

Abstract
Quantitative and/or qualitative alteration of actin cytoskeletal molecules, involved in the regulation of cellular dynamic functions, should be intimately related with cancer phenotypes. Based on several lines of experimental evidence from our group, and others, this report proposes a strategy to simultaneously attack cancer cells and protect the host from cancer invasion, with one molecule. Calponin h1, an actin‐stabilizing protein that is also intimately related to signal transduction, is very often suppressed in vascular smooth muscle cells of malignant human tumors and in mesothelial cells by coexisting cancer cells. We generated mice deficient for calponin h1, exhibiting fragility in blood vessels and peritoneal membranes. Hematogenous cancer metastasis occurred more easily in the calponin h1‐deficient mice than in wild‐type mice, and the peritoneal dissemination was extremely enhanced. The fragility was rescued by the exogenous introduction of the calponin h1 gene into mesothelial cells of the peritoneum. Furthermore, calponin h1 gene transfer into several transformed cell lines resulted in a suppression of malignancy. The peritoneal dissemination of intraperitoneally‐injected B16‐F10 cells was suppressed by the calponin h1 gene, given to target both cancer cells and the mesothelial cells of the host. The multifunctional nature of the molecule, as a machinery player of cytoskeleton and mediator of signal transduction, probably resulted in a favorable recipient‐discriminating effect on cancerous and normal cells. Thus, we believe that if we use adequate multifunctional molecules for therapy, it is possible to simultaneously suppress cancer phenotypes and protect normal cells from the attack of cancer cells. (Cancer Sci 2005; 96: 738 –746)
Abbreviations:
- CARD
caspase recruitment domain
- CH
calponin homology
- ERK
extracellular regulated kinase
- PKC
protein kinase C
- PYD
pyrin domain
- SMα
smooth muscle α actin.
It is a well‐known fact that metastatic phenotypes make it difficult to cure cancer. Without metastasis, any tumor is completely resectable by surgery and/or radiation. However, the efficiency of metastasis (number of metastatic nodules/number of cancer cells in a primary tumor) is low, less than 10−7, so another rational way for the host to coexist with cancer cells could be through milder treatments to inhibit metastasis rather than a severe total‐killing of cancer cells. The molecules responsible for metastatic phenotypes have been getting attention as a source of cancer‐specific probes for an ideal treatment to completely eradicate cancer cells.
Metastatic phenotypes depend on cell adhesiveness, motility and deformability, in addition to other relating phenotypes, such as protease activity to degrade the extracellular matrix and an infinite proliferation ability. Cytoskeletal molecules are involved in the regulation of cell morphology, motility and adhesion and other dynamic cellular functions, so that it is to be expected that quantitative and/or qualitative alterations of cytoskeletal molecules are involved in metastasis‐related phenotypes. The functions of cytoskeletal proteins are generally regulated by Ca++‐binding and phosphorylation, which are also essential for various signal transductions, indicating that cytoskeletal systems are interrelated with signal transducing systems. Cytoskeletal systems function as static skeletons of cell architecture, as dynamic machineries, in cellular motility and adhesion, and also as a medium for signal transduction.
Cellular skeletal systems consist of, from the cellular outside to the nuclear inside, an extracellular matrix, a membrane skeletal system, the cytoskeleton, and a nuclear matrix. They play their specific roles and are also intimately interrelated. According to a popular definition, cytoskeleton consists of actin filaments (microfilaments), intermediate filaments and microtubules.
In this review, we direct special attention to the actin cytoskeletal system, which is the most widely involved system in the regulation of cellular motility, adhesiveness, and morphology and dynamics of organella.
Based on several lines of experimental evidence from the actin cytoskeletal system, we will propose a new idea for coping with cancer diseases: a possibility of protecting the host and simultaneously attacking cancer cells with one molecule, which seemingly functions as a cytoskeletal protein and also as a regulator of signal transductions. We will also briefly refer to a cytoskeletal‐like protein discovered by us, which acts as a scaffold regulator in signal transductions, and in particular for inflammation and apoptosis.
Suppression of cancer phenotypes by actin‐related molecules
We observed that when mammalian cells were transformed or progressed to acquire metastatic potential, the expression of actin and/or actin‐related proteins changed. We have reported that the mutation of actin itself, or an alteration in the expression of actin, affected the metastatic phenotypes and/or growth potential of cancer cells. Variant β‐actins found in human fibrosarcoma (244th amino acid: glycine to aspartic acid)( 1 ) and mouse malignant melanoma (28th amino acid: arginine to leucine) induced destabilization or inverse stabilization of actin filaments,( 2 ) respectively. Thus, each variant β‐actin affected the dynamics of actin filaments, leading to the progression or suppression of malignant phenotypes.( 3 ) A rat fibroblast cell line, 3Y1, expresses smooth muscle α actin (SMα), which is downregulated in the transformed counterparts, such as src‐transformed 3Y1 cells.( 4 ) When exogenous SMα was expressed by cDNA transfer into src‐transformed 3Y1 cells, cell motility, invasiveness, growth potential, and metastatic potential were suppressed.( 5 )
In addition to alteration of actin itself, we have also observed several cases where a change in expression of actin‐binding proteins affected cancer phenotypes. A low molecular type of tropomyosin, for instance, that destabilizes actin filaments, was more expressed in highly metastatic cells than in low metastatic ones, and the suppression of the low molecular tropomyosin in the highly metastatic cell line resulted in suppression of cell motility.( 6 ) As another example, vinculin, a connecter between actin filaments and cell adhesion molecules, was suppressed in highly metastatic B16 melanoma cell lines.( 7 ) A number of studies noted that actin‐related molecules modify cancer phenotypes, such as gelsolin,( 8 ) a protein severing or capping actin filaments.
We observed that the overexpression of calponin h1, which stabilizes actin filaments, suppressed cell motility, cell growth, and/or angiogenesis, in several transformed cell lines.( 9 , 10 , 11 ) In this review, we will mainly refer to several lines of evidence and our ideas obtained through our experiments with calponin h1, and also discuss the possibility of applying these ideas to human cancer diseases, with special reference to the cure and protection of peritoneal cancer dissemination.
