

Abstract
Small guanosine triphosphatase RhoA has been known to re‐organize cytoskeletons and regulate cell migration. The present authors have previously reported that expression of RhoA is significantly increased in advanced ovarian carcinomas and also in the peritoneal disseminated lesions. The present study investigated whether overexpression of RhoA could alter the progressive behavior of ovarian cancer cells. The effect of various Rho inhibitors on the biological behavior of ovarian cancer cells in vitro and in vivo was also examined. A stable RhoA‐transfectant of an ovarian cancer cell line SKOV3 was generated and examined in vitro for alterations of proliferative activity and invasiveness, and also in the nude mice model for peritoneal dissemination. In addition, the effect of a specific Rho inhibitor (C3 exoenzyme), Rho kinase inhibitor (Y27632) and hydroxymethylglutaryl coenzyme A (HMG–CoA) reductase inhibitor (Lovastatin and Pravastatin) were studied in vitro and in vivo. Forced overexpression of RhoA did not alter proliferative activity but significantly increased the invasiveness in vitro, which was suppressed by addition of C3 exoenzyme, Y27834, Lovastatin and Pravastatin. In the nude mice model, the frequency of dissemination and the number of disseminated lesions were significantly increased in the RhoA transfectant than in the control. In addition, oral administration of Lovastatin significantly decreased the number of metastatic sites compared with the control. These findings suggest that upregulation and/or activation of RhoA play an important role in the peritoneal dissemination of ovarian carcinoma, and that Lovastatin might be a candidate for the possible, novel treatment for ovarian carcinoma patients with peritoneal dissemination. (Cancer Sci 2008; 99: 2532–2539)
Epithelial ovarian carcinoma is the leading cause of death from female genital malignancies and more than half of patients are diagnosed at the advanced stage of disease.( 1 , 2 ) The poor prognosis in patients with ovarian carcinoma is most likely related to the degree of peritoneal dissemination. Peritoneal dissemination is a unique metastatic process in which the behavior of carcinoma cells is possibly influenced by their microenvironment, such as hypoxia( 3 ) and various growth factors being present in the ascites of carcinoma patients.( 4 ) Epidermal growth factor (EGF), fibroblast growth factor (FGF), hepatocyte growth factor (HGF) and lysophosphatidic acid (LPA) have been reported to exist in ascitic or tumorous fluids and play roles in cell proliferation and migration.( 5 , 6 ) Recently, we have demonstrated that ovarian carcinoma cells express and secrete a calcium‐binding protein S100A4, also known as mts‐l/metastasin /pEL98/p9ka, which is an important autocrine/paracrine factor for tumor progression and a poor prognostic factor for patients.( 7 ) In addition, we showed that S100A4 enhances the invasiveness of ovarian cancer cells via upregulation and activation of RhoA,( 7 ) which also transduces the signal of EGF, FGF, HGF and LPA.( 8 , 9 , 10 )
The Rho family are ras‐related small guanosine triphosphatases (GTPases), consisting of Rho, Rac and CDC42 subfamilies.( 11 ) Rho family proteins have been known to re‐organize cytoskeletons and regulate cell migration via activation of effector proteins, Rho Kinase (ROCK).( 12 ) Upregulation of Rho has recently been reported in several human malignancies, such as breast, lung, pancreas and colon carcinomas, and testicular germ cell tumors.( 13 , 14 , 15 , 16 , 17 ) We previously reported that the expression of RhoA is significantly increased at both mRNA and protein levels in peritoneal metastatic lesions compared with primary tumors of advanced ovarian carcinomas.( 18 ) These findings strongly suggest that RhoA plays a central role in the tumor progression of ovarian carcinoma cells, and is a possible molecular target for novel treatment. Therefore, in the present study, we investigated further the role of RhoA in the peritoneal dissemination of ovarian carcinoma cells. After we confirmed the expression levels of RhoA in various ovarian cancer cell lines and normal ovarian surface epithelial (OSE) cells, we established the RhoA transfectant of an ovarian cancer cell line and analyzed whether forced overexpression of RhoA could alter the progressive behavior of cancer cells in vitro and in vivo in the nude mice model. Then, we examined the in vitro effect of Rho inhibitors, such as specific inhibitor of Rho (C3 exoenzyme), Rho kinase inhibitor (Y27632) and hydroxymethylglutaryl coenzyme A (HMG‐CoA) reductase inhibitor, (Lovastatin and Pravastatin), which modifies the carboxy termini of small GTPase proteins and decreased the activity of Rho.( 19 ) Finally, we tested whether Lovastatin could suppress the peritoneal dissemination of ovarian cancer cells in the nude mice model.
Materials and Methods
Cell culture.
Ovarian cancer cells: The ovarian cancer cell lines SKOV3 and OVCAR3 were purchased from the American Type Culture Collection (Rockville, MD, USA). The ovarian cancer cell lines A2780 and A2780/CDDP (a cisplatin‐resistant cell line derived from A2780) were kind gifts from Dr Takashi Tsuruo (Cancer Chemotherapy Center, Tokyo, Japan) with the permission of Dr Thomas C. Hamilton (Fox Chase Cancer Institute, Philadelphia, PA, USA). A2780, A2780/CDDP and OVCAR3 were maintained in Roswell Park Memorial Institute medium (RPMI 1640; Sigma‐Aldrich, St. Louis, MO) supplemented with 10% inactivated fetal bovine serum (FBS) (Biomeda, Foster City, CA, USA). SKOV3 was cultured in Dulbecco's modified Eagle's medium (DMEM) (Sigma‐Aldrich) with 10% FBS. Incubation was carried out at 37°C under 5% CO2 in air. For RT‐PCR and Western blotting, the cultured cells were transferred to a 100‐mm cell culture dish (8 × 105 cells/dish) and incubated at 37°C under 20% O2.
