use of the combination of these drugs in fludarabine‐resistant CLL.
Although the clinical evaluation of ERCC1 as a biomarker of platinum resistance has been successful so far, it is apparent that platinum sensitivity can not be satisfactorily predicted by ERCC1 measurement alone. Potential candidate markers would emerge once the role of ERCC1 in ICL repair is understood. Targeted gene replacement was drastically impaired in ERCC1‐null mouse embryonic stem (ES) cells, defining a novel role for ERCC1 in HR.( 11 ) The DNA adducts formed by ICL agents are converted to DSB (Fig. 1), leading to a hypothesis that proteins involved in HR may be predictable markers of platinum resistance. Consistent with its role in the repair of ICL, Rad51 has been shown to be a good marker of cisplatin resistance in NSCLC.( 12 ) This is supported by a function analysis showing that overexpression of Rad51 is well correlated with DNA damage resistance.( 13 )
To identify a gene responsible for regulating chemotherapeutic drug sensitivity, targeted disruption of genes of interest has proven to be a powerful tool. The roles of the genes involved in HR in the regulation of sensitivity to DNA‐damaging agents have been extensively examined in the chicken B cell line DT40, which is highly proficient in gene targeting.( 14 , 15 ) In accord with previous studies in knockout mouse demonstrating the early embryonic lethality of the Rad51 mutation, Rad51 is essential for cell viability in DT40 cells.( 14 ) Subsequently, DT40 cells deficient in Rad51B, Rad51C, Rad51D, XRCC2, or XRCC3 were generated and exposed to several DNA‐damaging agents.( 15 ) The most dramatic hypersensitivity was found in cells treated with ICL agents, suggesting that Rad51 paralogs play a critical role in ICL repair. These findings were also supported by studies in Chinese hamster ovary (CHO) cells defective in XRCC2, XRCC3, or Rad51C.( 16 ) Further evidence for the role of Rad51 paralogs in ICL repair came from the identification of XRCC3′s role in melphalan resistance.( 17 ) This drug's cytotoxic effects are thought to be exerted by alkylation and ICL.
The p53 protein is defective in DT40 and CHO cells, in which the functions of Rad51 paralogs were examined. In order to understand the roles of Rad51 paralogs in human cancer cells with intact p53, we have used the colon cancer cell line HCT116, because targeted recombination is more proficient in this cell line than in other human cells.( 18 , 19 ) A defect in mismatch repair (MMR) due to MLH1 mutation in HCT116 emphasizes the importance of this cell line from the viewpoint of clinical oncology, because MMR is often defective in human colorectal cancers. In addition, because MMR status was shown to affect sensitivity to cisplatin, HCT116 provides an optimal tool for understanding the mechanisms underlying the drug sensitivity without the effect of the MMR pathways.( 20 ) Consistent with studies in other species, mutations in XRCC3 or Rad51B caused hypersensitivity to mitomycin C and cisplatin, indicating that Rad51 paralogs play a critical role in ICL repair in human cancers with defective MMR even in the presence of intact p53.( 18 , 19 )
Like changes in expression levels, genetic polymorphisms are likely to affect sensitivity to ICL agents. XRCC2 R188H was shown to induce resistance to ICL agents in DT40 cells, suggesting that individuals with this variation may tolerate cisplatin treatment.( 21 ) Unlike XRCC2 R188H, XRCC3 M241T does not affect sensitivity to DNA‐damaging agents, although an association between this variation and cancer risk has been proposed. Instead, the XRCC3 variation seems to be associated with aneuploidy.( 18 ) Thus, each genetic variation in the ICL repair genes is likely to have a distinct role.
In addition to proteins involved in the basal processes of HR, accessory proteins also play critical roles in the regulation of sensitivity to ICL agents. The BLM protein, whose mutation is responsible for Bloom syndrome, is also involved in HR by catalyzing branch migration of Holliday junctions. BLM expression is upregulated in chronic myelogenous leukemia (CML), in which BCR/ABL plays a causal role. The BCR/ABL protein was shown to promote the interaction between BLM and Rad51 to induce cisplatin resistance in CML.( 22 )
The physical interaction between BRCA2 and Rad51 suggests that a defect in HR is involved in the genesis of hereditary breast cancers.( 23 ) This was supported by the findings that BRCA1 and BRCA2 play roles in HR. BRCA1 expression is upregulated in cisplatin‐resistant breast cancer cell lines.( 24 ) BRCA1 promotes cisplatin resistance in association with Rad51.( 25 ) From a clinical point of view, BRCA1‐negative ovarian cancer patients who received cisplatin‐based chemotherapy after surgical resection have significantly longer median survival times and disease‐free intervals than do age‐ and treatment‐course‐matched controls.( 26 ) Similarly, lung cancer patients with low levels of BRCA1 benefit from cisplatin‐based neoadjuvant chemotherapy more so than patients with high levels of BRCA1.( 27 ) These data clearly indicate that BRCA1 expression can be used as a predictor of cisplatin resistance. Based on a similar hypothesis, a clinical trial comparing the efficacy between carboplatin and the microtubule poison docetaxel is underway to assess the benefits of ICL agents in patients with metastatic breast cancer harboring BRCA1 or BRCA2 mutations.( 23 )
The Fanconi anemia‐BRCA (FA‐BRCA) pathway is also involved in cisplatin resistance. A protein complex consisting of FANC proteins (A, B, C, E, F, G, L, M) monoubiquitinates FANCD2.( 28 ) Subsequently, FANCD2 interacts with FANCD1/BRCA2, which is required for HR. FANCI is also monoubiquitinated and associates with FANCD2. FANCN/PALB2 is a binding partner of FANCD1/BRCA2 and stabilizes BRCA2. FANCJ/BRIP1 associates with BRCA1. In addition to BRCA1 and BRCA2, BRIP1 and PALB2 have been shown to be hereditary breast cancer susceptibility proteins. Furthermore, the FANC/BRCA pathway was shown to be defective in cisplatin‐sensitive ovarian cancers.( 29 ) These findings led to the idea that inhibitors of the FANC/BRCA pathway may sensitize cancer cells to cisplatin. Based on this idea, small‐molecule inhibitors of the FANC/BRCA pathway are under investigation.( 30 )
Recent evidence suggests that ICL is converted to DSB by the Mus81‐Eme1 complex (Fig. 1).( 31 ) Mus81 was identified as a member of the XPF family of endonucleases, sharing an active motif, VERKxxxD (Fig. 2).( 32 ) Like the XPF‐ERCC1 complex, Mus81 forms a heterodimeric structure‐specific endonuclease by interacting with Eme1.( 33 ) Mus81‐Eme1 resembles XPF‐ERCC1, because only one partner possesses endonuclease activity. In yeast, mus81 and eme1 mutants demonstrate hypersensitivity to UV radiation, methylmethane sulfonate, hydroxyurea, and camptothecin, but not to ionizing radiation.( 32 , 34 ) In contrast, Mus81‐ or Eme1‐defective murine cells are hypersensitive to mitomycin C and cisplatin, suggesting that Mus81‐Eme1 plays a role in ICL repair in mammals.( 35 ) We observed that the haplo‐insufficiency of either Mus81 or Eme1 led to hypersensitivity to mitomycin C and cisplatin but not to other DNA‐damaging agents in HCT116.( 36 ) Thus, like XPF‐ERCC1, Mus81‐Eme1 might be a potential target of cisplatin‐based chemotherapy.