Recently, a group at Massachusetts Institutes of Technology and Harvard University noted that actin and calponin h1 were highly rated molecular markers for metastatic potential by microarray examinations.( 12 )
Calponin
Calponin is a family of actin regulatory proteins. Three isoforms of separate gene products have been reported: (i) a basic calponin, h1, specific for smooth muscle;( 13 ) (ii) a neutral calponin, h2, that is localized primarily in cardiac muscle;( 14 ) and (iii) an acidic variant,( 15 ) which is not tissue‐specific, but largely expressed in the brain. They are different from each other mainly in the C‐terminal region of acidic amino acids.( 16 , 17 )
Calponin h1 was originally found as a protein in smooth muscle, interacting with F‐actin and inhibiting actomyosin Mg‐ATPase activity in vitro. It has also been reported that calponin binds to calmodulin,( 18 ) myosin,( 19 ) desmin,( 20 ) and phospholipids.( 21 ) It is a substrate for PKC, Ca2+/calmodulin‐dependent kinase II.( 22 )
The structure of calponin (Fig. 1) contains the calponin homology (CH) domain,( 23 ) which has a high homology among the different isoforms, an actin binding region( 24 ) with homology to troponin I, and three C‐terminal repeats that are also involved in actin binding.( 25 ) The function of CH domains at the N terminal in proteins is identified as an extracellular regulated kinase (ERK) binding domain.( 26 ) The coexistence of CH domains with other actin binding domains indicates that calponin functions as a cytoskeletal protein and also as a mediator of signal transductions.
Figure 1.
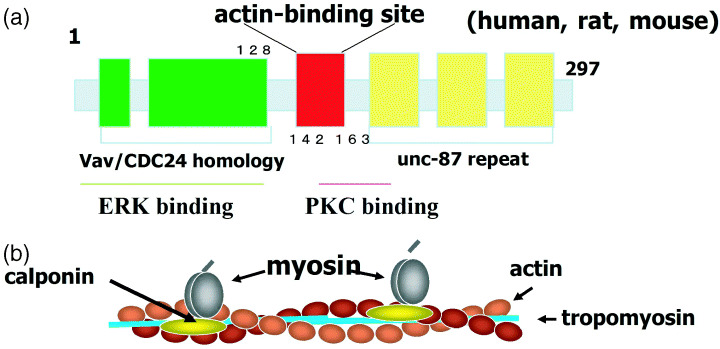
(a) Characteristics of the structure of calponin h1. In addition to the actin‐binding region, the protein has regions that react with molecules involved in signal transduction. (b) An image of the interaction between calponin and actin filaments. Calponin binds and stabilizes actin filaments and inhibits actomyosin ATPase. ERK, extracellular regulated kinase; PKC, protein kinase C.
Regarding the role of calponin h1 in muscle contractility, two controversial mechanisms have been proposed for its function. The first, based on the in vitro properties of calponin, is that binding to actin may stabilize the filaments and directly inhibit the actin‐activated Mg‐ATPase activity of myosin.( 22 ) However, it has also been reported that the effect of calponin on the contractile force in permeabilized preparations of smooth muscle cells is relatively small.( 27 )
The other proposed mechanism for the function of calponin h1 is that calponin may be involved in agonist‐induced signal transduction, based on the observations that ERK and PKC coimmunoprecipitate and cotranslocate with calponin in the vicinity of the plasmalemma( 28 ) and that ERK and PKC directly bind to calponin in vitro. ( 29 ) Furthermore, the binding of calponin appears to activate PKC.( 29 ) These results suggest a role for calponin as a signaling molecule that recruits and/or activates PKC and ERK as an adaptor protein.( 26 , 28 ) It was further reported that calponin was a target of Rho kinase, being phosphorylated in Thr‐170, Ser‐175, Thr‐180, Thr‐184, and Thr‐259.( 30 )
In calponin‐deficient mice, a faster unloaded shortening velocity was observed in the phasic smooth muscle tissues of the bladder and vas deferens,( 31 ) consistent with the hypothesis that calponin is a direct regulator of actomyosin interactions. On the other hand, we observed that the vascular response to phenylephrine in calponin‐deficient mice was reduced to 40–63% compared to that of wild‐type mice.( 32 ) This appears to be consistent with the hypothesis that calponin is involved in agonist‐induced signal transduction. Taken together, calponin is suggested to be a molecule with multiple functions, being both a machinery player and a mediator for signal transductions (Fig. 2).
Figure 2.
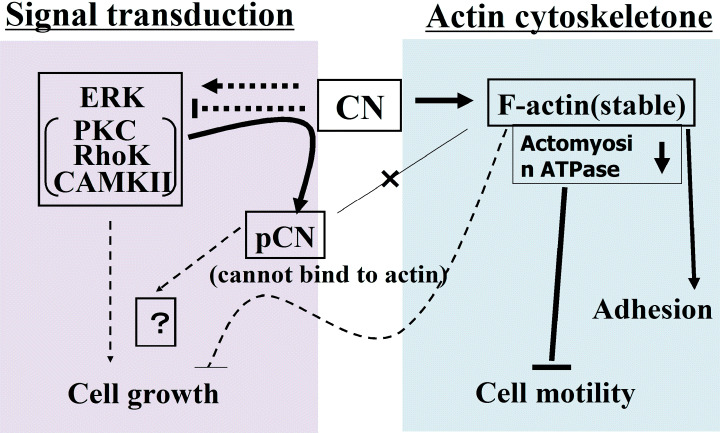
Hypothesis of calponin (CN) functioning as a cytoskeletal machine or as a mediator for signal transduction. Because CN reacts with both actin and kinases for signal transductions, it could affect the downstream, for example a signal for growth regulation, one of the reactions between CNh1 and kinases, or through stabilization of actin filaments by CN. (––) Established flows; (‐‐‐) speculated flows. ERK, extracellular regulated kinase; PKC, protein kinase C.