Normal ovarian surface epithelial (OSE) cells: Ovarian surface epithelium was obtained from three women who were treated surgically for benign gynecologic disease, after obtaining written consent. At the operation, OSE cells were collected by scraping the surface of ovaries with a surgical blade and immediately culturing on collagen type I‐coated plastic dishes (Iwaki Glass, Chiba, Japan) in a 1 : 1 mixture of Medium 199 (GibcoBRL, Grand Island, NY, USA) and MCDB 105 (Sigma‐Aldrich) containing 15% FBS.
DNA transfection. Wild‐type human RhoA complementary DNA (cDNA) subcloned into a pcDNA3 (Invitrogen, San Diego, CA, USA) vector was a kind gift from Dr Kazuyuki Itoh (Department of Tumor Biochemistry, Osaka Medical Center for Cancer and Cardiovascular Diseases, Japan)( 20 ) Twenty µg of the human RhoA cDNA construct were transfected into ovarian cancer cell lines SKOV3 (4 × 106 cells) by Effectene Transfection Reagent (Qiagen, Valencia, CA, USA). To enable the selection of neomycin‐resistant clones, transfected cells were maintained at 37°C in DMEM with 10% FBS containing 500 mg/mL of G418 (Wako, Osaka, Japan) in SKOV3. After 14 days, G418‐resistant colonies were harvested and maintained at 37°C in DMEM with 10% FBS under 5% CO2 in air.
Reverse transcription–polymerase chain reaction (RT‐PCR). Total RNA was extracted by the acid guanidinium–phenol–chloroform method. One microgram of total RNA was treated with 1 U/10 µL DNase I (Life Technologies, Gaithersburg, MD, USA). RT was performed using an RNA PCR Kit (Takara Shuzo, Otsu, Japan) as described previously.( 18 ) Using a thermal cycler (Applied Biosystems, GeneAmp PCR System 9700, Foster City, CA, USA), the reaction mixture was incubated at 42°C for 30 min, heated at 99°C for 5 min, and then cooled to 5°C for 5 min.
One microliter of the RT products, containing 50 ng reverse transcribed total RNA, was amplified by adding 20 µL of PCR reaction mixture containing 10 mm Tris‐HCl (pH 8.3), and 50 mm KCl, 2.5 units/100 µL of TaKaRa Taq DNA polymerase, to 0.2 µM of a set of oligonucleotide primers for RhoA or glyceraldehyde‐3‐phosphate dehydrogenase (G3PDH) cDNA.( 18 ) The corresponding cDNA fragments were denatured at 94°C for 30 s, annealed at 58°C for 1 min, and extended at 72°C for 1 min. After 30 cycles of amplification, the PCR products were analyzed on a 2% agarose gel, and the bands were visualized using ethidium bromide during exposure to an ultraviolet transilluminator.
Western blot analysis. Extracts equivalent to 30 µg of total protein were separated by sodium dodecylsulfate–polyacrylamide gel electrophoresis (SDS‐PAGE; 12% acrylamide) and transferred onto nitrocellulose membranes (Hybond‐C super, Amersham Biosciences, Piscataway, NJ, USA). The membranes were blocked in TBST (0.2 M NaCl, 10 mm Tris, pH 7.4, 0.2% Tween‐20) containing 5% non‐fat dry milk and 0.02% NaN3 for 1 h and then incubated with mouse monoclonal antibodies against against RhoA, Rho guanosine diphosphate dissociation inhibitor (GDI) and ROCK (Santa Cruz Biotechnology, Inc, Santa Cruz, CA), or β‐actin (Biomakor, Rehovot, Israel) in TBST containing 5% non‐fat dry milk. Membranes were then incubated with sheep anti‐mouse or rabbit immunoglobulin G (IgG; Amersham Biosciences) in TBST containing 2% non‐fat dry milk. Bound antibodies were detected with an enhanced chemiluminescence (ECL) system (Amersham Biosciences).
Cell proliferation assay. Cell number and viability were further assessed by using the reagent WST‐1 (Roche, Indianapolis, IN, USA). This colorimetric assay measures the metabolic activity of viable cells based on cleavage of the tetrazolium salt WST‐1 into formazan by mitochondrial dehydrogenase in live cells.
Transfected cells were plated at a density of 4 × 103 cells into a 96‐well flat‐bottomed tissue‐culture plate (Corning Laboratory Sciences, Corning, NY, USA). The cells were incubated for 48 and 72 h at 37°C with 5% CO2 in air in DMEM with 10% fetal calf serum. This was followed by incubation with WST‐1 reagent at a dilution of 1:10 in the original conditioned media for 4 h. After thorough shaking, the formazan produced by metabolically active cells in each sample was measured at a wavelength of 450 nm with Multiscan JX (Thermo Labsystems, Vantee, Finland). Absorbance readings were normalized against control wells.
In vitro invasion assay. Cell invasion through a reconstituted basement membrane, Matrigel (Becton Dickinson Labware, Bedford, MA, USA), was assayed by a method reported previously.( 21 ) Briefly, polycarbonate membranes (8.0 µM pore size) of the upper compartment of transwell culture chambers were coated with 5% Matrigel. Subconfluent cells were starved for 24 h and harvested with 0.05% trypsin containing 0.02% ethylenediaminetetraacetic acid (EDTA), washed twice with phosphate‐buffered saline (PBS) and resuspended at 1 × 106 cells/mL in serum‐free medium with 0.1% bovine serum albumin (BSA) as the control, C3 exoenzyme (ALEXIS Biochemicals, Lausen, Switzerland), Y27632 (Calbiochem, La Jolla, CA) and hydroxymethylglutaryl coenzyme A (HMG–CoA) reductase inhibitor (Lovastatin and Pravastatin; Wako, Osaka, Japan). The cell suspension 100 µL was placed in the upper compartment, and the lower compartment was immediately filled with 500 µL of serum‐free medium containing 0.1% BSA. After 22 h of incubation, the cells on the upper surface of the filter were removed carefully with a cotton swab, the membranes were stained with Diff‐Quik solution (Kokusai‐Shiyaku, Kobe, Japan) and cells that had migrated through the membrane to the lower surface were counted in ten different fields under a light microscope at ×400 magnification. Each experiment was performed in triplicate wells and repeated three times.