Figure 2.
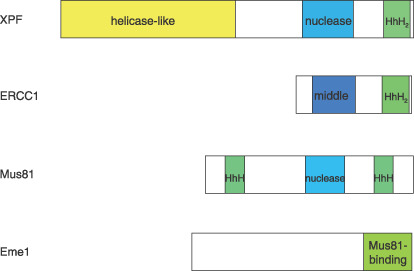
Schematic representation of the protein structure of members of the xeroderma pigmentosum group F (XPF) nuclease family in mammals.( 32 , 33 ) Interactions of the subunits are mediated by C‐terminal helix‐hairpin‐helix (HhH) domains. Both XPF and ERCC1 contain C‐terminal tandem HhH domains (HhH2). XPF contains an N‐terminal helicase‐like domain, whereas Mus81 does an N‐terminal HhH domain instead. Both XPF and Mus81 contain the nuclease domains harboring conserved metal‐binding residues of a VERKxxxD motif. Remnants of nuclease domains are present in the middle domain of ERCC1.
The expression levels of the proteins described here have been extensively examined in primary cancer tissues (Table 1). To promote individualized platinum‐based chemotherapy, an understanding of the biological basis of the response to ICL agents, together with information on the expression patterns of proteins associated with the HR machinery in cancer, will be greatly helpful.
Table 1.
Altered expression levels of molecules involved in homologous recombination in primary sporadic cancer tissues
| Gene/protein | Increased expression | Decreased expression | References |
|---|---|---|---|
| Rad51 | Pancreatic cancer | Colorectal cancer | ( 12, 13, 56, 70, 71, 72, 73 ) |
| Breast cancer | Breast cancer | ||
| Non‐small‐cell lung cancer | |||
| Head and neck cancer | |||
| Soft tissue sarcoma | |||
| Rad51C | Breast cancer | ( 74 ) | |
| Rad52 | Colorectal cancer | ( 72 ) | |
| Brca1 | Lung cancer | Breast cancer | ( 23, 27, 73 ) |
| Ovarian cancer | |||
| Lung cancer | |||
| Brca2 | Ovarian cancer | ( 23, 75 ) | |
| Mre11 | Breast cancer | ( 66, 67 ) | |
| Colorectal cancer | |||
| Rad50 | Breast cancer | ( 66 ) | |
| NBS1 | Head and neck cancer | Breast cancer | ( 66, 67, 76, 77 ) |
| Melanoma | Colorectal cancer | ||
| Melanoma | |||
| BLM | Chronic myelogenous leukemia | ( 22, 78 ) | |
| Lymphoma | |||
| Breast cancer | |||
| Colon cancer | |||
| Lung cancer | |||
| Renal cell cancer | |||
| Seminoma | |||
| WRN | Chronic myelogenous leukemia | Gastric cancer | ( 22, 49 ) |
| Colorectal cancer | |||
| FANCF | Ovarian cancer | ( 79, 80 ) | |
| Cervical cancer | |||
| ERCC1 | Non‐small‐cell lung cancer | Non‐small‐cell lung cancer | ( 4, 5, 6, 7, 12 ) |
| Ovarian cancer | Gastric cancer | ||
| Colorectal cancer | Colorectal cancer |
Expression has been confirmed at mRNA and/or protein levels. Studies using cultured cancer cells are excluded. Cancers in hereditary chromosome instability syndromes and familial cancers are also excluded. Only molecules whose main functions are associated with homologous recombination are listed.
Tyrosine kinase inhibitors as sensitizers to chemotherapy and radiotherapy
After ATM is activated in response to DNA damage induced by ionizing radiation, Rad51 is phosphorylated by the c‐Abl kinase.( 37 ) A fusion tyrosine kinase BCR/ABL, arising from chromosomal translocation in CML, has been shown to enhance the Rad51 level through STAT5‐dependent transcriptional control, although the precise molecular mechanism underlying this enhancement remains to be demonstrated (Fig. 3).( 38 ) Similarly, other fusion tyrosine kinases, such as TEL/ABL, TEL/JAK2, TEL/PDGFβR, and NPM/ALK, enhance Rad51 levels in a STAT5‐dependent manner.( 39 ) Imatinib, an inhibitor of these tyrosine kinases, is now used in the treatment of BCR/ABL‐positive leukemias and c‐Kit‐positive gastro‐intestinal stromal tumors. Since BCR/ABL enhances resistance to cisplatin and mitomycin C by promoting the HR activity mediated by Rad51 and its associated proteins, inhibitors of c‐Abl such as imatinib are expected to sensitize cancer cells to DNA‐damaging agents. Consistent with this hypothesis, imatinib treatment enhanced sensitivity to cisplatin and mitomycin C in BCR/ABL‐expressing myeloid cells.( 38 ) From a mechanistic point of view, it is interesting to note that, in addition to Rad51, c‐Abl also regulates Rad52 activity.( 40 ) Thus, inhibitors of c‐Abl can be used as sensitizers to cisplatin‐based chemotherapy.
Figure 3.
