

Abstract
Vaccination with heat shock proteins (HSP) protects mice from challenge with the tumor from which the HSP were isolated. The antigenicity of HSP vaccination is thought to result from HSP‐associated endogenous major histocompatibility complex class I peptides or their precursors. The vaccination effect can be achieved in an adjuvant‐free manner and is mediated by CD8+ T cells, indicating that HSP can act as a natural adjuvant and cross‐prime T cells in vivo. We previously devised a recombinant vaccine composed of a CD8+ T cell epitope fused to the carboxyl‐terminus of hsc70 and demonstrated efficient generation of antigen‐specific cytotoxic T lymphocyte (CTL) after vaccination with a few micrograms of the hsc70‐CTL epitope fusion protein. The present study aimed to determine if the fusion protein vaccine could control tumor growth in vivo and whether simultaneous fusion of a CD4+ T cell epitope to the amino terminus of the hsc70‐CTL epitope would be a more potent vaccine compared to the CTL epitope alone. Ovalbumin (OVA)–derived 8 mer peptide, OVA257‐264, and 16mer peptide, OVA265‐280, were used as CD8+ and CD4+ T cell epitopes, respectively. Vaccination with hsc70‐OVA257‐264 generated peptide specific CTL more effectively than a peptide plus incomplete Freund's adjuvant combination, and suppressed growth of OVA expressing EL4 (E.G7) and B16 melanoma tumor cells. Addition of OVA265‐280 to the amino‐terminus of hsc70‐OVA257‐264 (OVA265‐280‐hsc70‐OVA257‐264) enhanced the generation of the OVA257‐264‐specific CTL population, leading to better eradication of MO5 lung metastasis compared to hsc70‐OVA257‐264. Our results suggest that fusion of both CD4+ and CD8+ T cell epitopes to hsc70 enhances tumor immunity beyond the effect of the CD8+ T cell epitope alone. (Cancer Sci 2008; 99: 1008–1015)
Heat shock proteins (HSP) have been implicated as tumor rejection antigens in vivo, for example vaccination with HSP gp96, hsp90, and hsc70, even without adjuvants, protects mice from challenge by the tumors from which the HSP were originally isolated.( 1 , 2 , 3 ) The immunogenicity of HSP vaccines has been suggested to result from carry‐over of endogenous peptides that were associated with HSP in cells, and not due to HSP per se. ( 1 ) Indeed, we previously identified major histocompatibility complex (MHC) class I ligands or their precursors that were acid‐eluted from gp96, hsp90, and hsc70 derived from the radiation leukemia, RL♂1.( 4 ) An important observation from those studies was that the quantity of tumor antigen peptides associated with HSP was no more abundant than we expected. Thus, it was surprising that such a tiny amount of peptide was sufficient to prime CD8+ T cells that enabled tumor rejection in vivo. These findings raise the fascinating possibility that HSP may serve as potent and safe natural adjuvants, better than any now available.
The recent observation that toll‐like receptors (TLR) 4 and 2 interact with gp96 or hsp70, resulting in production of pro‐inflammatory cytokines by antigen presenting cells (APC)( 5 , 6 ) may provide the rationale behind the strong HSP adjuvant effect, since activation of APC is essential for the full‐priming of T cells. In addition, HSP seemed to target a number of receptors such as CD91,( 7 ) LOX‐1,( 8 ) and SRA( 9 ) on dendritic cells (DC) or macrophages in vivo. HSP are efficiently captured by these receptors on DC and internalized into endosome/lysosome compartments in which peptides associated with HSP are channeled into the cytosol, probably through a retro‐translocation mechanism via a translocon consisting of Sec61 and p97.( 10 ) The peptides released into the cytosol are on a track to a classical MHC class I antigen‐processing pathway to be presented to CD8+ T cells. This conceptual mechanism for HSP‐induced immunity encouraged us to construct a novel recombinant vaccine in which MHC class I ligands are covalently fused to either the carboxyl‐ or amino‐terminus of hsc70. In these fusion proteins, each hsc70 molecule contained at least one T cell epitope, thus the number of specific peptides associated with HSP was dramatically increased, in comparison with the natural HSP purified from tumor cells. Moreover, presentation of HSP‐bound self‐peptides that might evoke autoimmunity could in principle be avoided. We previously showed that specific CTL were easily generated against five distinct CD8+ T cell epitopes by vaccination with a few micrograms of those hsc70‐peptide complexes,( 11 ) but the protective efficacy of this approach against in vivo tumor growth has not been investigated. Another issue to be addressed is how CD4+ T cells modulate HSP‐mediated immunity. HSP‐induced cross‐priming of CD8+ T cell does not require CD4+ T cells.( 11 , 12 ) Nonetheless, it is also true that antigen‐specific CD4+ T cells are activated by vaccination with antigen‐HSP complexes.( 13 )
In the present study, we investigated whether vaccination with CTL epitopes fused to hsc70 could protect against tumor growth in vivo. We also examined whether simultaneous fusion of a helper epitope in addition to CTL epitope to hsc70 gives rise to the enhanced protection over the effect of CTL epitope alone.