Rho–GTP pull‐down assay. The Rho–GTP pull‐down assay was performed using a Rho Activation Assay Biochem Kit (Cytoskeleton, Denver, CO, USA). In brief, all cells were washed with ice‐cold PBS and lysed with lysis buffer. After centrifugation at 4000 g for 5 min, Rhotekin (Rho effector)‐Rho binding domain (RBD) protein, which has high affinity for activated GTP–Rho, bound to glutathione‐agarose, was added to the cell lysate and incubated for 1 h at 4°C. After centrifugation, Rho–GTP bound to beads was washed with lysis buffer and eluted in Laemmli sample buffer. Proteins were analyzed by Western blotting using antibodies against RhoA.
Nude mice tumorigenicity assay. Transfected ovarian cancer cells were trypsinized, and then resuspended in DMEM and 5% FCS at a concentration of 1 × 107/mL. Six‐week‐old‐nude mice (BALB/c Slc‐nu; Japan SLC Co, Hamamatsu, Japan) were injected with 5 × 106 cells in the abdominal cavity. Two similar experiments were conducted 8 weeks apart, the only difference being the number of mice involved. The care and use of these experimental animals was in accordance with institutional guidelines. The mice were observed weekly, and sacrificed after 2 months. Individual disseminated tumors were counted and excised.
Drug administration. Pharmacokinetic studies have shown that HMG–CoA reductase inhibitors exhibit liver selectivity in inhibiting sterol synthesis and have elimination half‐lives of 1.2–3 h;( 22 ) however, Lovastatin did not show such selectivity. Therefore, we examined the in vivo effect of Lovastatin on a mouse dissemination model of ovarian cancer. Lovastatin (Wako, Osaka, Japan) was freshly prepared as a 300‐mg/mL solution in ethanol, and pH was adjusted to 7.4. Animals injected with ovarian cancer cells into the abdominal cavity were randomly divided into two different groups: control and Lovastain 3 ng/g. Lovastain was administered to 10‐week‐old mice orally per day at a level corresponding to the human dosage. The control group was given an equal volume of 0.05% ethanol vehicle.
Histology and immunohistochemical analysis. The tumors were fixed in 4% paraformaldehyde for 24 h and then embedded in paraffin. Sections were prepared for hematoxylin and eosin staining. Immunohistochemical staining was performed on formalin‐fixed paraffin‐embedded sections by the streptavidin–biotin–peroxidase complex method using a Histofine SAB‐PO kit (Nichirei, Tokyo, Japan). The primary antibody was an anti‐Ki‐67 mouse monoclonal antibody (Dako, Grostrup, Denmark). Three‐µm‐thick sections were deparaffinized and boiled in 0.01 M citrate buffer (pH 6.0) for 25 min in a microwave oven. They were then treated with 0.3% hydrogen peroxide and incubated with 10% normal mouse serum to block non‐specific binding. The sections were then incubated with a primary antibody for ki‐67 (MIB‐1) (DAKO, Glostrup, Denmark) at 4°C overnight. After washing in PBS, they were incubated with biotinylated goat anti‐mouse IgG, followed by treatment with peroxidase‐conjugated streptavidin and stained with diaminobenzidine and 0.15% hydrogen peroxidase. Counterstaining was performed with hematoxylin. Immunoreactive tumor cells were quantitatively estimated. Immunostaining was evaluated by two independent observers (A.H. and N.K.) unaware of the fate of the tissue site.
Statistical analyses. Statistical analysis was performed with the Kruskal–Wallis rank test, Scheffe and Mann–Whitney's U‐test. Differences were considered significant when P < 0.05.
Results
Expression of RhoA was increased in ovarian cancer cells compared with OSE cells. First, we examined mRNA and protein expressions of RhoA in cultured normal OSE cells and four ovarian cancer cell lines (SKOV3, OVCAR3, A2780, A2780/CDDP). An intense band for RhoA mRNA was observed in ovarian cancer cells, whereas a faint or weak band was observed in normal OSE cells, which is thought to be the histogenetic origin of ovarian cancer (Fig. 1a). A specific band for G3PDH‐mRNA was also detected at the predicted 226 base pair in all cultured cells (Fig. 1a).
Figure 1.
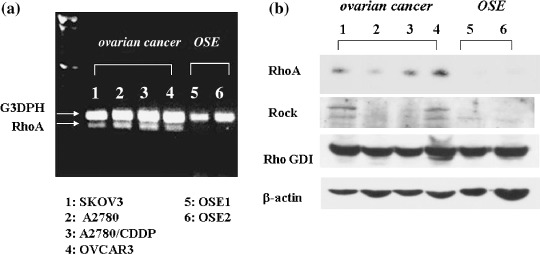
Expression of RhoA mRNA and protein analyzed in ovarian cancer cell lines and ovarian surface epithelium (OSE) cells. (a) An intense band for RhoA mRNA was observed in all four ovarian cancer cells (lane 1–4), whereas a faint or weak band was observed in normal OSE cells (lane 5–6). A specific band for G3PDH–mRNA was also detected at the predicted 226 base pair in all cultured cells. (b) The bands of RhoA protein and the effector protein, ROCK are observed in ovarian cancer cell lines (lane 1–4), but not in OSE cells (lane 5–6). In contrast, bands of Rho–GDI were observed in all ovarian cancer cells and OSE cells.