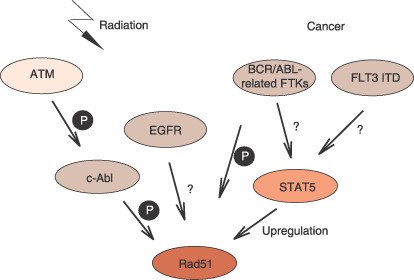
Regulation of Rad51 by tyrosine kinases. When DNA is damaged by ionizing radiation, ATM activates c‐Abl, whereby Rad51 is phosphorylated.( 37 ) epidermal growth factor receptor (EGFR) upregulates Rad51 in response to ionizing radiation by an unknown mechanism.( 45 ) BCR/ABL‐related fusion tyrosine kinases (FTKs) arising from chromosomal translocation in cancer and FLT3 ITD mutations activate the transcriptional activator STAT5, which upregulates Rad51.( 38 , 39 , 43 , 44 ) BCR/ABL also phosphorylates Rad51.( 38 )
The HR pathway also regulates sensitivity to radiotherapy. When glioma cells were treated with imatinib, they became modestly sensitive to ionizing radiation, whereas normal fibroblasts did not.( 41 ) The mechanism of this sensitization was explained by the finding that Rad51 levels were reduced by imatinib treatment. Similarly, imatinib treatment was shown to sensitize primary CLL cells to chlorambucil by reducing damage‐induced Rad51 activation.( 42 ) These data suggest that c‐Abl inhibitors can sensitize tumor cells to DNA‐damaging therapies whose sensitivity is regulated by the HR pathway.
FLT3 (fms‐like tyrosine kinase 3) is activated in about 30% of acute myeloid leukemia (AML) cases. Internal tandem duplication (ITD) mutations in FLT3 are associated with the risk of relapse in AML.( 43 ) Some patients with AML have dual mutations of ITD and the tyrosine kinase domain. The dual mutations induce resistance to FLT3 inhibitors and chemotherapeutic agents. The mechanism underlying the resistance was shown to be mediated by STAT5 activation, leading to upregulation of Bcl‐x(L) and Rad51. Another study has shown that the FLT3 inhibitor PKC412 and the silencing of FLT3 by RNA interference repress Rad51 in cells with FLT3‐ITD mutations but not in cells with intact FLT3. These data suggest that Rad51‐mediated HR activity contributes to resistance to therapy in AML with FLT ITD mutations.( 44 )
Accumulating evidence suggests that concurrent radiotherapy with epidermal growth factor receptor (EGFR) inhibitors provides a survival benefit in a variety of cancers, such as those of the lung, head, and neck. The EGFR inhibitor Erlotinib was shown to inhibit radiation‐dependent activation of Rad51, indicating that repressed Rad51 contributes to the effect of the concurrent therapy.( 45 )
Thus, some tyrosine kinase inhibitors may not only inhibit growth‐promoting signals but also overcome resistance to chemotherapy and radiotherapy by downregulating the HR pathways mediated by Rad51 and its associated proteins.
Werner protein regulates sensitivity to camptothecin
Topoisomerase I (Top1) is essential for unwinding supercoiled DNA structures for DNA metabolic functions, such as transcription, replication, and repair.( 46 ) Camptothecin, a Top1 inhibitor, is frequently used in cancer therapy. However, symptoms caused by its toxicity, in particular diarrhea, are severe in some individuals, so a marker of sensitivity to the drug is needed. Single‐strand breaks (SSB), which are normally transient, are stabilized by the interaction of camptothecin with Top1. These SSB are converted to DSB during replication, triggering the HR repair pathway. Furthermore, a recent study using single‐molecule nanomanipulation revealed an unexpected novel role of topotecan, an analog of camptothecin, in impeding Top1‐dependent uncoiling.( 47 ) This finding suggests that topotecan induces DNA damage by the accumulation of positive supercoils during replication.
The Werner protein (WRN) is a member of the RecQ helicase superfamily with helicase and exonuclease activities. WRN mutations are responsible for the premature aging Werner syndrome, characterized by genome instability, atherosclerosis, myocardial infarction, and cancer predisposition. Cells derived from Werner syndrome patients and WRN‐deficient murine cells are hypersensitive to DNA‐damaging agents, particularly camptothecin.( 48 ) In some human cancer cells, WRN expression is repressed by CpG island hypermethylation.( 49 ) These cells are more sensitive to camptothecin than hypomethylated cells are. The introduction of the exogenous WRN gene in hypermethylated cells abolished the hypersensitivity. These findings led to a hypothesis that WRN may be a predictable marker of camptothecin‐based chemotherapy. Irinotecan (CPT‐11), a camptothecin analog, has been used in the treatment of colon cancers, in which CpG island hypermethylation frequently silences WRN. In one retrospective study assessing the survival of patients treated with irinotecan, the median survival time of patients with WRN‐hypermethylated cancers was significantly longer than that of patients with unmethylated cancers.( 49 ) This suggests the possibility that low levels of WRN in cancer cells may predict a good response to irinotecan‐based therapy.
Since Top1 inhibition results in DSB, in addition to WRN, proteins regulating the HR repair pathway are likely to play a role in the regulation of sensitivity to camptothecin. This idea is also supported by the finding that WRN physically associates with Rad51, Rad54, and Rad54B.( 50 ) However, the roles of these proteins in the regulation of sensitivity to camptothecin are complicated, unlike those to other DNA‐damaging agents. Impairment of BLM, another member of the RecQ helicase superfamily, led to camptothecin resistance in mouse ES cells.( 51 ) In contrast, deletion of BRCA2, like that of Rad54, led to hypersensitivity to camptothecin in the same mouse ES cells, suggesting that the role of BLM is different from those of BRCA2 and Rad54 in terms of sensitivity to camptothecin. Furthermore, there appear to be functional differences among Rad51 paralogs in the regulation of sensitivity to this drug. Each mutation of XRCC2, XRCC3, or Rad51C led to hypersensitivity to camptothecin in CHO cells. However, a mutation of XRCC3 exhibited the most drastic sensitivity to camptothecin without rapid induction of apoptosis, while a mutation of XRCC2 or Rad51C showed milder sensitivity to the drug, with rapid induction of apoptosis.( 52 ) This finding suggests that XRCC3 plays a role in the induction of apoptosis in response to camptothecin‐induced DNA damage. Thus, the mechanisms underlying sensitivity to camptothecin are regulated by complex pathways associated with the HR machinery.