Materials and methods
Cell lines. EL‐4 is a methylchoranthlene‐induced thymoma of C57BL/6(H‐2b) origin. E.G7 is an OVA cDNA transfected EL‐4 cell line. B16 is a melanoma cell line derived from C57BL/6, and MO5 is an OVA cDNA transfected B16 cell line.
Media and cell cultures. RPMI‐1640 was supplemented with nonessential amino acids (Gibco/BRL, Tokyo, Japan), glutamine, sodium pyruvate, antibiotics (all from Sigma, St Louis, MO, USA), 10% fetal calf serum, and 5 × 10−5 M 2‐Mercaptoethanol. This complete RPMI media was used for CTL induction and tumor cell culturing. E.G7 was cultured in the presence of 500 µg/mL G418.
Reagents. Synthetic peptides OVA265‐280 (TEWTSSNVMEERKIKV), OVA257‐264 (SIINFEKL), and TRP2180‐188 (SVYDFFVWL) were purchased from Sawady (Tokyo, Japan). Their sequences were confirmed by a 490 Procise protein sequencer (Perkin‐Elmer, Tokyo, Japan). Expression and purification of synthetic peptides fused with hsc70 were done as described previously.( 11 , 12 , 13 , 14 ) Tetramer consisting of H‐2Kb plus SIINFEKL (OVA257‐264) labeled with phycoerythrin (PE) was purchased from MBL (Nagoya, Japan).
Preparation of recombinant fusion proteins. The recombinant hsc70 and fusion proteins described in Figure 1 were produced as previously described.( 14 ) For the hsc70‐OVA257‐264, hsc70‐TRP2180‐188, OVA265‐280‐hsc70, OVA265‐280‐hsc70‐OVA257‐264, and OVA265‐280‐hsc70‐TRP2180‐188, mini‐genes encoding OVA257‐264, TRP2180‐188, and OVA265‐280 were amplified by polymerase chain reaction using forward or reverse primers containing 5′BamHI and 3′KpnI restriction sites and subcloned into the pQE31 vector (Qiagen, Germantown, MD, US). Escherichia coli (E. coli) strain M15 was transformed by the constructed plasmids and grown in Lysogeny broth (LB) medium containing ampicillin (50 µg/mL) and kanamycin (20 µg/mL). Protein expression was induced by 0.1 M isopropyl‐β‐D‐thiogalactoside. The protein was solubilized in buffer B (8 M urea, 0.1 M sodium phosphate, 0.01 M Tris/HCl, pH 8.0) and, after centrifugation of the lysate at 10 000 g, the supernatant was applied to an Ni2 ± NTA (nitrilotriacetic acid) agarose column and extensively washed with buffer C (8 M urea, 0.1 M sodium phosphate, 0.01 M Tris/HCl, pH 6.3). The Ni2 ± NTA resin‐bound 6× His‐tagged protein was refolded rapidly by washing with 35 column volumes of urea‐free Tris buffer (pH 7.5) and eluted with Tris buffer containing 200 mM imidazole. The eluate was extensively dialyzed against phosphate‐buffered saline (PBS) (pH 7.4) to remove imidazole, and then it was concentrated using an Amicon Ultra‐15 centrifugal filter device (Millipore, Bedford, MA, USA). The recovered proteins were subjected to Detoxi‐gel Endotoxin Removing Columns (Pierce, Rockford, IL, USA) to get rid of contaminated endotoxin.
Figure 1.