The amount of actin was reduced in calponin h1‐deficient smooth muscles,( 33 ) probably due to the destabilization of actin filaments. Microarray analysis of calponin‐deficient cells indicated that some adhesion molecules and caspases were suppressed (data not shown), indicating reduced cell‐adhesiveness and apoptotic potential. These lines of evidence indicate a possibility that calponin is involved in the regulation of cell growth signals in addition to the regulation of cell motility. The phosphorylated calponin h1, by PKC or Rho kinase, cannot bind to actin. Thus, the down stream effects of calponin binding to PKC, ERK and/or Rho kinase remain to be determined, except the reduction of the reaction between phosphorylated calponin and actin. The destabilization of actin filaments through the phosphorylation of calponin, consequently, could induce various reactions, presumably including an increase in cellular growth potential (Fig. 2). Currently, there is no distinction between the direct effects of calponin on signal transduction and the effects through the cytoskeletal system. Studies have been undertaken to discriminate between the functions of calponin as a reactor with actin or as a mediator of signal transductions, by using various calponin mutants and various cellular systems.
In the regulatory region of the calponin h1 gene,( 34 ) including the first intron,( 35 ) there is a GATA box, a CARG box (in the first intron) and CpG islands,( 36 ) indicating that GATA6, which is downregulated by growth stimulation,( 37 ) and SRF, which is regulated by a G‐actin pool through its coactivator Mal,( 38 ) could induce calponin h1 gene expression and methylation of CpG islands, leading to a suppression of calponin h1. ( 39 ) Neither a TATAA nor an initiator consensus element is found in the calponin h1 promoter. The absence of such elements is often associated with multiple transcription initiation sites. It is noted that the upstream activators between −115 and −549 play a crucial role in the initiation of mouse calponin h1 transcription, and sequence analysis of this region reveals two E‐boxes and one GATA site.( 40 ) In addition, there is an interesting sequence in the regulatory region of the calponin gene,( 40 ) purine/pyrimidine dinucleotide repeats promote the formation of Z DNA, which has recently been shown to regulate the transcription of the c‐myc proto‐oncogene.
Suppression of cancer phenotypes by gene transfer of calponin h1
In leiomyosarcomas, the expression of endogenous calponin h1 proved to be decreased compared with those in leiomyoma and normal uterus smooth muscle.( 41 ) When we transferred a calponin h1 expression vector into leiomyosarcoma cells to induce expression, suppression of growth and induction of differentiated phenotypes were observed.( 9 ) The inhibition of cell motility and growth was also observed in a human fibrosarcoma cell line, HT1080( 10 ) by calponin expression. In this case, the expression of α5β1 integrin was increased, thus the cell morphology became flat through enhanced adhesion to fibronectin. A rat fibroblast cell line, 3Y1, expressed calponin h1, while calponin h1 expression was decreased in its src‐induced transformants. The tumorigenicity of the cells transferred with the exogenous calponin h1 gene was reduced in immunosuppressed nude mice. In this case, the suppression of growth or cell motility was minor, and the main effect was the suppression of angiogenesis through an inhibition of the expression of VEGF.( 11 )
The effects of calponin h1 gene transfer were dependent on the recipient cells, which probably attributed to the difference in actin binding proteins, or to the different main pathways of signal transductions for growth in each cell line. There is a paper, in addition to ours, on the suppressive effects of calponin h1 on tumorigenesis and/or malignant progression.( 42 )
As well as the change in calponin expression in cancer cells, we also found calponin suppression in blood vessels of malignant tumor tissue, and also in peritoneal mesothelial cells, which coexisted with cancer cells.
Altered expression of actin‐related genes in smooth muscle cells of vessels close to, or inside, malignant tumors
We observed the downregulation of SMα inside or close to human malignant melanoma and ovary cancer tissue( 43 , 44 ) and there are similar reports consistent with our results.( 45 , 46 )
Furthermore, we identified factors excreted by malignant tumor cells that suppressed calponin expression, for example PDGF‐BB (a product of sis oncogene).( 43 ) Other known suppressors are growth factors,( 47 ) which induce a dedifferentiation state of smooth muscle cells, and inflammatory cyotokines, such as INFγ.( 48 ) We also recognized that calponin expression was not detected even in cells expressing SMα, that is, calponin was more easily downregulated than SMα.( 48 ) When screening the expressions of smooth muscle‐related genes, calponin h1 proved to be extremely suppressed even in the early stages of tumors, while h caldesmon was gradually decreased correlating to metastatic frequency and prognosis of the patients.( 49 )
We believed that such an alteration of molecules relating to the actin cyotoskeleton with blood vessels could become markers for the diagnosis of malignant tumors, while the change would result in a fragility of blood vessels, thus leading to enhancement of metastasis. Several other groups have also recognized the suppression of calponin in malignant tumors. Observations with electronic microscopy suggested to us that both endothelial cells and smooth muscle cells exhibited fragility.( 49 )
Augmentation of blood‐born metastasis and peritoneal dissemination in calponin‐deficient mice
To understand the significance of the downregulation of calponin in vessels of malignant tissues, we examined calponin h1‐deficient mice. In the mice that were back‐crossed with B6 mice for syngeneic transplant experiments, the fragility of blood vessels and cancer metastasis occurred more easily in calponin‐deficient mice than in wild‐type mice( 50 ) (Fig. 3). That smooth muscles became fragile in calponin‐deficient mice is in agreement with our expectation, based on our previous studies. However, it remains to be elucidated why endothelial cells also become fragile. In endothelial cells, calponin protein expression was not detected but mRNA was detectable by reverse transcription‐polymerase chain reaction.( 51 ) Thus, although expression of the calponin h1 protein is low, the small number of protein molecules might play a very important role in the cellular adhesion of endothelial cells. An alternative explanation could be that the alteration in smooth muscle by the suppression of calponin h1 could induce paracrine effects in endothelial cells, such as changes in the extracellular matrix.
Figure 3.