Analysis of RhoA protein expression by Western blotting revealed a band at 21 kDa (Fig. 1b). Consistent with RhoA mRNA expression, the bands of RhoA protein in OSE cells were either below the limit of detection or only faintly detectable. In contrast, all cancer cell lines exhibited RhoA protein expression. Accordingly, the levels of RhoA mRNA and protein in ovarian cancer cells were higher than that of normal OSE cells.
To compare the activity of in vivo disseminated metastasis among the four ovarian cancer cell lines, we examined the successful rate of dissemination after intraperitoneal injection of these cells into nude mice. The dissemination rate was 1/11 mice (9%) in OVCAR3, 3/11 (27%) in SKOV3, 10/12 (83%) in A2780 and 10/12 (83%) in A2780/CDDP, respectively. Accordingly, the disseminated metastatic activity in vivo was correlated with the expression levels of RhoA mRNA of ovarian cancer cells.
Overexpression of RhoA increased the invasiveness of ovarian cancer cells. Wild‐type RhoA and control vector were introduced into SKOV3 cells. Wild‐type RhoA‐transfected cells (RhoA‐transfected) and vector‐transfected cells were analyzed for RhoA transcripts by RT‐PCR amplification (Fig. 2a). Although the band for G3PDH was the same level for all these cell lines, a band for RhoA was elevated in RhoA‐transfected cells compared with parent cells and vector‐transfected cells. Analysis of Western blotting also showed elevated RhoA protein expression in RhoA‐transfected cells compared with parent cells and vector‐transfected cells (Fig. 2a). In addition, Rho–GTP pull‐down assay revealed that RhoA‐transfected cells increased the amount of the GTP‐binding form of RhoA (Fig. 2a). These results confirmed that the expression of RhoA protein was increased and activated in RhoA‐transfected cells.
Figure 2.
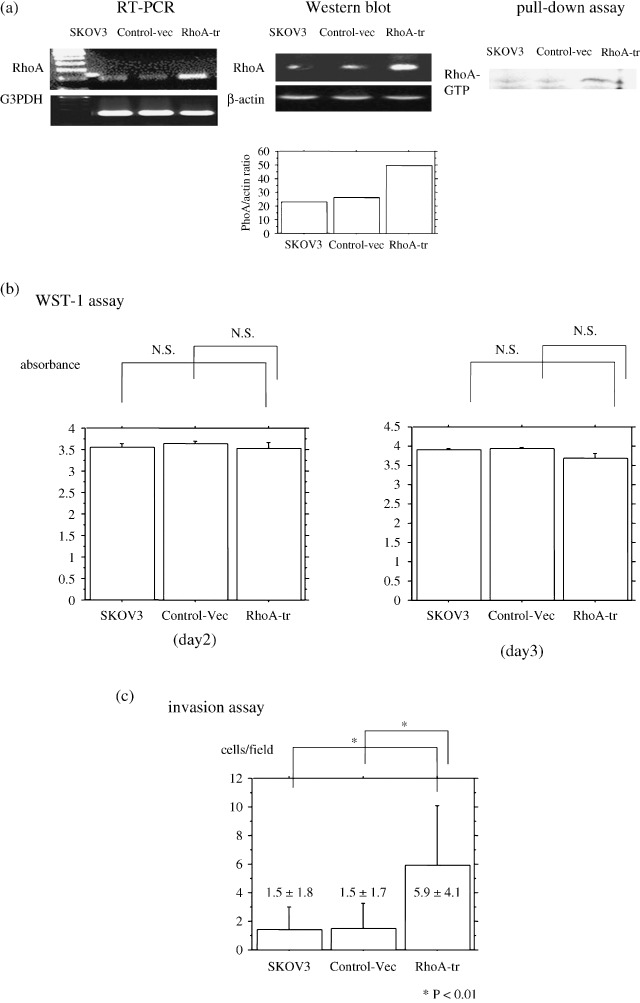
(a) Reverse transcriptase–polymerase chain reaction (RT‐PCR) for RhoA. An intense band for RhoA was observed in RhoA‐transfected ovarian cancer cells. The 226 bp band for G3PDH was obtained for all RT reactions. Analysis of RhoA protein by Western blotting. A stronger band of protein at 21 kDa for RhoA was revealed in RhoA‐transfected ovarian cancer cells compared with vector‐transfected cells and non‐transfected cells. A band (42 kDa) for β‐actin was observed in protein extracts from all cell lines. In addition, a Rho–GTP pull‐down assay revealed that RhoA‐transfected cells increased the amount of the GTP‐binding form of RhoA. (b) Effect of RhoA on the proliferation rate of cells cultured from RhoA‐transfected cells or from vector‐transfected cells. WST1 assay showed that the number of cultured cells per 96‐well plate was not changed by RhoA transfection at 3 day's incubation. Data are expressed as the means with 95% confidence intervals. P‐value is two‐sided and significant at P < 0.05. (c) Effect of RhoA on cell invasiveness. The Matrigel invasion assay showed that the number of invaded cells increased by RhoA transfection.
First, we analyzed the effect of RhoA overexpression on cell growth; however, a WST‐1 assay showed that the number of cultured cells per 96‐well plate did not change following RhoA transfection of SKOV3 cells at 48, and 72 h (Fig. 2b). To analyze in vitro invasiveness, the cells were incubated for 22 h on Matrigel‐coated membranes of culture chambers. The count of cells that passed through the Matrigel‐coated membranes showed that the invasive activity of RhoA‐transfected cells was significantly increased compared with that of parent cells and vector‐transfected cells (Fig. 2c).