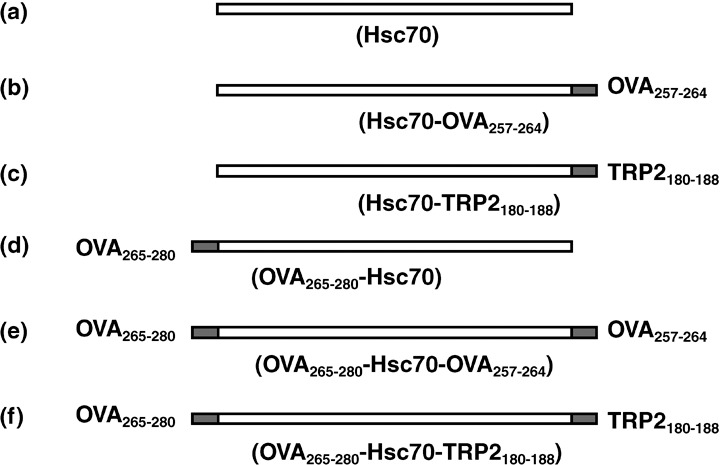
Recombinant hsc70 and fusion proteins used in this study. Cytotoxic T lymphocyte epitopes OVA257‐264 and TRP2180‐188 were fused to the C‐terminus of hsc70. (d–f) In some constructs, the helper T cell epitope OVA265‐280 was fused to the N‐terminus of hsc70. Recombinant proteins also contained an N‐terminal 6 × His‐tag. They were expressed in E. coli, M15, and purified with Ni‐NTA columns.
Vaccination and induction of CTL. C57BL/6 mice were vaccinated intraperitoneally twice in a 1‐week interval with the indicated amount of synthetic peptides plus incomplete Freund's adjuvant (IFA) or with 10 µg (or the indicated dose) of hsc70 fusion protein. Two weeks after the second immunization, the mice were sacrificed and spleen cells were stimulated in vitro with each peptide at 10−6 M final concentration for 5 days.
51Cr‐release assay. The cytolytic activity of the induced CTL was determined by a standard 51Cr‐release assay. Target cells were labeled with 50 µCi 51Cr‐labeled sodium chromate in RPMI with 10% fetal calf serum for 1 h at 37°C and then washed twice with unsupplemented RPMI. Target cells (5 × 103) were added to titrated CTL effectors in 96‐well round‐bottom plates. Plates with final volume of 200 µL/well were centrifuged to promote cell contact and incubated at 37°C for 4 h. Then 100 µL of supernatant from each well was harvested manually and the radioactivity released into the supernatant was measured in a γ‐counter. The percent specific release was calculated from the mean of duplicate cultures according to the following formula: percent specific 51Cr release = (experimental release – spontaneous release) × 100/[maximal release (2% Triton X) – spontaneous release].
Enzyme‐linked immunospot (ELISPOT) assay. For the ELISPOT assay, HA‐Multiscreen plates (Millipore, Burlington, MA, USA) were coated with 50 µL of an antimouse γ‐interferon (IFN‐γ) antibody (10 µg/mL; RMMG1) in PBS, incubated overnight at 4°C, washed with PBS/0.25% Tween20 to remove unbound antibody and blocked with 100 µL/well PBS containing 5% bovine serum albumin for 1 h at 37°C. Immunized spleen cells were stimulated with 10−6 M peptide for 5 dyas and effector cells were obtained. The resulting cells (1 × 104 or 2 × 103 cells/well, 96‐well plate) were incubated with mitomycin C (50 µg/mL for 45 min) treated, 5 × 105 naïve spleen cells/well pulsed with or without 1 µg/mL peptides in triplicate form. After incubation for 19 h at 37°C with 5% CO2, the plates were extensively washed with PBS/0.25% Tween 20, and 100 µL/well biotinylated detecting antibody against mouse IFN‐γ (1 µg/mL; R46A2) was added. After 2 h incubation at room temperature, the plates were washed with PBS/0.25% Tween 20, and 50 µL/well avidin solution (alkaline phosphatase conjugated) was added and incubated for 2 h. Spots were developed using nitro blue tetrazolium chloride/5‐bromo‐4‐chloro‐3‐indolyl phosphate (NBT/BCIP) and counted using a zoom stereo microscope at ×40 magnification.
Depletion of CD8+ cells by fluorescence‐activated cell sorting (FACS). In vitro stimulated spleen cells with peptides were stained with fluorescein‐isothiocyanate conjugated antimouse CD8α (53–6.7; eBioscience, San Diego, CA, USA). By using FACS Vantage, we sorted those stained cells into CD8− and CD8+ cells. As a control, stimulated spleen cells were mock sorted and non‐depleted effector cells were obtained. Those CD8+ depleted and non‐depleted cells were used for CTL assay and ELISPOT assay.
Immunotherapy of solid tumors. On day 0, mice were inoculated intradermally with 1 × 106 E.G7 cells on the right side of the back. On days –14 and –7, days 0 and 7, or days 2 and 9, mice were vaccinated with 10 µg hsc70 fusion proteins. Mice challenged with 2 × 105 MO5 cells were vaccinated with 10 µg hsc70 fusion proteins on days 0 and 7. Tumor size was measured every 2–3 days, and the average tumor diameter was determined. Average tumor diameter = [longest tumor diameter + shortest tumor diameter]/2.
