

Abstract
Although some kinds of bile acids have been implicated in colorectal cancer development, the mechanism of cancer progression remains unexplored in hepatobiliary cancer. From our personal results using complementary DNA microarray, we found that chenodeoxycholic acid (CDCA) induced Snail expression in human carcinoma cell lines derived from hepatocellular carcinoma and cholangiocarcinoma. Snail expression plays an important role in the regulation of E‐cadherin and in the acquisition of invasive potential in many types of human cancers including hepatocellular carcinoma. We found that CDCA and lithocholic acid (LCA) induced Snail expression in a concentration‐dependent manner and down‐regulated E‐cadherin expression in hepatocellular carcinoma and cholangiocarcinoma cell lines. Moreover, Snail short interference RNA (siRNA) treatment reduced the down‐regulation of E‐cadherin by CDCA or LCA. Luciferase analysis demonstrated that the promoter region from –111 to –24 relative to the transcriptional start site was necessary for this induction and, at least in part, nuclear factor Y (NF‐Y) and stimulating protein 1 (Sp1) might be an inducer of Snail expression in response to bile acids. In addition, using an in vitro wound healing assay and invasion assay, we observed that CDCA and LCA induced cell migration and invasion. These results suggest that bile acids repress E‐cadherin through the induction of transcription factor Snail and increase cancer invasiveness in human hepatocellular carcinoma and cholangiocarcinoma. Inhibition of this bile acid‐stimulated pathway may prove useful as an adjuvant in the therapy of hepatocellular carcinoma. (Cancer Sci 2008; 99: 1785–1792)
Bile acids are commonly considered as physiological detergents that facilitate the absorption, transport and distribution of lipid‐soluble vitamins and dietary fats. Recently, bile acids have also been shown to exert signaling activities leading to the modulation of the expression of genes involved in their own synthesis and transport.( 1 , 2 , 3 , 4 ) Moreover, previous reports have indicated that bile acids can promote carcinogenesis and cancer progression, especially colon cancer, by stimulating a variety of signaling pathways.( 5 , 6 , 7 ) Deoxycholic acid (DCA) has been shown to modulate p53 gene expression in a colonic adenoma cell line.( 8 ) It was reported that the prolonged deregulated expression of activator protein‐1 (AP‐1) activity in colonic cells by certain bile acids contributed to tumor promotion in the colon.( 9 , 10 ) Bile acids were also reported to be involved in the induction of cyclooxygenase‐2 (COX‐2), the secretion of matrix metalloproteinase‐2 (MMP‐2) and the migration of colorectal cancer cells.( 11 , 12 ) Thus, some kinds of bile acids have been shown to act as tumor promoters in colon cancer. More recently, chenodeoxycholic acid (CDCA) has also been shown to activate the epidermal growth factor receptor (EGFR) and to induce COX‐2 expression in hepatocellular carcinoma and cholangiocarcinoma cell lines.( 13 , 14 ) These results have suggested that bile acid can also promote carcinogenesis and cancer progression in hepatocytes and cholangiocytes. However, in hepatocytes and cholangiocytes, which are always exposed to bile acids, the mechanisms of bile acid‐induced carcinogenesis and cancer progression are poorly understood. Moreover, from our personal results using complementary DNA (cDNA) microarray gene expression profiling approach to identify novel genes regulated by primary bile acids (CDCA), we found that CDCA induced Snail expression in hepatocellular carcinoma and cholangiocarcinoma cell lines.
Increased cell invasion is a key phenotypic advantage of malignant cells favoring metastasis. The invasion process involves the loss of cell–cell interactions together with the gain of proteolytic and migratory properties and is often referred to as epithelial‐mesenchymal transition (EMT).( 15 ) E‐cadherin is a cell–cell adhesion molecule especially expressed on the membrane of epithelial cells, and a decrease of its expression has been reported in the invasion and metastasis of cancers.( 16 , 17 , 18 ) It was reported that DCA caused a significant loss of E‐cadherin binding with beta‐catenin, which is associated with an increase in cancer cell invasiveness as reflected by increase in the number of migrating cells.( 19 ) The zinc finger transcriptional factor Snail represses E‐cadherin transcription in vitro and in vivo by binding to E‐boxes of the E‐cadherin promoter. Snail was found to evoke tumorigenic and invasive properties in epithelial cells.( 20 ) Nevertheless, the mechanisms governing the expression of human Snail are starting to be identified.( 21 , 22 )
Our purpose of this study is to identify the mechanism of Snail gene induction by bile acids and whether this induction participated in the progression of cancer cells.
Materials and Methods
Materials. Cholic acid (CA), deoxycholic acid (DCA), chenode‐oxycholic acid (CDCA), lithocholic acid (LCA), ursodeoxycholic acid (UDCA), taurocholic acid (TCA), taurodeoxycholic acid (TDCA), taurochenodeoxycholic acid (TCDCA), taurolithocholic acid (TLCA), tauroursodeoxycholic acid (TUDCA), glycocholic acid (GCA), glycodeoxycholic acid (GDCA) and glycochenodeoxycholic acid (GCDCA) were obtained from Sigma Chemical (St. Louis, MO, USA), glycolithocholic acid (GLCA) was obtained from Calbiochem (San Diego, CA, USA) and glycoursodeoxycholic acid (GUDCA) was obtained from Mitsubishi Pharma Corporation (Osaka, Japan), and all of the bile acids were maintained as 100‐mM stock solutions in dimethyl sulfoxide (DMSO). The stock solution was stored at –20°C. Mouse monoclonal anti‐E‐cadherin antibody (G‐10), goat polyclonal antiactin antibody (I‐19) and horseradish peroxidase (HRP)‐linked antibody (goat antimouse for E‐cadherin; donkey antigoat for actin) were purchased from Santa Cruz Biotechnology, Inc (Santa Cruz, CA, USA).
Cell culture and treatment. The THLE‐3 (American Type Culture Collection [ATCC], Bethesda, MD, USA), which was derived from human primary normal liver cells( 23 ) were cultured in BEGM® BulletKit® (Clonetic corp, MD, USA) supplemented with 10% dialyzed fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA, USA), 5 ng/mL epidermal growth factor (EGF), and 70 ng/mL phosphoethanolamine. Human tumor‐derived cells, Hep3B hepatocellular carcinoma and HuCCT‐1 cholangiocarcinoma, were cultured in Roswell Park Memorial Institute medium (RPMI)‐1640 (Sigma Chemical) supplemented with 10% dialyzed FBS, 100 mg/mL penicillin, and 100 U/mL streptomycin. The cells were maintained at 37°C in a humidified atmosphere of 5% CO2. The cell culture was passed every three or four days.