Fragile structure of (a) blood vessel and (c) peritoneal membrane in calponin (CN)‐deficient mice.( 50 ) Augmentation of (b) blood‐born metastasis and (d) peritoneal dissemmination of B16‐F10 cells in calponin‐deficient mice.( 50 ) (a) Top: Electronmicroscopic image of lung arterioles. The walls of the vessels are thinner in the calponin‐deficient mice than in the normal mice. Scale bar = 5 µm. (a) Bottom: Scanning electronmicroscopic image of the surface of lung capillaries. The capillaries of the calponin‐deficient mice have an irregular surface. Scale bar = 100 nm.
Furthermore, we showed that mesothelial cells expressed calponin h1, and the expression was suppressed by co‐culturing with cancer cells.( 52 ) In calponin‐deficient mice, the cellular adhesion of mesothelial cells appeared to be weakened, and peritoneal dissemination was extremely enhanced( 50 ) (Fig. 3).
When we injected adenovirus vectors carrying a GFP‐fused calponin gene into the peritoneal cavity of calponin‐deficient mice, or to cultured mesothelial cells, cell‐to‐cell or cell‐to‐substrate adhesion of mesothelial cells was strengthened and the invasion of cancer cells suppressed (Fig. 4), consequently increasing the lifespan of mice that had received an intraperitoneal (IP) injection of B16‐F10 cells( 52 ) (Fig. 4). The suppression of calponin h1 was also observed in mesothelial cells from normal mice prior to the cellular retraction by co‐culturing with cancer cells.( 52 )
Figure 4.
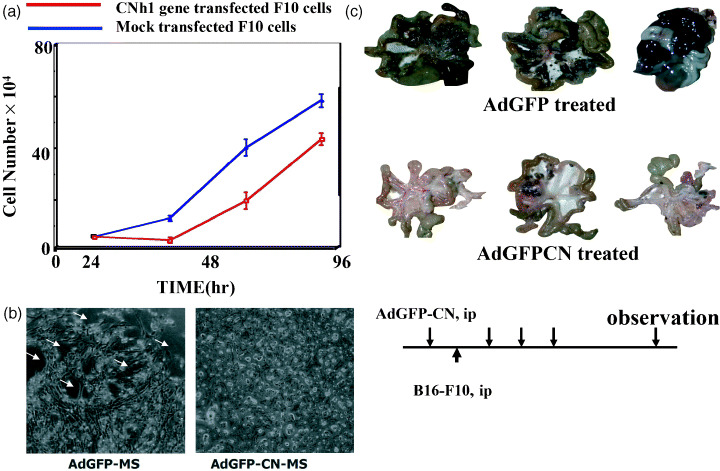
(a) Calponin (CN)h1 functions to suppress the proliferation of F10 cells by gene transfer. (b) The protective effect of calponin h1 on the retraction in calponin‐deficient mesothelial cells caused by B16‐F10 cells in vitro. B16‐F10 cells were overlaid on AdGFP or AdGFP‐CN‐I infected calponin‐deficient mesothelial cells cultured to confluence. The images were obtained 6 h after the overlaying of B16‐F10 cells. Bright and round cells indicate the representative B16‐F10 cells and stars indicate the exposed substrate after the retraction. Scale bar = 3 m. (c) Prevention of peritoneal dissemination of B16‐F10 cells by the expression of CNh1 using adenovirus vectors. (Top) B16‐F10 cells were injected into the peritoneal cavity of calponin‐deficient mice infected with AdGFP or AdGFP‐CN. Virus infection was performed every 3 days. Fourteen days after the intraperitoneal (ip) injection of B16‐F10 cells, the mice were killed and the extent of peritoneal dissemination was examined.
Calponin h1 suppressed the phenotypes of malignant tumor cells as already noted. Thus, calponin protects host cells, in this case mesothelial cells, from cancer invasion, and simultaneously inhibits malignant phenotypes of cancer cells. Side‐effects were dose‐limiting factors in hitherto‐known cancer therapies, but this is not the case for the method presented here. This beneficial effect of calponin h1 could be derived from the multiple functions of calponin. The overexpression of calponin h1 has never induced apoptosis of normal mesothelial cells (data not shown). To induce calponin h1 expression by biomaterials would be another way to treat peritoneal dissemination of cancer cells.( 53 )
In summary, the speculated mode for the actions of calponin is shown in Fig. 5. We would like to propose the possibility of a new therapeutic method that attacks cancer cells and simultaneously protects host cells from the invasion of cancer cells.
Figure 5.
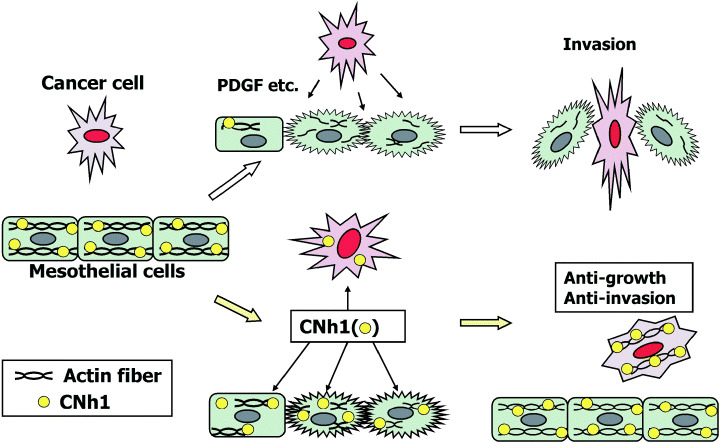
Hypothetical mechanism of calponin (CN) to suppress peritoneal dissemination of cancer cells. This cartoon shows that calponin h1 exerts a bifunctional action, that is, to protect the host against cancer invasion by recovering the intercellular fragility of mesothelial cells and to inhibit the growth of cancer cells.