To further examine the effect on disseminated metastatic activity in vivo, we implanted RhoA‐transfected ovarian cancer cells into the abdominal cavity of nude mice. Although 3 of 11 (1/5 and 2/6) mice injected with vector‐transfected cells developed disseminated tumors, RhoA‐transfected cells did induce tumors in 9 of 12 (5/6 and 4/6) mice 2 months after injection (respectively in the two similar experiments) (Fig. 3a). The frequency of nude mouse dissemination of RhoA‐transfected SKOV3 cells was significantly elevated compared with vector cells after 2 months (P = 0.014).
Figure 3.
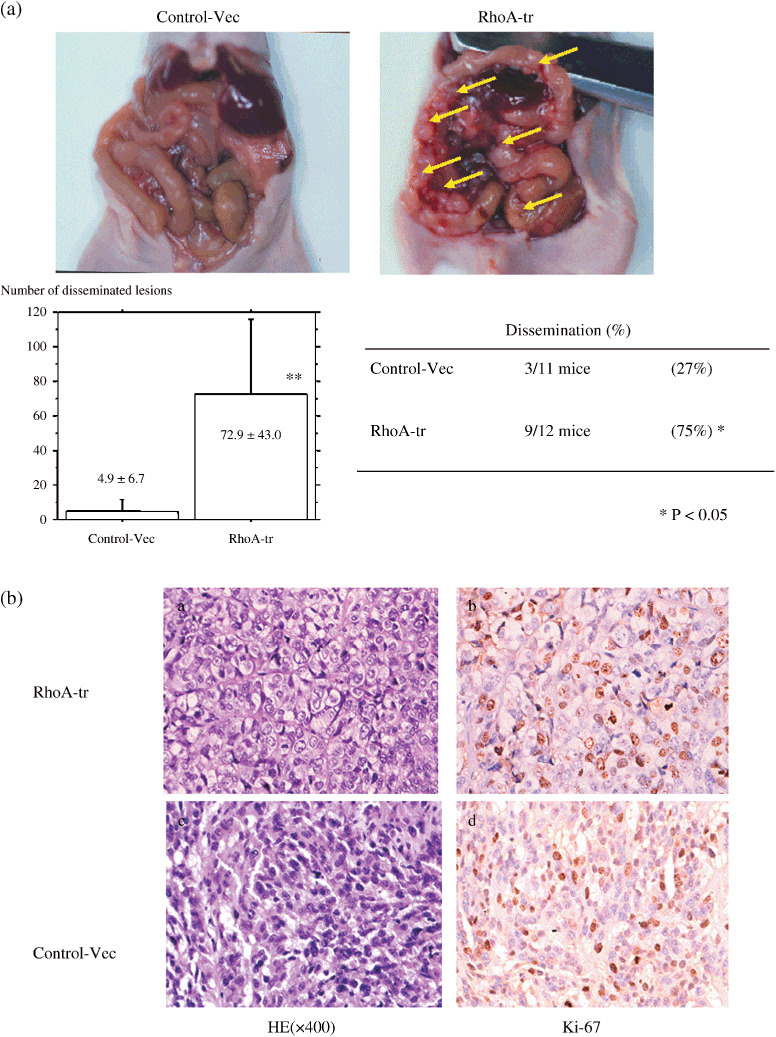
(a) Nude mouse tumorigenicity assay. RhoA‐transfected cells induced many small tumors 2 months after injection, whereas a small number of tumors developed in mice injected with vector‐transfected cells. The frequency of nude mouse dissemination of RhoA‐transfected SKOV3 cells was significantly elevated compared with vector cells after 2 months (P = 0.014). (b) Pathological analysis of tumors (Fig. 3a). Pathologically, tumors induced by RhoA‐transfected cells showed prominent nucleoli; however, nucleoli did not show a difference in Ki‐67 positivety in a histology examination of the sowing focus compared with the control, as for RhoA‐transfected cells (×400).
Moreover, the number of metastatic sites significantly increased in RhoA‐transfected cells (72.9 ± 43.0) compared with vector‐transfected cells (4.9 ± 6.7) (P < 0.0001). These results suggested that the dissemination capacity in RhoA‐transfected cells is much higher than that of vector‐transfected controls. Pathologically, tumors induced by RhoA‐transfected cells showed prominent nucleoli (Fig. 3b); however, they did not show a difference in Ki‐67 positivity compared with the control.
Rho inhibitor reduced the invasiveness of RhoA‐transfected cancer cells in vitro. To confirm the involvement of the Rho pathway in the stimulation of invasion, we analyzed the in vitro effect of Rho inhibitors, such as C3 exoenzyme, Y27632, Lovastatin and Pravastatin, on RhoA‐transfected cells. When RhoA‐transfected SKOV3 cells were treated with C3 exoenzyme, Y27632, Lovastatin and Pravastatin for 22 h, the GTP‐binding form of RhoA was decreased (Fig. 4a). In addition, the treatment with C3 exoenzyme, Y27632, Lovastatin and Pravastatin suppressed the number of invaded cells (Fig. 4b).
Figure 4.