Microarray. AceGene (Human oligo chip 30K, subset A; DNA Chip Research Inc. and Hitachi Software Engineering, Yokohama, Japan) containing 10 000 genes was used for mRNA expression profiling. (The protocol is available at http://www.dna‐chip.co.jp/thesis/AceGeneProtocol.pdf.) Four µg of total RNA was amplified with an Amino Allyl MessageAmp aRNA kit (Ambion, Austin, TX, USA). Amino‐allyl‐labeled cRNA was purified and then 5 µg of cRNA was labeled with Cy5 Mono‐reactive Dye (Amersham Biosciences Inc., Tokyo, Japan) or Cy3 Mono‐reactive Dye (Amersham Biosciences Inc.), according to the protocol of Hitachi Software Engineering. The arrays were scanned using a GenePix 4000B microarray scanner (Axon Instruments Inc., Union City, CA, USA).
Data were obtained from quadruplicates of each cell lines. The digitized image data were processed using GenePix Pro 4.1 software (Axon Instruments). The result files were imported into the GeneSpring 6.1 software (Silicon Genetics, Redwood City, CA, USA) and analyzed for gene expression differences. Data were normalized using per‐spot and per‐chip intensity dependent (Lowess) normalization. The Cross Gene Error model was activated and based on replicates. To find differentially expressed genes, one‐sample t‐tests and Benjamini and Hochberg false discovery rate multiple testing corrections were performed at 95% confidence levels on log transformed data.
Quantitative real‐time reverse transcription (RT)–polymerase chain reaction (PCR). The cells were treated with bile acids or vehicle (DMSO) for 24 h. Then, total RNA was isolated using TRIzol (Invitrogen) following standard procedures. cDNA was then synthesized from 4 µg total RNA, using the SuperScript III First‐Strand Synthesis System for RT‐PCR (Invitrogen) with oligo dT primer according to the manufacturer's instructions. The primer sequences were as fellows. For human Snail, forward: 5′‐TCT AAT CCA GAG TTT ACC TTC CAG C‐3′: reverse: 5′‐AGA TGA GCA TTG GCA GCG A‐3′. For human E‐cadherin, forward: 5′‐AGA ACG CAT TGC CAC ATA CAC TCT C‐3′; reverse: 5′‐CGG TTA CCG TGA TCA AAA TCT CCA‐3′. The PCR reactions were carried out in a 25‐µL volume containing 200 nM of each primer and 1 × QuantiTect SYBR Green PCR master mix (QIAGEN, Valencia, CA, USA). Real‐time PCR was performed on the ABI PRISM 7700 Sequence Detection System (PE Applied Biosystems) using the parameters recommended by the manufacturer (2 min at 50°C, 10 min at 95°C and 40 cycles of 95°C for 15 s and 62°C for 20 s). Each PCR reaction was performed in quadruplicate. The mRNA expression levels for all samples were normalized to the levels for the housekeeping gene GAPDH.
Plasmid construction. The human Snail promoter 5′ flanking region( 22 ) from –1824–1866 was amplified by PCR from human genomic DNA (Clontech Laboratories, Inc., Palo Alto, CA, USA) with the forward primer 5′‐CGT GAA GCT TTA GGA GCA AGA GAC GTA G‐3′ (–1834 to –1807) and the reverse primer 5′‐GTC GAA GCT TTG GGG TCG CCG ATT‐3′.( 76,53 ) For cloning purposes, a HindIII site was added to both primers. The PCR product was digested with HindIII, gel‐isolated and subcloned into the multiple cloning site of the pGL3–Basic Vector (Promega, Madison, WI, USA). The 5′ deletion mutants were generated by PCR and inserted into pGL3–Basic Vector. The forward primers were modified to contain a BglII restriction site. The sequences of primers were as follows: –966/66 forward: 5′‐AGC AGA TCT GAC CCC TCC GG‐3′ (–975 to –956), –339/66 forward: 5′‐CTC AGA TCT CCG GGC GCT GA‐3′ (–348 to –329), –111/66 forward: 5′‐CGG AGA TCT GCC TCC GAT TGG C‐3′ (–120 to –99), –66/66 forward: 5′‐CAG AGA TCT CCC CGC CCC TC‐3′ (–75 to –56), –24/66 forward: 5′‐GAG TAG ATC TGG GAG TTG GCG GC‐3′ (–34 to –13) and reverse (common): 5′‐GTC GAA GCT TTG GGG TCG CCG ATT‐3′ (76 to 53). The PCR product was digested with BglII and HindIII and subcloned into the multiple cloning site of the pGL3–Basic Vector. All reporter constructs were sequenced to confirm the correct sequence.
Site‐directed mutagenesis. The mutants were generated from the –1824/66 Snail pGL3 construct using the QuickChange Site‐Directed Mutagenesis kit (Stratagene). The following oligonucleotides were used as primers: 5′‐CTT CGG CGG AGA CGA GCC TCC GGT CCG CGC GGA GGT GAC AAA GGG GCG‐3′ and 5′‐CGC CCC TTT GTC ACC TCC GCG CGG ACC GGA GGC TCG TCT CCG CCG AAG‐3′ for mutation in the nuclear factor Y (NF‐Y) binding site, 5′‐GAT TGG CGC GGA GGC CCG AAA GGG GCG TGG CAG‐3′ and 5′‐CTG CCA CGC CCC TTT CGG GCC TCC GCG CCA ATC‐3′ for mutation in the AP‐1 binding site, and 5′‐GCA GAT AAG GCC CCC AAA CTC CCA CCC CCC AC‐3′ and 5′‐GTG GGG GGT GGG AGT TTG GGG GCC TTA TCT GC‐3′ for mutation in the stimulating protein 1 (Sp1) binding site. The presence of mutations was verified by sequencing.
Transient transfection and luciferase assay. DNA constructs were transiently transfected into cells using LipofectAMINE 2000 Reagent (Invitrogen). Twenty‐four hours before transfection, Hep3B cells were subcultured in 24‐well plates so that they would be at 70% confluence on the following day. Transfections were optimized for the amounts of DNA and LipofectAMINE per well culture as follows: 0.3 µg of reporter plasmids; 10 ng of pRL‐TK vector (Promega) as an internal control; and 2 µL of LipofectAMINE Reagent. 24 h after the transfection, the bile acid was added to the cell culture media and the cells were incubated for another 24 h. Luciferase assays were performed with the dual‐luciferase reporter assay system (Promega). All reported firefly luciferase values were normalized for transfection efficiency using the pRL‐TK, Renilla‐luciferase value. Graphs are representative for one of two experiments, each performed in quadruplicate.
Western blot analysis. The cells were treated with bile acids or vehicle (DMSO) for 24 h. Whole‐cell lysates were prepared and aliquots of 40 µg of protein lysates were applied for the sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS‐PAGE). Blots were probed with antibodies against E‐cadherin diluted 1:200 or actin diluted 1:500. Both were stained using secondary HRP‐conjugated antibodies diluted 1:1000 (goat antimouse for E‐cadherin and donkey antigoat for actin).