Cytoskeleton‐like protein ASC, a signal mediator that forms scaffolds, inflammasomes and apoptosomes
ASC is an apoptosis‐associated protein containing a caspase recruitment domain (CARD). It is a cyotosleketal‐like adaptor protein very similar to calponin, in that they are both mutifunctional. This speck‐forming protein is responsible for forming apoptosomes and inflammasomes. While trying to find and identify the molecules responsible for altering the nuclear morphology of HL‐60 cells associated with apoptosis and/or differentiation by treatment with retinoic acid, we accidentally discovered ASC, which formed a speck near the nucleus of HL‐60 cells in association with induced apoptosis. ASC had a C‐terminal CARD and a pyrin‐homologous region (PYD).( 54 ) Pyrin is known as a causative gene of familial Mediterranean fever.( 55 ) From its structural character, we assumed that ASC must be involved in both apoptosis and inflammation.
We showed PYD homotypic interaction to form filaments( 56 ) and determined the responsible amino acid regions.( 57 ) ASC was not expressed in the basal layer of the epidermis, but the expression became extensive towards the outer layers( 58 ) where the cells are exposed to external factors and are destined to die. This is also the case in bowel. Such an expression pattern led us to assume that ASC functions as a surveyor from pathogenic invaders and an inducer of programmed cell death. Furthermore, monocytes and neutrophil cells expressed ASC, and the expression was induced in the region of inflammation where ASC was usually not expressed, indicating that ASC is involved in inflammation. However, ASC was induced by apoptotic inducers, such as Fas ligands.( 59 ) Taken together, we speculated that ASC was related to both inflammation and apoptosis.
Meanwhile, a fast gene hunt of the PYD family was done in USA and Europe, and a number of genes were reported.( 60 ) Cryopyrin, for instance, was reported as a product of the causative gene of familial cold autoinflammatory syndrome.( 61 ) Surprisingly, many of those PYD proteins contained leucine rich repeats, which also exist in Toll‐like receptors as an antenna against pathogenic materials. Thus, it is likely that PYD molecules could be responsible for innate immunity in the cell cytosol, corresponding to Toll‐like receptors in the membrane. Furthermore, ASC proved to be a specific activator of procaspase 1,( 62 , 63 ) also known as interleukin‐1β converting enzyme. ASC was required for activation of proIL‐1β and proIL‐18 in in vitro experiments. PYD proteins bound to ASC first, recruiting procaspase 1 with CARD of ASC, then procaspase 1 was activated by proximity mode.( 64 ) Such a molecular aggregation has been named inflammasome.( 64 , 65 )
We generated ASC‐deficient mouse, and analyzed the phenotypes.( 66 ) We observed that in macrophages of the peritoneal cavity and kupffer cells of the liver, no activation of caspase 1 occurred and no activation of proIL‐1 β or proIL‐18. Thus, the mice were resistant to lipopolysaccharide‐induced endotoxin shock.( 66 ) Consistent results using ASC‐deficient mice were reported independently.( 67 )
When ASC is methylated, the expression is suppressed, and this was reported to act as a pro‐apoptotic and antitumor gene one year after our findings.( 68 , 69 ) We also observed methylation of the ASC gene and its downregulation correlated with the degree of methylation in the CpG islands.( 70 , 71 ) ASC was also reported to react with Bax and to function as a promoter of apoptosis.( 72 ) So far, no tumorigenesis is evident in the ASC‐deficient mice. We are planning to cross the ASC‐deficient mice with APC‐ and p53‐deficient mice, and to perform chemical carcinogenesis and infection experiments. ASC will be an effective probe to assess the relation of inflammation and apoptosis to carcinogenesis and/or malignant progression.
Concluding remarks
We have noted that actins and related molecules can suppress cancer phenotypes such as abnormal growth, motility and angiogenesis, focusing on calponin h1, a stabilizer of actin filaments. Simultaneously, we have shown the downregulation of calponin h1 in host normal cells to be induced by cancer cells, such as in blood vessels and the peritoneum. Although anti‐angiogenesis has received a lot of attention, the alteration in molecular changes in the smooth muscle of blood vessels could provide important information for the therapy of solid malignant tumors. When the calponin gene is transferred into cancer cells, growth and/or cell motility is suppressed. The overexpression of calponin in mesothelial cells of the peritoneum or into cultured mesothelial cells, however, did not induce any cell damage, but only strengthened cellular adhesion. Thus, we believe that it is possible to simultaneously suppress cancer phenotypes and strengthen the adhesiveness of normal cells. This means that one gene, or one biomolecule, can be useful to both treat the cancer and protect the host, if we use an adequate gene for therapy. Preclinical experiments with the calponin h1 gene have been done to treat the peritoneal dissemination of ovarian cancers in collaboration with Dr H. Kobayashi et al. at Kyushu University, showing data completely consistent with our published results (H. Ogura et al., manuscript in preparation).
Calponin h1 is an example of a multifunctional cytoskeletal molecule that is involved in cellular dynamics and signal transductions. ASC is an adaptor molecule responsible for apoptosis and inflammation. Both calponin and ASC are common in that they provide a scaffold for various intracellular reactions. Normally we try to understand various biological phenomena individually, but these reactions are so closely interrelated that they cannot be separated. So‐called cytoskeletal proteins should actually be called scaffold proteins, from a broad point of view. The chromosomal genes for calponin and ASC have CpG islands for methylation, thus their expression appears to be epigenetically changeable, and this could make these molecules useful probes to understand instability of cancer phenotypes in vivo.
Acknowledgments
The author thanks the staff and graduate students in the Department of Molecular Oncology, Institute on Aging and Adaptation, Shinshu University Graduate School of Medicine for their contributions to the work presented in this review. The author also wishes to thank many laboratories in the Shinshu University School of Medicine for their helpful collaborations, discussions and/or technical instructions. The generation of the calponin h1‐deficient mouse was performed under the guidance of Dr M. Katsuki, Director‐General, National Institute of Basic Biology. The generation of ASC‐deficient mouse was performed in collaboration with Dr T. Noda, Cancer Research Institute of the Japanese Foundation of Cancer Research, Tokyo. These studies were supported by Grant‐in‐Aids (13218055, 1659100, 126701109) from the Ministry of Education, Culture, Sports, Science and Technology, Japan, and by the President's Discretionary Fund of Shinshu University.