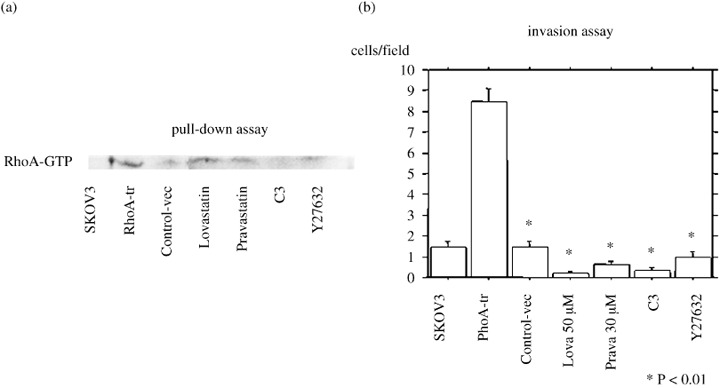
Effect of blocking the Rho signal transduction pathway in RhoA‐transfected cell invasion. (a) When RhoA‐transfected SKOV3 cells were treated with C3 exoenzyme, Y27632, Lovastatin or Pravastatin for 22 h, the GTP‐binding form of RhoA was decreased. (b) Treatment with C3 exoenzyme, Y27632, Lovastatin or Pravastatin suppressed the number of invaded cells. The invasive capacity of RhoA‐transfected cells depended on Rho signaling. Data are expressed as the mean ± SEM of three separate experiments in triplicate wells. *P < 0.01 versus control cells with RhoA‐transfected cells.
Lavastatin treatment was effective for peritoneal dissemination in nude mice. Finally, we examined the in vivo effect of Rho inhibitor using a dissemination model in nude mice. One month after injection of RhoA‐transfected cells into the mouse abdominal cavity, oral Lovastatin was administered by gastric tube for 4 weeks. Although there was no statistical difference in the presence or absence of peritoneal dissemination between with or without Lovastatin administration, the number of metastatic sites significantly were decreased with Lovastain oral administration (94.7 ± 23.0: mean ± SD) compared with the control (46.3 ± 17.0) (P = 0.0014). (Fig. 5)
Figure 5.
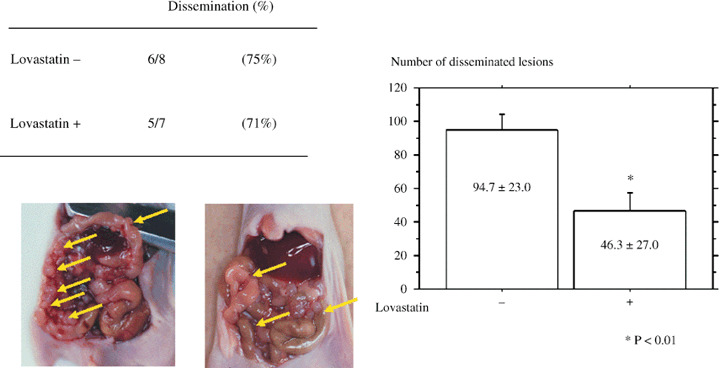
Nude mouse tumorigenicity assay. One month after injection of RhoA‐transfected cells into the mouse abdominal cavity, oral Lovastatin was administered by gastric tube for 4 weeks. The number of metastatic sites significantly decreased by Lovastain oral administration compared with the control (P = 0.0014). Values are the means ± SEM. (n = 5–6).
Discussion
Small GTPases Rho have been reported to control cell motility and adhesion rather than cell proliferation.( 23 ) Several studies indicated that a basal level of RhoA activity is necessary for adherens junction assembly; however, high RhoA activity disrupts adherens junction assembly and may lead to cell invasion.( 24 , 25 ) In the present study, we investigated whether overexpression of RhoA could alter the progressive behavior of ovarian cancer cells. We first confirmed that basal expression of RhoA in ovarian cancer cell lines is increased at mRNA and protein levels compared with that of normal OSE cells, and then established the stable transfectant of RhoA of an ovarian cancer cell line SKOV3, which expresses constitutively an increased expression of RhoA associated with an increase of activated form of RhoA. The transfection of RhoA in the ovarian cancer cell did not show the change in cell proliferation, but increased the in vitro invasiveness. Additionally, our experiment in vivo using the nude mice model for peritoneal dissemination showed that both the frequency of peritoneal dissemination and the number of disseminated lesions in the abdominal cavity were significantly increased in the RhoA‐transfected SKOV3 cells compared with those in vector‐transfected or original SKOV3 cells. These data strongly suggest that overexpression and/or activation of RhoA play an important role in the invasiveness and in the peritoneal dissemination of ovarian cancer cells.
We previously reported that the expression of RhoA was significantly increased at mRNA and protein levels in ovarian carcinomas compared with benign and borderline tumors.( 18 ) Among various histological types of ovarian carcinoma, the highest expression of RhoA was seen in serous papillary carcinoma, which is more frequently associated with peritoneal dissemination than other histological types. In addition, the expression levels of RhoA were significantly higher in tumors with peritoneal dissemination in abdominal cavity than in tumors localized in the pelvic cavity,( 18 ) suggesting an importance of RhoA in the disseminated metastasis. Moreover, we found that in vivo disseminated metastatic activity in nude mice was correlated with RhoA mRNA expression level of ovarian cancer cells. It is thought that ovarian carcinoma cells during the dissemination are under the influence of their microenvironment; the tumor hypoxia attenuates the expression of E‐cadherin and increases the invasiveness of ovarian cancer cells.( 3 ) Various growth factors such as EGF, FGF, HGF, LPA and S100A4 present in their microenvironment could affect the behavior of ovarian carcinoma cells, and we have shown that all of these growth factors upregulated and/or activated RhoA.( 9 ) Accordingly, it is likely that overexpression and activation of RhoA play an important role in the disseminated metastasis of ovarian carcinoma cells, and RhoA could be a novel therapeutic target in treating cancer cell invasion and metastasis.