Roles of homologous recombination in regulation of sensitivity to topoisomerase II inhibitors
Etoposide, an inhibitor of topoisomerase II (Top2), is a key drug in the treatment of small‐cell lung cancer (SCLC). However, sensitivity to etoposide is highly variable among SCLC cell lines. Because CHO mutant cells exhibiting defective HR were shown to be hypersensitive to etoposide, Rad51 levels were examined in SCLC cell lines.( 53 ) Rad51 levels were well correlated with resistance to etoposide.( 54 ) The introduction of the exogenous Rad51 gene in etoposide‐sensitive SCLC cells with a low level of Rad51 conferred resistance to etoposide. Conversely, the introduction of the antisense Rad51 gene in etoposide‐resistant SCLC cells with a high level of Rad51 conferred sensitivity to the drug. Thus, Rad51 appears to be involved in the regulation of sensitivity to etoposide in SCLC cells.
The role of the HR repair pathway in the regulation of sensitivity to etoposide was also revealed from studies in CHO mutant cells and DT40 cells. The genes that complement etoposide‐hypersensitive CHO mutants turned out to be XRCC2 and XRCC3.( 53 ) Although other proteins involved in the HR machinery, such as Rad54 and BLM, do not appear to play a role in the regulation of etoposide sensitivity, Rad51 and its paralogs may be potential targets of etoposide‐based chemotherapy.( 51 , 53 ) Unlike yeast Rad52, vertebrate Rad52 was assumed to exhibit no distinct effects on sensitivity to DNA‐damaging agents. However, the loss of Rad52 in DT40 cells increased sensitivity to etoposide, indicating a novel role for Rad52 in the repair of etoposide‐induced DNA damage.( 55 )
Despite the successful development of new chemotherapeutic agents, doxorubicin, a Top2 inhibitor, still plays a key role in the treatment of cancers such as lymphoma, sarcoma and breast cancer. The Rad51 level is increased in response to DNA damage induced by doxorubicin in soft tissue sarcoma (STS) cells.( 56 ) The silencing of Rad51 by RNA interference resulted in increased sensitivity to doxorubicin in STS cells. Rad51 overexpression has been found in a variety of cancers (Table 1). Overexpressed Rad51 may be therefore a potential target of doxorubicin‐based chemotherapy. Because Rad51 expression was shown to be negatively regulated by p53, p53 mutations frequently found in cancers may induce resistance to doxorubicin by Rad51 overexpression.( 56 )
Although Rad51's role in the regulation of sensitivity to doxorubicin in STS cells was clearly demonstrated, that in breast cancer cells appears to be rather complicated.( 57 ) Doxorubicin induced Rad51 expression in one breast cancer cell line and in normal breast cells, but reduced expression in another breast cancer cell line. The expression of Top2, the target of doxorubicin, was increased in all cell lines tested; suggesting that Rad51's role in the regulation of sensitivity to doxorubicin is dependent on the genetic background of cells. It is therefore highly likely that a complex pathway regulates sensitivity to the drug. Nevertheless, the HR repair pathway may be a potential target of doxorubicin in some cancer cells.
Roles of homologous recombination in regulation of sensitivity to replication inhibitors
Gemcitabine, 2′,2′‐difluorodeoxycytidine (dFdC), is a deoxycytidine analog that has antitumor activity against a broad spectrum of cancers by inhibiting DNA synthesis. Studies in cultured cells and mice have demonstrated cytotoxic synergy between gemcitabine and cisplatin, supporting the clinical use of the combination of these drugs.( 58 ) Because ERCC1 was shown to be involved in the regulation of sensitivity to cisplatin, the possible involvement of ERCC1 in the synergy has been investigated. ERCC1 repression in MMR‐deficient colon cancer cells abrogated the synergy, suggesting that gemcitabine‐mediated inhibition of ERCC1 activity plays a role in the drug's synergy with cisplatin.( 58 ) This finding supports the clinical evidence that the combination of gemcitabine with cisplatin was effective in some patients with ovarian cancers that had already been refractory to cisplatin‐based chemotherapy.( 59 )
Genetic polymorphisms in DNA repair genes have been proposed to affect the clinical outcome of gemcitabine‐based chemotherapy. The median survival time was significantly longer for NSCLC patients treated with gemcitabine and cisplatin harboring XRCC3 241MetMet than for patients with ThrMet or ThrThr.( 60 ) Because this variation was shown to have no effect on sensitivity to cisplatin in CHO and HCT116 cells, the biological significance of XRCC3 T241M is likely to be associated with the outcome of gemcitabine‐based therapy.( 18 ) Another study revealed that RecQ1 A159C, Rad54 C157T, XRCC1 R194W, and ATM T77C genotypes affected the overall survival of patients with pancreatic cancer treated with neoadjuvant concurrent gemcitabine and radiotherapy.( 61 ) It should be noted that Rad54 C157T is a silent polymorphism, suggesting that this variation's effect is associated with linkage disequilibrium with other polymorphisms. The median survival time was significantly longer for patients with none of the adverse genotypes than for those with one or more at‐risk alleles. Rad54 plays an essential role in HR. ATM functions as a sensor of DNA damages to promote DNA repair, cell‐cycle regulation, and apoptosis. RecQ1, a member of the RecQ helicase superfamily, was shown to interact with MMR factors that regulate HR. XRCC1 plays a critical role in base excision repair. These findings strongly suggest that variations in DNA repair activity, including HR, affect the clinical outcome of patients treated with concurrent gemcitabine and radiotherapy.
Despite the development of new drugs effective for leukemias, hydroxyurea, which is an inhibitor of ribonucleotide reductase, is still used for the treatment of myeloproliferative disorders. Inhibition of this enzyme reduces a pool of deoxyribonucleotide triphosphates, leading to replication block. Stalled replication forks induced by hydroxyurea ultimately generate DSB, which can be repaired by HR. CHO cells overexpressing Rad51 are resistant to hydroxyurea, suggesting that Rad51‐dependent HR plays a role in the repair of replication block‐induced DSB.( 53 ) Similarly, XRCC2 is involved in the repair of hydroxyurea‐induced damages at stalled replication forks, suggesting that HR repairs replication block‐induced DSB.( 62 ) However, unexpectedly, BLM‐deficient mouse ES cells retaining about 10% levels of endogenous BLM protein exhibited resistance to hydroxyurea, whereas sensitivity to hydroxyurea in human cells derived from Bloom syndrome patients is not different from that in normal cells.( 51 ) Thus, the amount of BLM protein appears to play a critical role in the regulation of sensitivity to hydroxyurea.