RNA interference. siGENOME SMARTpool reagent for Human Snail and siCONTROL Non‐Targeting siRNA Pool were obtained from Dharmacon, Inc (Lafayette, CO, USA). Short interference RNA (siRNA) was transiently transfected into cells using LipofectAMINE 2000 Reagent. Transfections were optimized for the amounts of siRNA, and LipofectAMINE per well culture as follows: 200 ng of siRNA and 4 µL of LipofectAMINE 2000 Reagent. The final concentration of siRNA used in each experiment was 100 nM.
In vitro wound healing assay. Cells were seeded in 24‐well culture plates at 1 × 105 cells/well. After 12 h, the cells were pretreated with bile acid for 24 h before wound formation. The in vitro‘scratch’ wounds were created by scraping the confluent cell monolayer with a sterile pipette tip. The cells were incubated with bile acid. After 6 h and 12 h, the cells were fixed with 100% methyl alcohol and stained with 0.5% solution of crystal violet and the distance of the wound closure (compared with control at t = 0 h) was measured in three independent wound sites per group. Relative cell motility was calculated as the percentage of the remaining cell‐free area compared with the area of the initial wound. Values from at least three independent experiments were pooled and expressed as mean ± standard deviation (SD).
Invasion assay. The cell invasion ability was determined using a BD Matrigel invasion chamber 24‐well Plate (BD Biosciences, Bedford, MA, USA), in which the chamber membrane filter (8 µm pore size) was coated with BD Matrigel Basement Membrane Matrix (BD Biosciences). The upper chamber was loaded with 2 × 104 cells in 0.2 mL of serum‐free medium with bile acid, and the lower chamber was filled with 0.7 mL of serum‐containing medium with bile acid. After 48 h, invading cells on the lower surface of the membrane were washed in phosphate‐buffered saline (PBS), fixed with 100% methyl alcohol and stained with 0.5% solution of crystal violet. The invading cells were counted under a microscope in six randomly selected fields for each membrane filter (×200). Each sample was assayed in duplicate in at least two independent experiments.
Data analysis. The obtained values are expressed as the mean ± SD. The data from different treated groups were compared by Welch t‐test. P‐values of <0.01 were considered to be statistically significant.
Results
Gene expression profiles induced by CDCA using cDNA microarray. To identify the changes in gene expression induced by CDCA, THLE‐3, Hep3B and HuCCT‐1 were treated with 100 µM of CDCA for 24 h. The cells treated with 0.1% DMSO were used as the control. Total RNA was isolated and gene expression profiling was performed by cDNA microarray using the AceGene (Human oligo chip 30K, subset A). The relative transcript abundance was expressed as Cy5/Cy3 ratios of signal intensities after background subtraction in each channel. Data analysis and quality control procedures are described in detail in section of Materials and methods. Seventy‐six genes were up‐regulated in the THLE‐3, 119 genes in the Hep3B and 53 genes in the HuCCT‐1. One‐hundred and nineteen genes were down‐regulated in the THLE‐3, 97 genes in the Hep3B and 18 genes in the HuCCT‐1. The Venn diagrams depict the number of overlapping and non‐overlapping genes (Fig. 1). The overlapping genes in all cell lines were listed as the common CDCA‐induced up‐ or down‐regulated genes (see Suppl. Table S1). In addition, E‐cadherin, which is a target gene of Snail, showed significant down regulation after 24‐h CDCA treatment in Hep3B and HuCCT‐1 (fold change; 0.73 and 0.62, P‐value; 0.003 and 0.037, respectively). However, the spot for E‐cadherin in several hybridizations of THLE‐3 was detected as a missing spot and E‐cadherin was eliminated from further analysis.
Figure 1.
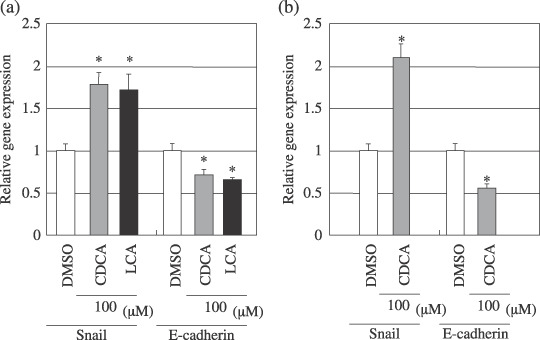
Chenodeoxycholic acid (CDCA) and lithocholic acid (LCA) induced expression of Snail mRNA and reduced expression of E‐cadherin mRNA. Cells were incubated with 100 µM of CDCA or LCA for 24 h. Then, total RNA was isolated and quantitative real‐time reverse transcription–polymerase chain reaction (RT‐PCR) was performed. (a) Hep3B cells were treated with 100 µM of CDCA or LCA. (b) HuCCT‐1 cells were treated with 100 µM of CDCA. Values for each gene were normalized to values obtained for GAPDH. Y‐axis represents a ratio for control (dimethyl sulfoxide [DMSO]). The data show the mean ± SD. *Significant difference (P < 0.01) from the respective control value.
CDCA‐ and LCA‐induced expression of Snail mRNA and reduced expression of E‐cadherin mRNA. To investigate alteration in the expression of Snail mRNA and E‐cadherin mRNA by bile acids, quantitative real‐time RT‐PCR was performed. Hep3B and HuCCT‐1 cells showed induced expression of Snail and reduced expression of E‐cadherin by 100 µM of CDCA or LCA (Fig. 1). The relative Snail gene expression resulted in an approximately 1.8‐fold increase in the Hep3B cells and about 2.2‐fold increase in the HuCCT‐1 cells, whereas the relative E‐cadherin expression resulted in a 0.7‐fold decrease in the Hep3B cells and about 0.5‐fold decrease in the HuCCT‐1 cells. These results demonstrated that E‐cadherin expression was inversely correlated with the expression of Snail, suggesting that up‐regulation of Snail by bile acids might be involved in the down‐regulation of E‐cadherin in hepatocellular carcinoma.
CDCA and LCA treatment induce Snail promoter activity in a concentration‐dependent manner. To determine whether the increased mRNA levels of Snail are associated with increased transcriptional activity, luciferase assays were performed. Hep3B cells were cotransfected with –1824/66 Snail pGL3 constructs and pRL‐TK vector and treated with 100 µM of various bile acids for 24 h. CDCA resulted in an approximately 2‐fold increase in luciferase activity compared with the control (DMSO). Furthermore LCA resulted in an approximately 3‐fold increase in luciferase activity compared with the control (DMSO). In contrast, the other free bile acids, glycine‐conjugated and taurine‐conjugated bile acids, caused no change in luciferase activity (Fig. 2a). Treatment with CDCA or LCA resulted in a concentration‐dependent increase in Snail promoter activities (Fig. 2b).