Although a number of strategies targeting Rho were searched over the last several years,( 26 ) relatively few drugs or inhibitors for the Rho GTPase signaling pathway have been developed. Specific inhibitor of Rho (C3 exoenzyme) and Rho kinase inhibitor (Y27632) are used only in the experiment in vitro. RhoA small interfering RNA (siRNA) gene therapy mediated by adenovirus has been tested on gastric cancer cells in vitro.( 27 ) For the administration in vivo, researchers have focused attention on the HMG–CoA reductase inhibitor, especially statins, which modifies the carboxy termini of small GTPase proteins and decreased the activity of Rho.( 19 )
Statins are the most widely used drugs to lower serum cholesterol levels. For cancer prevention, several epidemiological studies showed that people taking statins reduce their risk of pancreatic, prostate, colorectal and breast cancer,( 28 , 29 ) although a recent report claimed such protective effect of statins against cancer development. ( 30 ) For cancer treatment, Lovastatin has been reported to inhibit metastasis in breast, pancreatic and thyroid carcinoma and lymphoma cells.( 31 , 32 , 33 , 34 ) The most important prognostic factor in ovarian cancer patients is disseminated metastasis. The 5‐year survival of advanced ovarian cancer patients with peritoneal dissemination is around 30% and has not been improved for years. Therefore, we also tested Lovastatin in the nude mice model for peritoneal dissemination of RhoA‐transfected ovarian cancer cells. Oral administration of Lovastatin for 4 weeks resulted in a significant decrease in the number of metastatic lesions. In the present study, we administered Lovastatin for a limited period after the establishment of peritoneal dissemination. Therefore, further analyses using its earlier and longer administration are necessary to test whether this drug has the prophylactic effect for peritoneal dissemination or has the effect for improvement of survival.
This is the first report on the effect of statin for the treatment of peritoneal dissemination of cancer cells. Recently, a Phase I trial of Lovastatin in patients with recurrent or metastatic squamous cell carcinoma of the head and neck showed that 23% of patients had stable disease for more than 3 months.( 35 )
Based on the results of the current experiment and on the previously reported data, it is possible that small GTPase RhoA plays a central role in signal transduction for the peritoneal dissemination of ovarian carcinoma cells, and RhoA would be a powerful target in molecular target therapy for human ovarian carcinoma. Clinical trials using statins for advanced ovarian carcinoma patients are urgently required to develop a novel effective treatment for this disease.
Acknowledgments
We thank Dr Kazuyuki Itoh (Department of Tumor Biochemistry, Osaka Medical Center for Cancer and Cardiovascular Diseases, Japan) for providing the wild‐type human RhoA cDNA subcloned into a pcDNA3 (Invitrogen, San Diego, CA) vector.
This work was supported in part by a Grant‐in‐Aid for Scientific Research to I.K. (No. 19390426) and A.H. (No. 18591828) from the Ministry of Education, Science and Culture, Japan.
References
- 1. Ozols RF, Rubin SC, Thomas GM et al . Epithelial ovarian cancer. In: Hoskins WJ, Perez CA, Young RC, eds. Principles and Practice of Gynecologic Oncology. Philadelphia: Lippincott Williams & Wilkins, 2000: 981–1057. [Google Scholar]
- 2. Mutch DG, Williams S. Biology of epithelial ovarian cancer. Clin Obstet Gynecol 1994; 37: 406–22. [DOI] [PubMed] [Google Scholar]
- 3. Imai T, Horiuchi A, Wang C et al . Hypoxia attenuates the expression of E‐cadherin via up‐regulation of SNAIL in ovarian carcinoma cells. Am J Pathol 2003; 163: 1437–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Garofalo A, Naumova E, Manenti L et al . The combination of the tyrosine kinase receptor inhibitor SU6668 with paclitaxel affects ascites formation and tumor spread in ovarian carcinoma xenografts growing orthotopically. Clin Cancer Res 2003; 9: 3476–85. [PubMed] [Google Scholar]
- 5. Xu Y, Gaudette DC, Boynton JD et al . Characterization of an ovarian cancer activating factor in ascites from ovarian cancer patients. Clin Cancer Res 1995; 1: 1223–32. [PubMed] [Google Scholar]
- 6. Westermann AM, Havik E, Postma FR et al . Malignant effusions contain lysophosphatidic acid (LPA)‐like activity. Ann Oncol 1998; 9: 437–42. [DOI] [PubMed] [Google Scholar]
- 7. Kikuchi N, Horiuchi A, Osada R et al . Nuclear expression of S100A4 is associated with aggressive behavior of epithelial ovarian carcinoma: an important autocrine/paracrine factor in tumor progression. Cancer Sci 2006; 97: 1061–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Goetzl EJ, An S. Diversity of cellular receptors and functions for the lysophospholipid growth factors lysophosphatidic acid and sphingosine 1‐phosphate. FASEB J 1998; 12: 1589–98. [PubMed] [Google Scholar]
- 9. Kikuchi N, Horiuchi A, Imai T, Osada R, Konishi I. Nuclear expression of S100A4 protein is a maker for unfavorable outcomes in patients in patients with ovarian carcinoma. 11th Biennial Meeting of International Gynecologic Cancer Society. CA, USA: Santa Monica, 2006. [Google Scholar]