The MRN complex and radiotherapy
NBS1 is mutated in Nijmegen breakage syndrome, which is characterized by hypersensitivity to ionizing radiation, cancer predisposition, microcephaly, and immunodeficiency.( 63 ) The hypomorphic mutation of Mre11 is responsible for ataxia‐telangiectasia (AT)‐like disorder (ATLD).( 64 ) Patients with AT or ATLD are extremely sensitive to ionizing radiation. The murine ES cells harboring Rad50 mutation exhibit hypersensitivity to ionizing radiation.( 65 ) These findings lead to a hypothesis that the MRN complex may be a potential target of radiotherapy. The expression levels of the complex are reduced in a variety of cancers (Table 1).( 66 , 67 ) However, the association between a favorable outcome of radiotherapy and reduced levels of the MRN complex in cancer remains to be demonstrated. There is a paradoxical report showing that an intact level of the MRN complex is a predictable marker of good response to adjuvant radiotherapy in early breast cancer, inconsistent with the role of the complex in cultured cells.( 68 ) Further studies are therefore needed to determine the role of the MRN complex in radiotherapy.
Upregulation of Rad51 may be associated with the risk of therapy‐related leukemia
Therapy‐related acute myeloid leukemia (t‐AML) is a devastating complication of chemotherapy and/or radiotherapy for a primary cancer. The risk of the development of t‐AML was found to be associated with the G‐to‐C polymorphism at –135 of the 5′ untranslated region (135G/C‐5′UTR) of Rad51 and XRCC3 T241M.( 69 ) The promoter activity of the Rad51 gene is enhanced by the G‐to‐C substitution (135G/C‐5′UTR), resulting in high levels of Rad51 expression in individuals with the variation. Rad51′s role in t‐AML was also supported by an indirect finding that Rad51 was upregulated in MMR‐deficient murine ES cells. This process can be recapitulated by treatment with alkylating agents. MMR deficiency has been proposed to play an early role in therapy‐related carcinogenesis. These data indicate that high levels of Rad51 not only confer resistance to DNA‐damaging agents but also contribute to the development of therapy‐related cancers. Unlike the Rad51 polymorphism, XRCC3 T241M is unlikely to upregulate the protein level. Since we have shown that this variation promotes the formation of tetraploidy, aneuploidy may play a causal role in t‐AML associated with XRCC3 T241M.( 18 )
Conclusion
The HR machinery plays critical roles in the regulation of sensitivity to the majority of chemotherapeutic drugs currently used in cancer therapy. The multifunctional DNA repair protein ERCC1 has provided a paradigm for the clinical application of basic knowledge on the mechanisms of DNA repair. This success can be followed by the application of other information regarding the proteins discussed in this review as well as novel proteins characterized in the future. Furthermore, in addition to conventional therapeutics, a novel therapeutic strategy with the targeted inhibition of particular DNA repair pathways represents a new concept in cancer therapy. BRCA1 or BRCA2 dysfunction was shown to sensitize cells to the inhibition of poly(ADP‐ribose) polymerase 1 (PARP1) activity.( 23 ) PARP1 is involved in base excision repair, which plays a critical role in the repair of SSB. The inhibition of PARP1 increases the number of SSB, leading to DSB that can be repaired by HR mediated by BRCA1 and BRCA2. Based on these findings, inhibitors of PARP1 are in the early stages of clinical trials. Thus, knowledge gained from the study of DNA repair has considerable potential to impact the development of novel targeted cancer therapies.
Acknowledgments
Research on homologous recombination in our laboratory was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and from the Ministry of Health, Labor and Welfare of Japan.
References
- 1. Helleday T, Lo J, Van Gent DC, Engelward BP. DNA double‐strand break repair: from mechanistic understanding to cancer treatment. DNA Repair 2007; 6: 923–35. [DOI] [PubMed] [Google Scholar]
- 2. Sung P, Klein H. Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nat Rev Mol Cell Biol 2006; 7: 739–50. [DOI] [PubMed] [Google Scholar]
- 3. Dronkert MLG, Kanaar R. Repair of DNA interstrand cross‐links. Mutat Res 2001; 486: 217–47. [DOI] [PubMed] [Google Scholar]
- 4. Dabholkar M, Bostick‐Bruton F, Weber C, Bohr VA, Egwuagu C, Reed E. ERCC1 and ERCC2 expression in malignant tissues from ovarian cancer patients. J Natl Cancer Inst 1992; 84: 1512–17. [DOI] [PubMed] [Google Scholar]
- 5. Shirota Y, Stoehlmacher J, Brabender J et al . ERCC1 and thymidylate synthase mRNA levels predict survival for colorectal cancer patients receiving combination oxaliplatin and fluorouracil chemotherapy. J Clin Oncol 2001; 19: 4298–304. [DOI] [PubMed] [Google Scholar]
- 6. Kwon H‐C, Roh MS, Oh SY et al . Prognostic value of expression of ERCC1, thymidylate synthase, and glutathione S‐transferase P1 for 5‐fluorouracil/oxaliplatin chemotherapy in advanced gastric cancer. Ann Oncol 2007; 18: 504–9. [DOI] [PubMed] [Google Scholar]
- 7. Olaussen KA, Dunant A, Fouret P et al . DNA repair by ERCC1 in non‐small‐cell lung cancer and cisplatin‐based adjuvant chemotherapy. N Engl J Med 2006; 355: 983–91. [DOI] [PubMed] [Google Scholar]