Figure 2.
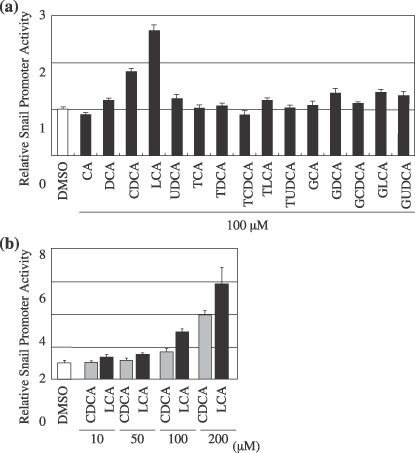
Chenodeoxycholic acid (CDCA) and lithocholic acid (LCA) increased Snail promoter activities in a concentration‐dependent manner. To investigate the effects of bile acids on the transcription of the Snail gene, 0.3 µg of the –1824/66 Snail pGL3 constructs and 10 ng of pRL‐TK vector were cotransfected into Hep3B cells. After 24 h, the bile acid was added to the cell culture media and the cells were incubated for 24 h. All reported firefly luciferase values were normalized for transfection efficiency using the pRL‐TK, Renilla‐luciferase value. Y‐axis represents the ratio for control (dimethyl sulfoxide [DMSO]). (a) The transfected cells treated with the various bile acids. The transfected cells treated with increasing amounts of (b) The transfected cells treated with CDCA or LCA at the concentration of 10–200 µM. The data show the mean ± SD of quadruplicate assay.
NF‐Y and Sp‐1 binding sites of Snail promoter are responsible for CDCA and LCA. To define the regulatory sequences required for the transcription of the Snail gene, Hep3B cells were cotransfected with a series of 5′‐deleted Snail pGL3 constructs and pRL‐TK vector and treated with 100 µM LCA for 24 h. In non‐LCA‐treated cells, deletion of nucleotides –1824 to –111 had no significant effect on the luciferase activity. However, deletion to nucleotide –66 resulted in significantly lower luciferase activity compared with that seen with Snail –1824/66‐Luc. Deletion to nucleotide –24 resulted in complete loss of the luciferase activity. In contrast, in LCA‐treated cells, luciferase activities were significantly higher than in untreated control cells and gradual deletion of the 5′ sequences from nucleotides –1875 to –111 resulted in no significant difference in the luciferase activity (Fig. 3a). The maximal activity was seen with Snail –111/66‐Luc. On further deletion to nucleotide –66, the luciferase activity fell to 47% of the maximal activity (P < 0.01). Deletion to nucleotide –24 resulted in complete loss of the luciferase activity. These results show that the region from nucleotide –111 to –24 is required for the maximal expression of Snail in Hep3B cells, both in the presence and absence of LCA.
Figure 3.

Deletion and mutagenesis analyzes of Snail promoter. 0.3 µg of each construct and 10 ng of pRL‐TK vector were cotransfected into Hep3B cells. After 24 h, the bile acid was added to the cell culture media and the cells were incubated for 24 h. All reported firefly luciferase values were normalized for transfection efficiency using the pRL‐TK, Renilla‐luciferase value activity and are shown as the relative activity compared to that for –1824/66 Snail pGL3 constructs treated with dimethyl sulfoxide (DMSO). The data show the mean ± SD of quadruplicate assay. (a) Deletion analysis of Snail promoter for the induction by LCA. (b) Effect of mutagenesis in NF‐Y, AP‐1 and Sp1 binding site on Snail promoter activity. The mutant promoter constructs used are schematically drawn.
To confirm which potential binding site from –111 to –24 was important for the induction of the Snail gene, we measured the luciferase activity by site‐directed mutagenesis. The vector mutated in the AP‐1 binding site showed no significant change compared with the wild‐type construct. The vector mutated in the NF‐Y or Sp1 binding site resulted in a marked decrease of the luciferase activity compared with the wild‐type construct in the both absence and presence of LCA (Fig. 3b). However, the luciferase activity of the vector mutated in the NF‐Y or Sp1 binding site treated with LCA still remained about 1.9‐ or 1.7‐fold higher than the activity of untreated cells, respectively. These results suggested that the NF‐Y and Sp1 binding sites were required for the basal activity of the Snail promoter, which is at least in part an inducer of Snail expression in response to bile acids.
Effects of E‐cadherin protein level and of subcellular localization of E‐cadherin by bile acid. To investigate alteration of subcellular localization of E‐cadherin by bile acid, immunofluorescence experiments with antibody to E‐cadherin were performed. Confocal microscopic studies revealed no significant difference in E‐cadherin protein pattern of subcellular distribution between Hep3B treated with DMSO, with 100 µM CDCA and with 100 µM LCA. In all treated cells, E‐cadherin staining was localized at areas of cell–cell contact. Moreover, in CDCA‐ and LCA‐treated cells, it seemed that E‐cadherin showed slightly weak staining, while strong membranous staining was observed in DMSO treated cells. Then, to verify the alteration in the expression of E‐cadherin protein by bile acid, Western blotting was performed. Treatment with CDCA or LCA resulted in a concentration‐dependent decrease in E‐cadherin protein expression. The expression of E‐cadherin in LCA‐treated cells was lower than that in CDCA‐treated cells (Fig. 4b). Western blotting analyzes confirmed the immunofluorescence results.
Figure 4.
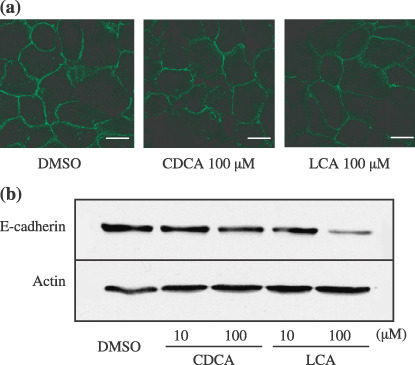
Effect of chenodeoxycholic acid (CDCA) or lithocholic acid (LCA) on E‐cadherin protein expression and subcellular distribution. Hep3B cells were incubated with bile acids or dimethyl sulfoxide (DMSO) for 24 h (a) Hep3B cells were fixed and probed with an anti‐E‐cadherin antibody followed by Alexa Fluor 488‐conjugated antimouse secondary antibody. Immunofluorescence showed the localization of E‐cadherin proteins (green fluorescence). Panel (left); DMSO (middle); CDCA 100 µM (right); LCA 100 µM treatmrnt. Bar, 20 µm (b) Protein levels were determined by Western blotting of whole cell extracts using mouse monoclonal anti‐E‐cadherin antibody. The expression of actin was analyzed in the same samples as a control for the amount of protein present in each sample.