References
- 1. Taniguchi S, Sagara J, Kakunaga T. Deficient polymerization in vitro of a point‐mutated beta‐actin expressed in a transformed human fibroblast cell line. J Biochem (Tokyo) 1988; 103: 707–13. [DOI] [PubMed] [Google Scholar]
- 2. Taniguchi S, Kawano T, Kakunaga T, Baba T. Differences in expression of a variant actin between low and high metastatic B16 melanoma. J Biol Chem 1986; 261: 6100–6. [PubMed] [Google Scholar]
- 3. Shimokawa‐Kuroki R, Sadano H, Taniguchi S. A variant actin (â m) reduces metastasis of mouse B16 melanoma. Int J Cancer 1994; 56: 689–97. [DOI] [PubMed] [Google Scholar]
- 4. Okamoto‐Inoue M, Taniguchi S, Sadano H et al. Alteration in expression of smooth muscle alpha‐actin associated with transformation of rat 3Y1 cells. J Cell Sci 1990; 96 (Pt 4): 631–7. [DOI] [PubMed] [Google Scholar]
- 5. Okamoto‐Inoue M, Kamada S, Kimura G, Taniguchi S. The induction of smooth muscle alpha actin in a transformed rat cell line suppresses malignant properties in vitro and in vivo . Cancer Lett 1999; 142: 173–8. [DOI] [PubMed] [Google Scholar]
- 6. Miyado K, Kimura M, Taniguchi S. Decreased expression of a single tropomyosin isoform, TM5/TM30nm, results in reduction in motility of highly metastatic B16‐F10 mouse melanoma cells. Biochem Biophys Res Commun 1996; 225: 427–35. [DOI] [PubMed] [Google Scholar]
- 7. Sadano H, Inoue M, Taniguchi S. Differential expression of vinculin between weakly and highly metastatic B16‐melanoma cell lines. Jpn J Cancer Res 1992; 83: 625–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sagawa N, Fujita H, Banno Y, Nozawa Y, Katoh H, Kuzumaki N. Gelsolin suppresses tumorigenicity through inhibiting PKC activation in a human lung cancer cell line, PC10. Br J Cancer 2003; 88: 606–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Horiuchi A, Nikaido T, Taniguchi S, Fujii S. Possible role of calponin h1 as a tumor suppressor in human uterine leiomyosarcoma. J Natl Cancer Inst 1999; 91: 790–6. [DOI] [PubMed] [Google Scholar]
- 10. Takeoka M, Ehara T, Sagara J, Hashimoto S, Taniguchi S. Calponin h1 induced a flattened morphology and suppressed the growth of human fibrosarcoma HT1080 cells. Eur J Cancer 2002; 38: 436–42. [DOI] [PubMed] [Google Scholar]
- 11. Kaneko M, Takeoka M, Oguchi M et al. Calponin h1 suppresses tumor growth of Src‐induced transformed 3Y1 cells in association with a decrease in angiogenesis. Jpn J Cancer Res 2002; 93: 935–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet 2003; 33: 49–54. [DOI] [PubMed] [Google Scholar]
- 13. Takahashi K, Abe M, Hiwada K, Kokubu T. A novel troponin T‐like protein (calponin) in vascular smooth muscle: interaction with tropomyosin paracrystals. J Hypertens 1988; 6: S40–S43. [DOI] [PubMed] [Google Scholar]
- 14. Strasser P, Gimona M, Moessler H, Herzog M, Small JV. Mammalian calponin: identification and expression of genetic variants. FEBS Lett 1993; 330: 13–18. [DOI] [PubMed] [Google Scholar]
- 15. Applegate D, Feng W, Green RS, Taubman MB. Cloning and expression of a novel acidic calponin isoform from rat aortic vascular smooth muscle. J Biol Chem 1994; 269: 10 683–90. [PubMed] [Google Scholar]
- 16. Danninger C, Gimona M. Live dynamics of GFP‐calponin: isoform‐specific modulation of the actin cytoskeleton and autoregulation by C‐terminal sequences. J Cell Sci 2000; 113 (Pt 21): 3725–36. [DOI] [PubMed] [Google Scholar]
- 17. Burgstaller G, Kranewitter WJ, Gimona M. The molecular basis for the autoregulation of calponin by isoform‐specific C‐terminal tail sequences. J Cell Sci 2002; 115 (Pt 10): 2021–9. [DOI] [PubMed] [Google Scholar]
- 18. Wills FL, McCubbin WD, Kay CM. Characterization of the smooth muscle calponin and calmodulin complex. Biochemistry 1993; 32: 2321–8. [DOI] [PubMed] [Google Scholar]
- 19. Shirinsky VP, Biryukov KG, Hettasch JM, Sellers JR. Inhibition of the relative movement of actin and myosin by caldesmon and calponin. J Biol Chem 1992; 267: 15 886–92. [PubMed] [Google Scholar]
- 20. Mabuchi K, Li B, Ip W, Tao T. Association of calponin with desmin intermediate filaments. J Biol Chem 1997; 272: 22 662–6. [DOI] [PubMed] [Google Scholar]
- 21. Bogatcheva NV, Gusev NB. Interaction of smooth muscle calponin with phospholipids. FEBS Lett 1995; 371: 123–6. [DOI] [PubMed] [Google Scholar]
- 22. Winder SJ, Walsh MP. Smooth muscle calponin: inhibition of actomyosin MgATPase and regulation by phosphorylation. J Biol Chem 1990; 265: 10 148–55. [PubMed] [Google Scholar]
- 23. Stradal T, Kranewitter W, Winder SJ, Gimona M. CH domains revisited. FEBS Lett 1998: 431: 134–7. [DOI] [PubMed] [Google Scholar]
- 24. Mezgueldi M, Mendre C, Calas B, Kassab R, Fattoum A. Characterization of the regulatory domain of gizzard calponin interactions of the 145–163 region with F‐actin, calcium‐binding proteins, and tropomyosin. J Biol Chem 1995; 270: 8867–76. [DOI] [PubMed] [Google Scholar]
- 25. Mino T, Yuasa U, Nakamura F, Naka M, Tanaka T. Two distinct actin‐binding sites of smooth muscle calponin. Eur J Biochem 1998; 251: 262–8. [DOI] [PubMed] [Google Scholar]