- 10. Horiuchi A, Kikuchi N, Imai T et al . Molecular target therapy in peritoneal dissemination of ovarian cancer. Jap J Gynecol Oncol 2005; 23: 234–8. [Google Scholar]
- 11. Jaffe AB, Hall A. Rho GTPases in transformation and metastasis. Adv Cancer Res 2002; 84: 57–80. [DOI] [PubMed] [Google Scholar]
- 12. Raftopoulou M, Hall A. Cell migration: Rho GTPases lead the way. Dev Biol 2004; 265: 23–32. [DOI] [PubMed] [Google Scholar]
- 13. Suwa H, Ohshio G, Imamura T et al . Overexpression of the rhoC gene correlates with progression of ductal adenocarcinoma of the pancreas. Br J Cancer 1998; 77: 147–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fritz G, Just I, Kaina B. Rho GTPases are over‐expressed in human tumors. Int J Cancer 1999; 81: 682–7. [DOI] [PubMed] [Google Scholar]
- 15. Van Golen KL, Wu ZF, Qiao XT, Bao LW, Merajver SD. RhoC GTPase, a novel transforming oncogene for human mammary epithelial cells that partially recapitulates the inflammatory breast cancer phenotype. Cancer Res 2000; 60: 5832–8. [PubMed] [Google Scholar]
- 16. Kamai T, Arai K, Tsujii T, Honda M, Yoshida K. Overexpression of RhoA mRNA is associated with advanced stage in testicular germ cell tumour. BJU Int 2001; 87: 227–31. [DOI] [PubMed] [Google Scholar]
- 17. Kleer CG, Van Golen KL, Zhang Y, Wu ZF, Rubin MA, Merajver SD. Characterization of RhoC expression in benign and malignant breast disease: a potential new marker for small breast carcinomas with metastatic ability. Am J Pathol 2002; 160: 579–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Horiuchi A, Imai T, Wang C et al . Up‐regulation of small GTPases, RhoA and RhoC, is associated with tumor progression in ovarian carcinoma. Lab Invest 2003; 83: 861–70. [DOI] [PubMed] [Google Scholar]
- 19. Laufs and Liao . 2000 Fritz GHMG‐CoA reductase inhibitors (statins) as anticancer drugs. Int J Oncol 2005; 27: 1401–9. [PubMed] [Google Scholar]
- 20. Yoshioka K, Matsumura F, Akedo H, Itoh K. Small GTP‐binding protein Rho stimulates the actomyosin system, leading to invasion of tumor cells. J Biol Chem 1998; 273: 5146–54. [DOI] [PubMed] [Google Scholar]
- 21. Albini A, Iwamoto Y, Kleinman HK et al . A rapid in vitro assay for quantitating the invasive potential of tumor cells. Cancer Res 1987; 47: 3239–45. [PubMed] [Google Scholar]
- 22. Plosker GL, Wagstaff AJ. Fluvastatin: a review of its pharmacology and use in the management of hypercholesterolaemia. Drugs 1996; 51: 433–59. [DOI] [PubMed] [Google Scholar]
- 23. Adamson P, Marshall CJ, Hall A, Tilbrook PA. Post‐translational modifications of p21rho proteins. J Biol Chem 1992; 267: 20033–8. [PubMed] [Google Scholar]
- 24. Cavallaro U, Christofori G. Cell adhesion and signalling by cadherins and Ig‐CAMs in cancer. Nat Rev Cancer 2004; 4: 118–32. [DOI] [PubMed] [Google Scholar]
- 25. Cáceres M, Guerrero J, Martínez J. Overexpression of RhoA‐GTP induces activation of the epidermal growth factor receptor, dephosphorylation of focal adhesion kinase and increased motility in breast cancer cells. Exp Cell Res 2005; 309: 229–38. [DOI] [PubMed] [Google Scholar]
- 26. Walker K, Olson MF. Targeting Ras and Rho GTPases as opportunities for cancer therapeutics. Curr Opin Genet Dev 2005; 15: 62–8. [DOI] [PubMed] [Google Scholar]
- 27. Pillé JY, Denoyelle C, Varet J et al . Anti‐RhoA and anti‐RhoC siRNAs inhibit the proliferation and invasiveness of MDA‐MB‐231 breast cancer cells in vitro and in vivo. Mol Ther 2005; 11: 267–74. [DOI] [PubMed] [Google Scholar]
- 28. Poynter JN, Gruber SB, Higgins PD et al . Statins and the risk of colorectal cancer. N Engl J Med 2005; 352: 2184–92. [DOI] [PubMed] [Google Scholar]
- 29. Boudreau DM, Gardner JS, Malone KE, Heckbert SR, Blough DK, Daling JR. The association between 3‐hydroxy‐3‐methylglutaryl conenzyme A inhibitor use and breast carcinoma risk among postmenopausal women: a case‐control study. Cancer 2004; 100: 2308–16. [DOI] [PubMed] [Google Scholar]
- 30. Caruso MG, Osella AR, Notarnicola M et al . Prognostic value of low density lipoprotein receptor expression in colorectal carcinoma. Oncol Rep 1998; 5: 927–30. [DOI] [PubMed] [Google Scholar]
- 31. Matar P, Matar P, Rozados VR, et al . Inhibitory effect of Lovastatin on spontaneous metastases derived from a rat lymphoma. Clin Exp Metastasis 1999; 17: 19–25. [DOI] [PubMed] [Google Scholar]
- 32. Zhong WB, Liang YC, Wang CY, Chang TC, Lee WS. Lovastatin suppresses invasiveness of anaplastic thyroid cancer cells by inhibiting Rho geranylgeranylation and RhoA/ROCK signaling. Endocr Relat Cancer 2005; 12: 615–29. [DOI] [PubMed] [Google Scholar]
- 33. Kusama T, Mukai M, Iwasaki T et al . 3‐hydroxy‐3‐methylglutaryl‐coenzyme a reductase inhibitors reduce human pancreatic cancer cell invasion and metastasis. Gastroenterology 2002; 122: 308–17. [DOI] [PubMed] [Google Scholar]
- 34. Alonso DF, Farina HG, Skilton G, Gabri MR, De Lorenzo MS, Gomez DE. Reduction of mouse mammary tumor formation and metastasis by lovastatin, an inhibitor of the mevalonate pathway of cholesterol synthesis. Breast Cancer Res Treat 1998; 50: 83–93. [DOI] [PubMed] [Google Scholar]
- 35. Knox JJ, Siu LL, Chen E et al . A Phase I trial of prolonged administration of lovastatin in patients with recurrent or metastatic squamous cell carcinoma of the head and neck or of the cervix. Eur J Cancer 2005; 41: 523–30. [DOI] [PubMed] [Google Scholar]