Snail expression modulates E‐cadherin levels. To examine whether the increased Snail expression by CDCA and LCA led to reduced E‐cadherin levels, we used RNA interference to reduce the Snail expression in Hep3B cells. Transfection with Snail siRNA resulted in an 80% reduction of the Snail mRNA levels compared with Hep3B cells transfected with Non‐Targeting siRNA (Fig. 5a). Concomitantly, transfection with Snail siRNA also led to a 55% increase in the levels of E‐cadherin mRNA. Transfection with Non‐Targeting siRNA treated with 100 µM of CDCA or LCA showed 22% or 47% reductions in the E‐cadherin mRNA levels, respectively, compared with Hep3B cells treated with DMSO. However, Snail siRNA treatment reduced the down‐regulation of E‐cadherin (about 30% of that by transfection with Non‐Targeting siRNA) by 100 µM of CDCA or LCA (Fig. 5b). These results demonstrated that CDCA and LCA activated the Snail signaling pathway and reduced the E‐cadherin levels in hepatocellular carcinoma cell lines.
Figure 5.
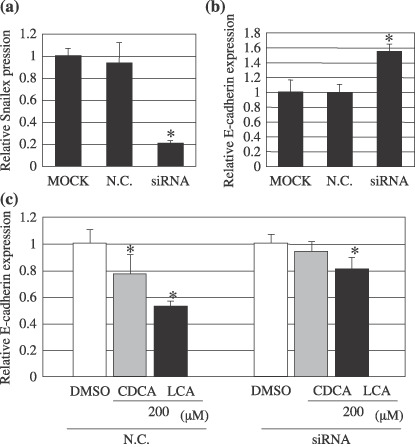
Snail short interference RNA (siRNA) treatment reduces down‐regulation of E‐cadherin by chenodeoxycholic acid (CDCA) or lithocholic acid (LCA). 200 ng of siRNA (final concentration: 100 nM) were transfected into Hep3B cells. After 72 h total RNA was isolated and quantitative real‐time reverse transcription–polymerase chain reaction (RT‐PCR) was performed. (a) The mRNA expression level of Snail. (b) The mRNA expression level of E‐cadherin. Mock: treated with only transfection reagent and N.C.: Non‐Targeting siRNA used as negative control. *Significant difference (P < 0.01) from N.C. (c) After transfection, Hep3B cells were incubated for 48 h and then cells were incubated with CDCA or LCA for 24 h. Total RNA was isolated. As a control, the same volume of dimethyl sulfoxide (DMSO) was used. The data show the mean ± SD. *Significant difference (P < 0.01) from the respective control value.
CDCA and LCA induces cell migration and invasion. To test whether the increased Snail expression by CDCA or LCA induced cell migration and invasion, cell motility and invasiveness were assessed using an in vitro wound healing assay and cell invasion assay. Our results showed that CDCA and LCA induced the ability of Hep3B cells to close the wound (Fig. 6a). MTS assay showed that the cell growth of Hep3B was not affected by concentrations of CDCA or LCA lower than 100 µM as compared with control (data not shown). The increased motility induced by CDCA and LCA treatment was not the result of increased cell proliferation. Invasion assays were carried out using Matrigel‐coated transwell culture chambers. Hep3B cells treated with CDCA showed significantly increased cell invasion compared with cells treated with vehicle. Furthermore, cells treated with LCA exhibited markedly increased cell invasion compared with the cells treated with vehicle (Fig. 6b). Transfection with Snail siRNA resulted in marked decreases in cell invasion compared with Hep3B cells transfected with Non‐Targeting siRNA. Moreover, there was a loss of the increase in cell invasion induced by CDCA and LCA (Fig. 6c).
Figure 6.
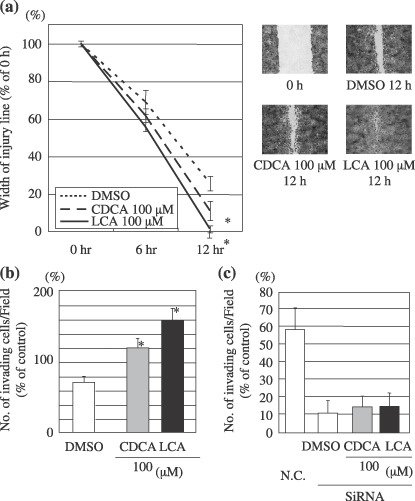
Chenodeoxycholic acid (CDCA) and lithocholic acid (LCA) induce cell migration. (a) The motility behavior of bile acid treated cells was analyzed in an in vitro wound model. Confluent cultures of Hep3B cells treated with bile acids were gently scratched with a pipette tip to produce a wound. Quantitative analysis was performed as described in Materials and methods. Data are mean ± SD. *Significant different (P < 0.01) from the respective control (dimethyl sulfoxide [DMSO]). (right panels) Photographs of cell migration were taken immediately after the incision and after 12 h. (b) The cell invasion assays were performed as described in Materials and methods. The invading cells were counted under the microscope in 6 randomly selected fields for each membrane filter (×200). Each sample was assayed in duplicate in at least two independent experiments. Data are mean ± SD. *Significant difference (P < 0.01) from the respective control (DMSO). (c) The cell invasion assays were performed using Snail knocked down cells. Before plating, 200 ng of short interference RNA (siRNA) (final concentration: 100 nM) were transfected into Hep3B cells and incubated for 72 h. N.C.: Non‐Targeting siRNA used as negative control.
Discussion
Bile acids are natural detergents synthesized in the liver. High levels of certain bile acids, however, are known to promote carcinogenesis and cancer progression by stimulating a variety of signaling pathways.( 5 , 6 , 7 ) The mechanisms of the tumor‐promoting actions of bile acids remain poorly understood.
In this study, a DNA microarray gene expression profiling approach was used to identify novel genes regulated by primary bile acids (CDCA) in hepatocytes and cholangiocytes. From this screening, several genes were found to be up‐ or down‐regulated in the presence of CDCA. Snail was found to be up‐regulated and E‐cadherin, a target gene of Snail, was found to be down‐regulated by the addition of CDCA. This is the first report that bile acids induced Snail.
Bile acids have been shown to modulate gene expression by regulating its promoter activity. The bile acids were reported to bind to the nuclear hormone receptors including farnesoid X‐activated receptor (FXR).( 2 , 24 , 25 , 26 , 27 ) Some kinds of nuclear hormone receptors can recognize bile acid response element (BARE) such as direct repeat, inverted repeat and everted repeat.( 28 ) In addition, bile acids can also activate NF‐kappa B and AP‐1.( 10 , 29 ) In this study, we revealed that several kinds of bile acids were able to induce Snail expression by activating its promoter activity. The luciferase assays showed treatment with 100 µM of CDCA or LCA resulted in 2–3‐fold increase in Snail promoter activities. The deletion mutant series for the Snail promoter revealed that 111 bp upstream of the transcription start site of the Snail gene was essential for the maximal promoter activity in both LCA‐treated and LCA‐untreated Hep3B cells. By sequence analysis of the 5′ flanking region of Snail gene, potential binding sites for NF‐Y, AP‐1 and Sp1 were identified on the 111 bp upstream region.