- 26. Leinweber BD, Leavis PC, Grabarek Z, Wang CLA, Morgan KG. Extracellular regulated kinase (ERK) interaction with actin and the calponin homology (CH) domain of actin‐binding proteins. Biochem J 1999; 344: 117–23. [PMC free article] [PubMed] [Google Scholar]
- 27. Horowitz A, Clement‐Chomienne O, Walsh MP, Tao T, Katsuyama H, Morgan KG. Effects of calponin on force generation by single smooth muscle cells. Am J Physiol 1996; 270: H1858–H1863. [DOI] [PubMed] [Google Scholar]
- 28. Menice CB, Hulvershorn J, Adam LP, Wang CLA, Morgan KG. Calponin and mitogen‐activated protein kinase signaling in differentiated vascular smooth muscle. J Biol Chem 1997; 272: 25 157–61. [DOI] [PubMed] [Google Scholar]
- 29. Morgan KG, Gangopadhyay SS. Invited review: Cross‐bridge regulation by thin filament‐associated proteins. J Appl Physiol 2001; 91: 953–62. [DOI] [PubMed] [Google Scholar]
- 30. Kaneko T, Amano M, Maeda A et al. Identification of calponin as a novel substrate of Rho‐kinase. Biochem Biophys Res Commun 2000; 273: 110–16. [DOI] [PubMed] [Google Scholar]
- 31. Takahashi K, Yoshimoto R, Fuchibe K et al. Regulation of shortening velocity by calponin in intact contracting smooth muscles. Biochem Biophys Res Commun 2000; 279: 150–7. [DOI] [PubMed] [Google Scholar]
- 32. Masuki S, Takeoka M, Taniguchi S, Nose H. Enhanced baroreflex sensitivity in free‐moving calponin knockout mice. Am J Physiol Heart Circ Physiol 2003; 284: H939–46. [DOI] [PubMed] [Google Scholar]
- 33. Matthew JD, Khromov AS, McDuffie MJ et al. Contractile properties and proteins of smooth muscles of a calponin knockout mouse. J Physiol 2000; 529 (Pt 3): 811–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Samaha FF, Ip HS, Morrisey EE et al. Developmental pattern of expression and genomic organization of the calponin‐h1 gene. A contractile smooth muscle cell marker. J Biol Chem 1996; 271: 395–403. [DOI] [PubMed] [Google Scholar]
- 35. Miano JM, Carlson MJ, Spencer JA, Misra RP. Serum response factor‐dependent regulation of the smooth muscle calponin gene. J Biol Chem 2000; 275: 9814–22. [DOI] [PubMed] [Google Scholar]
- 36. Takahashi K, Tazunoki T, Okada T et al. The 5′‐flanking region of the human smooth muscle cell calponin gene contains a cis‐acting domain for interaction with a methylated DNA‐binding transcription repressor. J Biochem (Tokyo) 1996; 120: 18–21. [DOI] [PubMed] [Google Scholar]
- 37. Mano T, Luo Z, Malendowicz SL et al. Reversal of GATA‐6 downregulation promotes smooth muscle differentiation and inhibits intimal hyperplasia in balloon‐injured rat carotid artery. Circ Res 1999; 84: 647–54. [DOI] [PubMed] [Google Scholar]
- 38. Miralles F, Posern G, Zaromytidou AI, Treisman R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 2003; 113: 329–42. [DOI] [PubMed] [Google Scholar]
- 39. Yamamura H, Yoshikawa H, Takahashi K. Aberrant methylation and silencing of the calponin gene in human sarcoma cells. Anticancer Res 2003; 23: 107–14. [PubMed] [Google Scholar]
- 40. Miano JM, Olson EN. Expression of the smooth muscle cell calponin gene marks the early cardiac and smooth muscle cell lineages during mouse embryogenesis. J Biol Chem 1996; 271: 7095–103. [DOI] [PubMed] [Google Scholar]
- 41. Horiuchi A, Nikaido T, Ito K et al. Reduced expression of calponin h1 in leiomyosarcoma of the uterus. Lab Invest 1998; 78: 839–46. [PubMed] [Google Scholar]
- 42. Lener T, Burgstaller G, Gimona M. The role of calponin in the gene profile of metastatic cells: inhibition of metastatic cell motility by multiple calponin repeats. FEBS Lett 2004; 556: 221–6. [DOI] [PubMed] [Google Scholar]
- 43. Okamoto‐Inoue M, Nakayama J, Hori Y, Taniguchi S. Human malignant melanoma cells release a factor that inhibits the expression of smooth muscle alpha‐actin. J Dermatol Sci 2000; 23: 170–7. [DOI] [PubMed] [Google Scholar]
- 44. Kobayashi H, Tsuruchi N, Sugihara K et al. Expression of α‐smooth muscle actin in benign or malignant ovarian tumors. Gynecol Oncol 1993; 48: 308–13. [DOI] [PubMed] [Google Scholar]
- 45. Islam AH, Ehara T, Kato H et al. Calponin h1 expression in renal tumor vessels: correlations with multiple pathological factors of renal cell carcinoma. J Urol 2004; 171: 1319–23. [DOI] [PubMed] [Google Scholar]
- 46. Sasaki Y, Yamamura H, Kawakami Y et al. Expression of smooth muscle calponin in tumor vessels of human hepatocellular carcinoma and its possible association with prognosis. Cancer 2002; 94: 1777–86. [DOI] [PubMed] [Google Scholar]
- 47. Hayashi K, Saga H, Chimori Y, Kimura K, Yamanaka Y, Sobue K. Differentiated phenotype of smooth muscle cells depends on signaling pathways through insulin‐like growth factors and phosphatidylinositol 3‐kinase. J Biol Chem 1998; 273: 28 860–7. [DOI] [PubMed] [Google Scholar]