- 8. Zhou W, Gurubhagavatula S, Liu G et al . Excision repair cross‐complementation group 1 polymorphism predicts overall survival in advanced non‐small cell lung cancer patients treated with platinum‐based chemotherapy. Clin Cancer Res 2004; 10: 4939–43. [DOI] [PubMed] [Google Scholar]
- 9. Viguier J, Boige V, Miquel C et al . ERCC1 codon 118 polymorphism is a predictive factor for the tumor response to oxaliplatin/5‐fluorouracil combination chemotherapy in patients with advanced colorectal cancer. Clin Cancer Res 2005; 11: 6212–17. [DOI] [PubMed] [Google Scholar]
- 10. Pepper C, Lowe H, Fegan C et al . Fludarabine‐mediated suppression of the excision repair enzyme ERCC1 contributes to the cytotoxic synergy with the DNA minor groove crosslinking agent SJG‐136 (NSC 694501) in chronic lymphocytic leukaemia cells. Br J Cancer 2007; 97: 253–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Niedernhofer LJ, Essers J, Weeda G et al . The structure‐specific endonuclease Ercc1‐Xpf is required for targeted gene replacement in embryonic stem cells. EMBO J 2001; 20: 6540–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Takenaka T, Yoshino I, Kouso H et al . Combined evaluation of Rad51 and ERCC1 expressions for sensitivity to platinum agents in non‐small cell lung cancer. Int J Cancer 2007; 121: 895–900. [DOI] [PubMed] [Google Scholar]
- 13. Maacke H, Jost K, Opitz S et al . DNA repair and recombination factor Rad51 is over‐expressed in human pancreatic adenocarcinoma. Oncogene 2000; 19: 2791–5. [DOI] [PubMed] [Google Scholar]
- 14. Sonoda E, Sasaki MS, Buerstedde J‐M et al . Rad51‐deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO J 1998; 17: 598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Takata M, Sasaki MS, Tachiiri S et al . Chromosome instability and defective recombinational repair in knockout mutants of the five Rad51 paralogs. Mol Cell Biol 2001; 21: 2858–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Godthelp BC, Wiegant WW, Van Duijin‐Goedhart A et al . Mammalian Rad51C contributes to DNA cross‐link resistance, sister chromatid cohesion and genomic stability. Nucl Acids Res 2002; 30: 2172–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang Z‐M, Chen Z‐P, Xu Z‐Y et al . In vitro evidence for homologous recombinational repair in resistance to melphalan. J Natl Cancer Inst 2001; 93: 1473–8. [DOI] [PubMed] [Google Scholar]
- 18. Yoshihara T, Ishida M. Kinomura et al . XRCC3 deficiency results in a defect in recombination and increased endoreduplication in human cells. EMBO J 2004; 23: 670–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Date O, Katsura M, Ishida M et al . Haploinsufficiency of RAD51B causes centrosome fragmentation and aneuploidy in human cells. Cancer Res 2006; 66: 6018–24. [DOI] [PubMed] [Google Scholar]
- 20. Fink D, Aebi S, Howell SB. The role of DNA mismatch repair in drug resistance. Clin Cancer Res 1998; 4: 1–6. [PubMed] [Google Scholar]
- 21. Danoy P, Sonoda E, Lathrop M, Takeda S, Matsuda F. A naturally occurring genetic variant of human XRCC2 (R188H) confers increased resistance to cisplatin‐induced DNA damage. Biochem Biophys Res Commun 2007; 352: 763–8. [DOI] [PubMed] [Google Scholar]
- 22. Slupianek A, Gurdek E, Koptyra M et al . BLM helicase is activated in BCR/ABL leukemia cells to modulate responses to cisplatin. Oncogene 2005; 24: 3914–22. [DOI] [PubMed] [Google Scholar]
- 23. Turner N, Tutt A, Ashworth A. Targeting the DNA repair defect of BRCA tumours. Curr Opin Pharm 2005; 5: 388–93. [DOI] [PubMed] [Google Scholar]
- 24. Husain A, He G, Venkatraman ES, Spriggs DR. BRCA1 up‐regulation is associated with repair‐mediated resistance to cis‐diamminedichloroplatinum (II). Cancer Res 1998; 58: 1120–3. [PubMed] [Google Scholar]
- 25. Bhattacharyya A, Ear US, Koller BH, Weichselbaum RR, Bishop DK. The breast cancer susceptibility gene BRCA1 is required for subnuclear assembly of Rad51 and survival following treatment with the DNA cross‐linking agent cisplatin. J Biol Chem 2000; 275: 23 899–903. [DOI] [PubMed] [Google Scholar]
- 26. Aida H, Takakuwa K, Nagata H et al . Clinical features of ovarian cancer in Japanese women with germ‐line mutations of. Brca1 Clin Cancer Res 1998; 4: 235–40. [PubMed] [Google Scholar]
- 27. Taron M, Rosell R, Felip E et al . BRCA1 mRNA expression levels as an indicator of chemoresistance in lung cancer. Hum Mol Genet 2004; 13: 2443–9. [DOI] [PubMed] [Google Scholar]
- 28. Grompe M, Van De Vrugt H. The Fanconi family adds a fraternal twin. Dev Cell 2007; 12: 661–2. [DOI] [PubMed] [Google Scholar]
- 29. Taniguchi T, Tischkowitz M, Ameziane N et al . Disruption of the Fanconi anemia‐BRCA pathway in cisplatin‐sensitive ovarian tumors. Nature Med 2003; 9: 568–74. [DOI] [PubMed] [Google Scholar]
- 30. Chirnomas D, Taniguchi T, De La Vega M et al . Chemosensitization to cisplatin by inhibitors of the Fanconi anemia/BRCA pathway. Mol Cancer Ther 2006; 5: 952–61. [DOI] [PubMed] [Google Scholar]
- 31. Hanada K, Budzowska M, Modesti M et al . The structure‐specific endonuclease Mus81‐Eme1 promotes conversion of interstrand DNA crosslinks into double‐strands breaks. EMBO J 2006; 25: 4921–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Interthal H, Heyer W‐D. MUS81 encodes a novel Helix‐hairpin‐Helix protein involved in the response to UV‐ and methylation‐induced DNA damage in Saccharomyces cerevisiae . Mol General Genet 2000; 263: 812–27. [DOI] [PubMed] [Google Scholar]