The Sp1 family of proteins is ubiquitously expressed in many tissues. They have versatile functions and are involved in cell cycle or differentiation.( 30 ) Early studies indicated that Sp1 was responsible for recruiting TATA‐binding protein and guiding transcriptional initiation at promoters without a TATA box.( 31 , 32 , 33 ) Sp1 proteins are known to interact with general transcription factors and are involved in the assembly of general transcriptional complexes. In addition to their function as general transcriptional activators, recent studies indicate Sp1 proteins also interact with many unique transcription factors and synergistically stimulate the genes involved in cholesterol and fatty acids synthesis.( 34 , 35 ) RAR, a well‐known nuclear receptor that heterodimerizes with RXR and binds to RAR‐responsive element to regulate gene expression, was reported to modulate Sp1 transcriptional activity on GC‐rich promoters through direct interaction between the two proteins.( 36 , 37 ) As shown in Fig. 2(a), hydrophobic bile acids, CDCA and LCA could activate the promoter activity of Snail, whereas hydrophilic bile acids, taurin‐ or glycin‐ conjugated bile acids could never activate Snail promoter activity. In general, unconjugated hydrophobic bile acids have higher affinity with nuclear receptors such as FXR than conjugated hydrophilic bile acids. It is speculated that CDCA or LCA, which might be able to enter the cell by passive diffusion through the plasma membrane of the cancer cell, directly activate the nuclear receptors and modulate the Sp1 transcriptional activity on the Snail promoter.
The NF‐Y is known as a ubiquitous heterotrimeric transcription factor that consist of NF‐YA, NF‐YB and NF‐YC subunits.( 38 , 39 ) Functional analysis has shown that NF‐Y is crucial for transcriptional activation and reinitiation on genes that lack a TATA‐box.( 33 , 40 ) In addition, it was suggested that NF‐Y could functionally interact with Sp1 and synergistically regulate promoter activity. Wright et al. demonstrated that the half‐life of either NF‐Y or Sp1 binding is dramatically increased when both transcriptional factors are bound to the proximal promoter of the major histocompatibility complex class II‐associated invariant chain gene.( 41 ) Liang et al. reported that the type‐A natriuretic peptide receptor gene transcription was regulated through functional interactions of NF‐1 and Sp1.( 42 ) The present study showed that mutations on NF‐Y or Sp1 binding sites in the Snail promoter markedly reduced transcriptional activity, possibly suggesting that the transcriptional activity of Snail was regulated by the functional interaction of NF‐Y and Sp1. In addition, the promoter activity of mutant on NF‐Y or Sp1 was higher compared with the minimal activation level observed with deletion to nucleotide –24, indicating that either NF‐Y or Sp1 are indispensable for the transcription of Snail. We also demonstrated that the promoter activity of the 111 bp upstream region was dramatically up‐regulated by CDCA or LCA, suggesting that these bile acids regulate the transcriptional activity of Snail gene by interacting with putative NF‐Y and Sp1 binding sites. 100 µM of LCA could up‐regulate the promoter activity of both mutant on NF‐1 and mutant on Sp1. It was suggested that these bile acids activate both Sp1 and NF‐Y. The effect of these bile acids on the activation of NF‐Y or Sp1 remains to be explored. To clarify the molecular mechanism of transcriptional regulation of Snail gene by bile acids will require further study.
In this present study, we revealed that the expression of E‐cadherin was decreased in CDCA‐ or LCA‐treated Hep3B cells. Alterations affecting cell adhesion molecules are considered to play a critical role in the invasive process. The cell–cell adhesion molecule E‐cadherin has been shown to execute important functions in embryogenesis and tissue architecture by forming intercellular junction complexes and establishing cell polarization.( 43 ) Loss of E‐cadherin results in dedifferentiation, invasiveness and lymph node or distant metastasis in a variety of human neoplasms, including hepatocellular carcinoma.( 16 , 17 , 18 , 44 ) Recently, the up‐regulation of the transcription factor Snail was reported to mediate significant negative regulation of E‐cadherin expression.( 20 , 45 ) The Snail mRNA levels have been reported to be independently correlated with capsular invasion in hepatocellular carcinoma tissues.( 46 , 47 ) Moreover, transfection of Snail in epithelial cells decreases E‐cadherin levels and induces changes resembling EMT.( 20 , 45 ) Interference with Snail expression leads to increased levels of E‐cadherin.( 21 , 45 , 48 ) The results from our present study showed that Snail expression was induced and E‐cadherin expression was reduced by CDCA or LCA. Moreover, it was revealed that these bile acids could never reduce the expression of E‐cadherin in Snail knocked down Hep3B cells. Together with these results, it is suggested that CDCA and LCA activate the Snail signaling pathway and reduce the expression of E‐cadherin in hepatocellular carcinoma cell lines. To address the hypothesis that Snail expression causes increased cancer invasion, the invasion activity was assessed using an in vitro wound healing assay and invasion assay. The invasive activity in Hep3B treated with 100 µM of CDCA or LCA was significantly increased compared with vehicle. Transfection with Snail siRNA resulted in marked decreases in cell invasion and a loss of the increase in cell invasion induced by CDCA and LCA. These results suggest that CDCA and LCA induce the expression of Snail gene, and as a consequence, promote invasiveness of the hepatocellular carcinoma cells. In the presented experiments, CDCA and LCA were applied at the concentration of 100 µM. Most of the biliary bile acids are conjugated and 100 µM of CDCA or LCA is excessively high as a concentration of physiological condition. However, in a pathological condition such as obstructive jaundice, hepatocellular carcinoma cells or cholangiocarcinoma, cells are probably exposed to these high concentrations of bile acids.
In summary, we found that CDCA and LCA positively regulate the transcriptional activity of the Snail gene. We showed that CDCA and LCA could activate the transcription of the Snail gene. Our data suggested that both Sp1 and NF‐Y are essential for this bile‐acid‐induced transcriptional activity. We revealed that CDCA or LCA could down‐regulate the expression of E‐cadherin by stimulating the Snail pathway and, as a consequence, these bile acids could promote cancer cell invasiveness. Hence, we conclude that CDCA and LCA are a novel inducer of Snail expression. It is suggested that CDCA and LCA play some role in promoting cancer invasiveness by stimulating the Snail pathway in hepatocelluler carcinoma. It is also suggested that inhibition of this bile‐acid‐stimulated Snail pathway may prove useful as an adjuvant in therapy for hepatocellular carcinoma.
Grant support
This study was supported by a Grant‐in Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan and Sagawa Foundation.
Supporting information
Table S1. Up‐ or down‐regulated genes in THLE‐3, Hep3B and HuCCT‐1 cells.