- 48. Hansson GK, Hellstrand M, Rymo L, Rubbia L, Gabbiani G. Interferon gamma inhibits both proliferation and expression of differentiation‐specific alpha‐smooth muscle actin in arterial smooth muscle cells. J Exp Med 1989; 170: 1595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Koganehira Y, Takeoka M, Ehara T et al. Reduced expression of actin‐binding proteins, h‐caldesmon and calponin h1, in the vascular smooth muscle inside melanoma lesions: an adverse prognostic factor for malignant melanoma. Br J Dermatol 2003; 148: 971–80. [DOI] [PubMed] [Google Scholar]
- 50. Taniguchi S, Takeoka M, Ehara T et al. Structural fragility of blood vessels and peritoneum in calponin h1‐deficient mice, resulting in an increase in hematogenous metastasis and peritoneal dissemination of malignant tumor cells. Cancer Res 2001; 61: 7627–34. [PubMed] [Google Scholar]
- 51. Sakihara C, Nishimura J, Kobayashi S, Takahashi S, Kanaide H. Expression of calponin mRNA in porcine aortic endothelial cells. Biochem Biophys Res Commun 1996; 222: 195–200. [DOI] [PubMed] [Google Scholar]
- 52. Hashimoto S, Takeoka M, Taniguchi S. Suppression of peritoneal dissemination through protecting mesothelial cells from retraction by cancer cells. Int J Cancer 2003; 107: 557–63. [DOI] [PubMed] [Google Scholar]
- 53. Horiuchi A, Nikaido T, Ya‐Li Z, Ito K, Orii A, Fujii S. Heparin inhibits proliferation of myometrial and leiomyomal smooth muscle cells through the induction of alpha‐smooth muscle actin, calponin h1 and p27. Mol Hum Reprod 1999; 5: 139–45. [DOI] [PubMed] [Google Scholar]
- 54. Masumoto J, Taniguchi S, Ayukawa K et al. ASC, a novel 22‐kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL‐60 cells. J Biol Chem 1999; 274: 33 835–8. [DOI] [PubMed] [Google Scholar]
- 55. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. The International FMF Consortium. Cell 1997; 90: 797–807. [DOI] [PubMed] [Google Scholar]
- 56. Masumoto J, Taniguchi S, Sagara J. Pyrin N‐terminal homology domain‐ and caspase recruitment domain‐dependent oligomerization of ASC. Biochem Biophys Res Commun 2001; 280: 652–5. [DOI] [PubMed] [Google Scholar]
- 57. Moriya M, Taniguchi S, Wu P, Liepinsh E, Otting G, Sagara J. Role of charged and hydrophobic residues in the oligomerization of the PYRIN domain of ASC. Biochemistry 2005; 44: 575–83. [DOI] [PubMed] [Google Scholar]
- 58. Masumoto J, Taniguchi S, Nakayama J et al. Expression of apoptosis‐associated speck‐like protein containing a caspase recruitment domain, a pyrin N‐terminal homology domain‐containing protein, in normal human tissues. J Histochem Cytochem 2001; 49: 1269–75. [DOI] [PubMed] [Google Scholar]
- 59. Shiohara M, Taniguchi S, Masumoto J et al. ASC, which is composed of a PYD and a CARD, is up‐regulated by inflammation and apoptosis in human neutrophils. Biochem Biophys Res Commun 2002; 293: 1314–8. [DOI] [PubMed] [Google Scholar]
- 60. Bertin J, DiStefano PS. The PYRIN domain: a novel motif found in apoptosis and inflammation proteins. Cell Death Differ 2000: 7: 1273–4. [DOI] [PubMed] [Google Scholar]
- 61. Feldmann J, Prieur AM, Quartier P et al. Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am J Hum Genet 2002; 71: 198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Stehlik C, Lee SH, Dorfleutner A, Stassinopoulos A, Sagara J, Reed JC. Apoptosis‐associated speck‐like protein containing a caspase recruitment domain is a regulator of procaspase‐1 activation. J Immunol 2003; 171: 6154–63. [DOI] [PubMed] [Google Scholar]
- 63. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL‐beta. Mol Cell 2002; 10: 417–26. [DOI] [PubMed] [Google Scholar]
- 64. Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell 2004: 117: 561–74. [DOI] [PubMed] [Google Scholar]
- 65. Stehlik C, Reed JC. The PYRIN connection: novel players in innate immunity and inflammation. J Exp Med 2004; 200: 551–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yamamoto M, Yaginuma K, Tsutsui H et al. ASC is essential for LPS‐induced activation of procaspase‐1 independently of TLR‐associated signal adaptor molecules. Genes Cells 2004; 9: 1055–67. [DOI] [PubMed] [Google Scholar]
- 67. Mariathasan S, Newton K, Monack DM et al. Differential activation of the inflammasome by caspase‐1 adaptors ASC and Ipaf. Nature 2004; 430: 213–8. [DOI] [PubMed] [Google Scholar]
- 68. McConnell BB, Vertino PM. TMS1/ASC: the cancer connection. Apoptosis 2004: 9: 5–18. [DOI] [PubMed] [Google Scholar]
- 69. Con Way KE, McConnell BB, Bowring CE, Donald CD, Warren ST, Vertino PM. TMS1, a novel proapoptotic caspase recruitment domain protein, is a target of methylation‐induced gene silencing in human breast cancers. Cancer Res 2000; 60: 6236–42. [PubMed] [Google Scholar]
- 70. Yokoyama T, Sagara J, Guan X et al. Methylation of ASC/TMS1, a proapoptotic gene responsible for activating procaspase‐1, in human colorectal cancer. Cancer Lett 2003; 202: 101–8. [DOI] [PubMed] [Google Scholar]
- 71. Guan X, Sagara J, Yokoyama T et al. ASC/TMS1, a caspase‐1 activating adaptor, is downregulated by aberrant methylation in human melanoma. Int J Cancer 2003; 107: 202–8. [DOI] [PubMed] [Google Scholar]
- 72. Ohtsuka T, Ryu H, Minamishima YA et al. ASC is a Bax adaptor and regulates the p53‐Bax mitochondrial apoptosis pathway. Nat Cell Biol 2004; 6: 121–8. [DOI] [PubMed] [Google Scholar]