- 33. Boddy MN, Gaillard P‐HL, McDonald WH, Shanahan P, Yate 3rd JR, Russell P. Mus81‐Eme1 are essential components of a Holliday junction resolvase. Cell 2001; 107: 537–48. [DOI] [PubMed] [Google Scholar]
- 34. Doe CL, Ahn JS, Dixon J, Whitby MC. Mus81‐Eme1 and Rqh1 involvement in processing stalled and collapsed replication forks. J Biol Chem 2002; 277: 32 753–9. [DOI] [PubMed] [Google Scholar]
- 35. Dendouga N, Gao H, Moechars D, Janicot M, Vialard J, McGowan CH. Disruption of murine Mus81 increases genomic instability and DNA damage sensitivity but does not promote tumorigenesis. Mol Cell Biol 2005; 25: 7569–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hiyama T, Katsura M, Yoshihara T et al . Haploinsufficiency of the Mus81‐Eme1 endonuclease activates the intra‐S‐phase and G2/M checkpoints and promotes rereplication in human cells. Nucl Acids Res 2006; 34: 880–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen G, Yuan S‐SF, Liu W et al . Radiation‐induced assembly of Rad51 and Rad52 recombination complex requires ATM and c‐Abl. J Biol Chem 1999; 274: 12 748–52. [DOI] [PubMed] [Google Scholar]
- 38. Slupianek A, Schmutte C, Tombline G et al . BCR/ABL regulates mammalian RecA homologs, resulting in drug resistance. Mol Cell 2001; 8: 795–806. [DOI] [PubMed] [Google Scholar]
- 39. Slupianek A, Hoser G, Majsterek I et al . Fusion tyrosine kinases induce drug resistance by stimulation of homology‐dependent recombination repair, prolongation of G2/M phase, and protection from apoptosis. Mol Cell Biol 2002; 22: 4189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kitao H, Yuan Z‐M. Regulation of ionizing radiation‐induced Rad52 nuclear foci formation by c‐Abl‐mediated phosphorylation. J Biol Chem 2002; 277: 48 944–8. [DOI] [PubMed] [Google Scholar]
- 41. Russell JS, Brady K, Burgan WE et al . Gleevec‐mediated inhibition of Rad51 expression and enhancement of tumor cell radiosensitivity. Cancer Res 2003; 63: 7377–83. [PubMed] [Google Scholar]
- 42. Aloyz R, Grzywacz K, Xu Z‐Y, Loignon M, Alaoui‐Jamali MA, Panasci L. Imatinib sensitizes CLL lymphocytes to chlorambucil. Leukemia 2004; 18: 409–14. [DOI] [PubMed] [Google Scholar]
- 43. Bagrintseva K, Geisenhof S, Kern R et al . FLT3‐ITD‐TKD dual mutants associated with AML confer resistance to FLT3 PTK inhibitors and cytotoxic agents by overexpression of Bcl‐x (L). Blood 2005; 105: 3679–85. [DOI] [PubMed] [Google Scholar]
- 44. Seedhouse CH, Hunter HM, Lloyd‐Lewis B et al . DNA repair contributes to the drug‐resistant phenotype of primary acute myeloid leukaemia cells with FLT3 internal tandem duplications and is reversed by the FLT3 inhibitor PKC412. Leukemia 2006; 20: 2130–6. [DOI] [PubMed] [Google Scholar]
- 45. Chinnaiyan P, Huang S, Vallabhaneni G et al . Mechanisms of enhanced radiation response following epidermal growth factor receptor signaling inhibition by Erlotinib (Tarceva). Cancer Res 2005; 65: 3328–35. [DOI] [PubMed] [Google Scholar]
- 46. Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nature Rev Cancer 2006; 6: 789–802. [DOI] [PubMed] [Google Scholar]
- 47. Koster DA, Palle K, Bot ESM, Bjornsti M‐A, Dekker NH. Antitumour drugs impede DNA uncoiling by topoisomerase I. Nature 2007; 448: 213–17. [DOI] [PubMed] [Google Scholar]
- 48. Lebel M, Leder P. A deletion within the murine Werner syndrome helicase induces sensitivity to inhibitors of topoisomerase and loss of cellular proliferative capacity. Proc Natl Acad Sci USA 1998; 95: 13 097–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Agrelo R, Cheng W‐H, Setien F et al . Epigenetic inactivation of the premature aging Werner syndrome gene in human cancer. Proc Natl Acad Sci USA 2006; 103: 8822–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Otterlei M, Bruheim P, Ahn B et al . Werner syndrome protein participates in a complex with RAD51, RAD54, RAD54B and ATR in response to ICL‐induced replication arrest. J Cell Sci 2006; 119: 5114–23. [DOI] [PubMed] [Google Scholar]
- 51. Marple T, Kim TM, Hasty P. Embryonic stem cells deficient for Brca2 or Blm exhibit divergent genotoxic profiles that support opposing activities during homologous recombination. Mutat Res 2006; 602: 110–20. [DOI] [PubMed] [Google Scholar]
- 52. Hinz JM, Helleday T, Meuth M. Reduced apoptotic response to camptothecin in CHO cells deficient in XRCC3. Carcinogenesis 2003; 24: 249–53. [DOI] [PubMed] [Google Scholar]
- 53. Lundin C, Schultz N, Arnaudeau C, Mohindra A, Hasen LT, Helleday T. RAD51 is involved in repair of damage associated with DNA replication in mammalian cells. J Mol Biol 2003; 328: 521–35. [DOI] [PubMed] [Google Scholar]
- 54. Hansen LT, Lundin C, Spang‐Thomsen M, Petersen LN, Helleday T. The role of RAD51 in etoposide (VP16) resistance in small cell lung cancer. Int J Cancer 2003; 105: 472–9. [DOI] [PubMed] [Google Scholar]
- 55. Adachi N, Iiizumi S, Koyama H. Evidence for a role of vertebrate Rad52 in the repair of topoisomerase II‐mediated DNA damage. DNA Cell Biol 2005; 24: 388–93. [DOI] [PubMed] [Google Scholar]
- 56. Hannay JAF, Liu J, Zhu Q‐S et al . Rad51 overexpression contributes to chemoresistance in human soft tissue sarcoma cells: a role for p53/activator protein 2 transcriptional regulation. Mol Cancer Ther 2007; 6: 1650–60. [DOI] [PubMed] [Google Scholar]