Please note: Blackwell Publishing are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
References
- 1. Pandak WM, Li YC, Chiang JY et al . Regulation of cholesterol 7 alpha‐hydroxylase mRNA and transcriptional activity by taurocholate and cholesterol in the chronic biliary diverted rat. J Biol Chem 1991; 266: 3416–21. [PubMed] [Google Scholar]
- 2. Wang H, Chen J, Hollister K, Sowers LC, Forman BM. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell 1999; 3: 543–53. [DOI] [PubMed] [Google Scholar]
- 3. Grober J, Zaghini I, Fujii H et al . Identification of a bile acid‐responsive element in the human ileal bile acid‐binding protein gene. Involvement of the farnesoid X receptor/9‐cis‐retinoic acid receptor heterodimer. J Biol Chem 1999; 274: 29749–54. [DOI] [PubMed] [Google Scholar]
- 4. Pineda Torra I, Claudel T, Duval C, Kosykh V, Fruchart JC, Staels B. Bile acids induce the expression of the human peroxisome proliferator‐activated receptor alpha gene via activation of the farnesoid X receptor. Mol Endocrinol 2003; 17: 259–72. [DOI] [PubMed] [Google Scholar]
- 5. Narisawa T, Magadia NE, Weisburger JH, Wynder EL. Promoting effect of bile acids on colon carcinogenesis after intrarectal instillation of N‐methyl‐N′‐nitro‐N‐nitrosoguanidine in rats. J Natl Cancer Inst 1974; 53: 1093–7. [DOI] [PubMed] [Google Scholar]
- 6. Reddy BS, Watanabe K, Weisburger JH, Wynder EL. Promoting effect of bile acids in colon carcinogenesis in germ‐free and conventional F344 rats. Cancer Res 1977; 37: 3238–42. [PubMed] [Google Scholar]
- 7. Hirano F, Tanada H, Makino Y et al . Induction of the transcription factor AP‐1 in cultured human colon adenocarcinoma cells following exposure to bile acids. Carcinogenesis 1996; 17: 427–33. [DOI] [PubMed] [Google Scholar]
- 8. Palmer DG, Paraskeva C, Williams AC. Modulation of p53 expression in cultured colonic adenoma cell lines by the naturally occurring lumenal factors butyrate and deoxycholate. Int J Cancer 1997; 73: 702–6. [DOI] [PubMed] [Google Scholar]
- 9. Matheson H, Branting C, Rafter I, Okret S, Rafter J. Increased c‐fos mRNA and binding to the AP‐1 recognition sequence accompanies the proliferative response to deoxycholate of HT29 cells. Carcinogenesis 1996; 17: 421–6. [DOI] [PubMed] [Google Scholar]
- 10. Glinghammar B, Holmberg K, Rafter J. Effects of colonic lumenal components on AP‐1‐dependent gene transcription in cultured human colon carcinoma cells. Carcinogenesis 1999; 20: 969–76. [DOI] [PubMed] [Google Scholar]
- 11. Halvorsen B, Staff AC, Ligaarden S, Prydz K, Kolset SO. Lithocholic acid and sulphated lithocholic acid differ in the ability to promote matrix metalloproteinase secretion in the human colon cancer cell line CaCo‐2. Biochem J 2000; 349: 189–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Glinghammar B, Rafter J. Colonic luminal contents induce cyclooxygenase 2 transcription in human colon carcinoma cells. Gastroenterology 2001; 120: 401–10. [DOI] [PubMed] [Google Scholar]
- 13. Qiao L, Studer E, Leach K et al . Deoxycholic acid (DCA) causes ligand‐independent activation of epidermal growth factor receptor (EGFR) and FAS receptor in primary hepatocytes: inhibition of EGFR/mitogen‐activated protein kinase‐signaling module enhances DCA‐induced apoptosis. Mol Biol Cell 2001; 12: 2629–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yoon JH, Higuchi H, Werneburg NW, Kaufmann SH, Gores GJ. Bile acids induce cyclooxygenase‐2 expression via the epidermal growth factor receptor in a human cholangiocarcinoma cell line. Gastroenterology 2002; 122: 985–93. [DOI] [PubMed] [Google Scholar]
- 15. Stetler‐Stevenson WG, Aznavoorian S, Liotta LA. Tumor cell interactions with the extracellular matrix during invasion and metastasis. Annu Rev Cell Biol 1993; 9: 541–73. [DOI] [PubMed] [Google Scholar]
- 16. Oka H, Shiozaki H, Kobayashi K et al . Expression of E‐cadherin cell adhesion molecules in human breast cancer tissues and its relationship to metastasis. Cancer Res 1993; 53: 1696–701. [PubMed] [Google Scholar]
- 17. Llorens A, Rodrigo I, Lopez‐Barcons L et al . Down‐regulation of E‐cadherin in mouse skin carcinoma cells enhances a migratory and invasive phenotype linked to matrix metalloproteinase‐9 gelatinase expression. Laboratory Invest 1998; 78: 1131–42. [PubMed] [Google Scholar]
- 18. Luo J, Lubaroff DM, Hendrix MJ. Suppression of prostate cancer invasive potential and matrix metalloproteinase activity by E‐cadherin transfection. Cancer Res 1999; 59: 3552–6. [PubMed] [Google Scholar]
- 19. Pai R, Tarnawski AS, Tran T. Deoxycholic acid activates beta‐catenin signaling pathway and increases colon cell cancer growth and invasiveness. Mol Biol Cell 2004; 15: 2156–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cano A, Perez‐Moreno MA, Rodrigo I et al . The transcription factor snail controls epithelial‐mesenchymal transitions by repressing E‐cadherin expression. Nat Cell Biol 2000; 2: 76–83. [DOI] [PubMed] [Google Scholar]
- 21. Fujita N, Jaye DL, Kajita M, Geigerman C, Moreno CS, Wade PA. MTA3, a Mi‐2/NuRD complex subunit, regulates an invasive growth pathway in breast cancer. Cell 2003; 113: 207–19. [DOI] [PubMed] [Google Scholar]
- 22. Barbera MJ, Puig I, Dominguez D et al . Regulation of Snail transcription during epithelial to mesenchymal transition of tumor cells. Oncogene 2004; 23: 7345–54. [DOI] [PubMed] [Google Scholar]
- 23. Pfeifer AM, Cole KE, Smoot DT et al . Simian virus 40 large tumor antigen‐immortalized normal human liver epithelial cells express hepatocyte characteristics and metabolize chemical carcinogens. Proc Natl Acad Sci USA 1993; 90: 5123–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Laffitte BA, Kast HR, Nguyen CM, Zavacki AM, Moore DD, Edwards PA. Identification of the DNA binding specificity and potential target genes for the farnesoid X‐activated receptor. J Biol Chem 2000; 275: 10638–47. [DOI] [PubMed] [Google Scholar]
- 25. Song CS, Echchgadda I, Baek BS et al . Dehydroepiandrosterone sulfotransferase gene induction by bile acid activated farnesoid X receptor. J Biol Chem 2001; 276: 42549–56. [DOI] [PubMed] [Google Scholar]
- 26. Kast HR, Goodwin B, Tarr PT et al . Regulation of multidrug resistance‐associated protein 2 (ABCC2) by the nuclear receptors pregnane X receptor, farnesoid X‐activated receptor, and constitutive androstane receptor. J Biol Chem 2002; 277: 2908–15. [DOI] [PubMed] [Google Scholar]
- 27. Ohtsuka H, Abe T, Onogawa T et al . Farnesoid X receptor, hepatocyte nuclear factors 1alpha and 3beta are essential for transcriptional activation of the liver‐specific organic anion transporter‐2 gene. J Gastroenterol 2006; 41: 369–77. [DOI] [PubMed] [Google Scholar]
- 28. Edwards PA, Kast HR, Anisfeld AM. BAREing it all: the adoption of LXR and FXR and their roles in lipid homeostasis. J Lipid Res 2002; 43: 2–12. [PubMed] [Google Scholar]
- 29. Hirano Y, Hirano F, Fujii H, Makino I. Fibrates suppress chenodeoxycholic acid‐induced RANTES expression through inhibition of NF‐kappaB activation. Eur J Pharmacol 2002; 448: 19–26. [DOI] [PubMed] [Google Scholar]
- 30. Suske G. The Sp‐family of transcription factors. Gene 1999; 238: 291–300. [DOI] [PubMed] [Google Scholar]
- 31. Kollmar R, Sukow KA, Sponagle SK, Farnham PJ. Start site selection at the TATA‐less carbamoyl‐phosphate synthase (glutamine‐hydrolyzing)/aspartate carbamoyltransferase/dihydroorotase promoter. J Biol Chem 1994; 269: 2252–7. [PubMed] [Google Scholar]
- 32. Weis L, Reinberg D. Accurate positioning of RNA polymerase II on a natural TATA‐less promoter is independent of TATA‐binding‐protein‐associated factors and initiator‐binding proteins. Mol Cell Biol 1997; 17: 2973–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wright KL, Vilen BJ, Itoh‐Lindstrom Y et al . CCAAT box binding protein NF‐Y facilitates in vivo recruitment of upstream DNA binding transcription factors. EMBO J 1994; 13: 4042–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bennett MK, Osborne TF. Nutrient regulation of gene expression by the sterol regulatory element binding proteins increased recruitment of gene‐specific coregulatory factors and selective hyperacetylation of histone H3 in vivo . Proc Natl Acad Sci USA 2000; 97: 6340–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wu Z, Chiang JY. Transcriptional regulation of human oxysterol 7 alpha‐hydroxylase gene (CYP7B1) by Sp1. Gene 2001; 272: 191–7. [DOI] [PubMed] [Google Scholar]
- 36. Husmann M, Dragneva Y, Romahn E, Jehnichen P. Nuclear receptors modulate the interaction of Sp1 and GC‐rich DNA via ternary complex formation. Biochem J 2000; 352 (Pt 3): 763–72. [PMC free article] [PubMed] [Google Scholar]
- 37. Shimada J, Suzuki Y, Kim SJ, Wang PC, Matsumura M, Kojima S. Transactivation via RAR/RXR–Sp1 interaction characterization of binding between Sp1 and GC box motif. Mol Endocrinol 2001; 15: 1677–92. [DOI] [PubMed] [Google Scholar]
- 38. Sinha S, Maity SN, Seldin MF, De Crombrugghe B. Chromosomal assignment and tissue expression of CBF‐C/NFY‐C, the third subunit of the mammalian CCAAT‐binding factor. Genomics 1996; 37: 260–3. [DOI] [PubMed] [Google Scholar]
- 39. Kim IS, Sinha S, De Crombrugghe B, Maity SN. Determination of functional domains in the C subunit of the CCAAT‐binding factor (CBF) necessary for formation of a CBF‐DNA complex: CBF‐B interacts simultaneously with both the CBF‐A and CBF‐C subunits to form a heterotrimeric CBF molecule. Mol Cell Biol 1996; 16: 4003–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liberati C, Ronchi A, Lievens P, Ottolenghi S, Mantovani R. NF‐Y organizes the gamma‐globin CCAAT boxes region. J Biol Chem 1998; 273: 16880–9. [DOI] [PubMed] [Google Scholar]
- 41. Wright KL, Moore TL, Vilen BJ, Brown AM, Ting JP. Major histocompatibility complex class II‐associated invariant chain gene expression is up–regulated by cooperative interactions of Sp1 and NF‐Y. J Biol Chem 1995; 270: 20978–86. [DOI] [PubMed] [Google Scholar]
- 42. Liang F, Schaufele F, Gardner DG. Functional interaction of NF‐Y and Sp1 is required for type a natriuretic peptide receptor gene transcription. J Biol Chem 2001; 276: 1516–22. [DOI] [PubMed] [Google Scholar]
- 43. Gumbiner B, Stevenson B, Grimaldi A. The role of the cell adhesion molecule uvomorulin in the formation and maintenance of the epithelial junctional complex. J Cell Biol 1988; 107: 1575–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Endo K, Ueda T, Ueyama J, Ohta T, Terada T. Immunoreactive E‐cadherin, alpha‐catenin, beta‐catenin, and gamma‐catenin proteins in hepatocellular carcinoma: relationships with tumor grade, clinicopathologic parameters, and patients’ survival. Hum Pathol 2000; 31: 558–65. [DOI] [PubMed] [Google Scholar]
- 45. Batlle E, Sancho E, Franci C et al . The transcription factor snail is a repressor of E‐cadherin gene expression in epithelial tumour cells. Nat Cell Biol 2000; 2: 84–9. [DOI] [PubMed] [Google Scholar]
- 46. Jiao W, Miyazaki K, Kitajima Y. Inverse correlation between E‐cadherin and Snail expression in hepatocellular carcinoma cell lines in vitro and in vivo . Br J Cancer 2002; 86: 98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sugimachi K, Tanaka S, Kameyama T et al . Transcriptional repressor snail and progression of human hepatocellular carcinoma. Clin Cancer Res 2003; 9: 2657–64. [PubMed] [Google Scholar]
- 48. Poser I, Dominguez D, De Herreros AG, Varnai A, Buettner R, Bosserhoff AK. Loss of E‐cadherin expression in melanoma cells involves up‐regulation of the transcriptional repressor Snail. J Biol Chem 2001; 276: 24661–6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Up‐ or down‐regulated genes in THLE‐3, Hep3B and HuCCT‐1 cells.
Please note: Blackwell Publishing are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item