- 57. Koehn H, Magan N, Isaacs RJ, Stowell KM. Differential regulation of DNA repair protein Rad51 in human tumour cell lines exposed to doxorubicin. Anti-Cancer Drugs 2007; 18: 419–25. [DOI] [PubMed] [Google Scholar]
- 58. Yang L‐Y, Li L, Jiang H, Shen Y, Plunkett W. Expression of ERCC1 antisense RNA abrogates gemcitabine‐mediated cytotoxic synergism with cisplatin in human colon tumor cells defective in mismatch repair but proficient in nucleotide excision repair. Clin Cancer Res 2000; 6: 773–81. [PubMed] [Google Scholar]
- 59. Lorusso D, Di Stefano A, Fanfani F, Scambia G. Role of gemcitabine in ovarian cancer treatment. Ann Oncol 2006; 17 (Suppl 5): v188–94. [DOI] [PubMed] [Google Scholar]
- 60. De Las Peñas R, Sanchez‐Ronco M, Alberola V et al . Polymorphisms in DNA repair genes modulate survival in cisplatin/gemcitabine‐treated non‐small‐cell lung cancer patients. Ann Oncol 2006; 17: 668–75. [DOI] [PubMed] [Google Scholar]
- 61. Li D, Frazier M, Evans DB et al . Single nucleotide polymorphisms of RecQ1, RAD54L, and ATM genes are associated with reduced survival of pancreatic cancer. J Clin Oncol 2006; 24: 1720–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Liu N, Lim C‐S. Differential roles of XRCC2 in homologous recombinational repair of stalled replication forks. J Cell Biochem 2005; 95: 942–54. [DOI] [PubMed] [Google Scholar]
- 63. Zhou J, Lim CUK, Li JJ, Cai L, Zhang Y. The role of NBS1 in the modulation of PIKK family proteins ATM and ATR in the cellular response to DNA damage. Cancer Lett 2006; 243: 9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Stewart GS, Maser RS, Stankovic T et al . The DNA double‐strand break repair gene hMRE11 is mutated in individuals with an ataxia‐telangiectasia‐like disorder. Cell 1999; 99: 577–87. [DOI] [PubMed] [Google Scholar]
- 65. Luo G, Yao MS, Bender CF et al . Disruption of mRad50 causes embryonic stem cell lethality, abnormal embryonic development, and sensitivity to ionizing radiation. Proc Natl Acad Sci USA 1999; 96: 7376–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Angele S, Treilleux I, Bremond A, Taniere P, Hall J. Altered expression of DNA double‐strand break detection and repair proteins in breast carcinomas. Histopathology 2003; 43: 347–53. [DOI] [PubMed] [Google Scholar]
- 67. Giannini G, Rinaldi C, Ristori E et al . Mutations of an intronic repeat induce impaired MRE11 expression in primary human cancer with microsatellite instability. Oncogene 2004; 23: 2640–7. [DOI] [PubMed] [Google Scholar]
- 68. Söderlund K, Stål O, Skoog L et al . Intact Mre11/Rad50/Nbs1 complex predicts good response to radiotherapy in early breast cancer. Int J Rad Oncol Biol Phys 2007; 68: 50–8. [DOI] [PubMed] [Google Scholar]
- 69. Seedhouse C, Russell N. Advances in the understanding of susceptibility to treatment–related acute myeloid leukaemia. Br J Haematol 2007; 137: 513–29. [DOI] [PubMed] [Google Scholar]
- 70. Maacke H, Opitz S, Jost K et al . Over‐expression of wild‐type RAD51 correlates with histological grading of invasive ductal breast cancer. Int J Cancer 2000; 88: 907–13. [DOI] [PubMed] [Google Scholar]
- 71. Connell PP, Jayathilaka K, Haraf DJ, Weichselbaum RR, Vokes EE, Lingen MW. Pilot study examining tumor expression of RAD51 and clinical outcomes in human head cancers. Int J Oncol 2006; 28: 1113–19. [PubMed] [Google Scholar]
- 72. Galamb O, Sipos F, Dinya E, Spisak S, Tulassay Z, Molnar B. mRNA expression, functional profiling and multivariate classification of colon biopsy specimen by cDNA overall glass microarray. World J Gastroenterol 2006; 12: 6998–7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yoshikawa K, Ogawa T, Baer R et al . Abnormal expression of BRCA1 and BRCA1‐interactive DNA‐repair proteins in breast carcinomas. Int J Cancer 2000; 88: 28–36. [PubMed] [Google Scholar]
- 74. Wu G‐J, Sinclair CS, Paape J et al . 17q23 amplifications in breast cancer involve the PAT1, RAD51C, PS6K, and SIGMA1B genes. Cancer Res 2000; 60: 5371–5. [PubMed] [Google Scholar]
- 75. Hilton JL, Geisler JP, Rathe JA, Hattermann‐Zogg MA, DeYoung B, Buller RE. Inactivation of BRCA1 and BRCA2 in ovarian cancer. J Natl Cancer Inst 2002; 94: 1396–406. [DOI] [PubMed] [Google Scholar]
- 76. Yang M‐H, Chiang W‐C, Chou T‐Y et al . Increased NBS1 expression is a marker of aggressive head and neck cancer and overexpression of NBS1 contributes to transformation. Clin Cancer Res 2006; 12: 507–15. [DOI] [PubMed] [Google Scholar]
- 77. Ehlers JP, Harbour JW. NBS1 expression as a prognostic marker in uveal melanoma. Clin Cancer Res 2005; 11: 1849–53. [DOI] [PubMed] [Google Scholar]
- 78. Turley H, Wu L, Canamero M, Gatter KC, Hickson ID. The distribution and expression of the Bloom's syndrome gene product in normal and neoplastic human cells. Br J Cancer 2001; 85: 261–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wang Z, Li M, Lu S, Zhang Y, Wang H. Promoter hypermethylation of FANCF plays an important role in the occurrence of ovarian cancer through disrupting Fanconi anemia‐BRCA pathway. Cancer Biol Ther 2006; 5: 256–60. [DOI] [PubMed] [Google Scholar]
- 80. Narayan G, Arias‐Pulido H, Nandula SV et al . Promoter hypermethylation of FANCF. disruption of Fanconi anemia‐BRCA pathway in cervical cancer. Cancer Res 2004; 64: 2994–7. [DOI] [PubMed] [Google Scholar]