Immunotherapy of lung metastases. On day 0, mice were inoculated with 5 × 105 MO5 cells via the tail vein. On days 0 and 2, or on days 2 and 9, those mice were vaccinated with 10 µg hsc70‐OVA257‐264. On day 15, the mice were sacrificed and the metastatic spots on the lung surface were counted using a zoom stereo microscope (DSZT‐44IF; Carton, Tokyo, Japan). In another experiment, MO5 inoculated mice were vaccinated with 10 µg hsc70 or hsc70‐OVA257‐264 or OVA265‐280‐hsc70‐OVA257‐264 on days 0, 2, 5, and 7. On day 21, the mice were sacrificed, and the spots were counted. For CD4+ cell depletion, on days –2, 0, 2, and 5, 100 µL GK1.5 (ascites, containing approximately 100 µg of antibody) was injected. Control mice were injected an equal volume of normal mouse ascites.
Immunotherapy of established tumor. On day 0, mice were inoculated intradermally with 1 × 106 E.G7 cells on the right side of the back. On day 6, 2 × 106 OTI CD8+ T cells were injected via tail vein, and simultaneously, 10 µg hsc70‐OVA257‐264 or equal molar PA28α‐OVA257‐264 was once peritoneally injected. For purification of OTI CD8+ T cells, OTI splenocytes were at first depleted of CD11b cells with BD IMag antimouse CD11b Magnetic Particles‐DM (negative selection) (BD Biosciences, San Jose, CA, USA) to remove splenic DC, and then, CD8+ T cells were positively purified with BD IMag antimouse CD8 Magnetic Particles‐DM (positive selection). Tumor diameter was monitored every 2–3 days. The average tumor diameter was determined. Average tumor diameter = [longest tumor diameter + shortest tumor diameter]/2.
Proliferation of transferred T cells. Purified OTI CD8+ T cells were stained with 5 µM carboxyfluorescein succinimidyl ester (CFSE) (Dojindo, Kumamoto, Japan). CFSE‐labeled 2 × 106 OTI CD8+ T cells were adoptively transferred into C57BL/6 mice via tail vein, and simultaneously vaccinated with 10 µg hsc70‐OVA257‐264 or equal molar PA28α‐OVA257‐264 via the peritoneal cavity. Control mice were not injected with any proteins. Three days later, splenocytes of those recipients were stained with PE‐conjugated antimouse CD44 (IM7; eBioscience) as a T‐cell activated marker, and then division of OTI CD8+ T cells was determined by CFSE dilution.
Results
Generation of CTL by hsc70 fusion proteins. Diagrams of the fusion proteins used in this study are shown in Figure 1. The H‐2Kb epitopes OVA257‐264 and TRP2180‐188 were genetically fused to the carboxyl terminus of hsc70 (Fig. 1). In addition, a presumed helper epitope OVA265‐280 that was previously suggested to augment OVA257‐264 specific CD8+T cell by DNA vaccination( 15 ) was fused to the amino terminus of hsc70 alone or hsc70 containing C‐terminal epitopes (Fig 1e,f). Fusion proteins were expressed in E. coli and recombinant proteins were purified as described previously.( 14 ) The endotoxin level in these proteins was less than 0.5 EU/mg as determined by the Limulus ES test (Mitsubishi Chemical Medicine Corporation, Tokyo, Japan).
We first investigated whether CD8+ cytotoxic T lymphocytes were generated by vaccination with hsc70 fused with both OVA257‐264 and OVA265‐280. Mice were vaccinated twice in a 1‐week interval with 10 µg hsc70 fusion protein, and 2 weeks later after the final imminization, spleen cells were stimulated with 10−6 M OVA257‐264 peptide for 5 days, and the resulting OVA257‐264‐specific CTL were confirmed by staining with tetramer comprising H‐2Kb + SIINFEKL (OVA257‐264) peptide, standard 51Cr‐release assay, and cytokine ELISPOT assay. We identified CD8+, tetramer positive cells by FACS analysis (Fig. 2a, upper panel). The CD8+ T cells killed EL‐4 cells pulsed with OVA257‐264 peptide, as well as E.G7 cells that express full length OVA, but not EL4 cells (Fig. 2b) and showed IFN‐γ production as determined by ELISPOT assay (Fig. 2b). The cytotoxicity and IFN‐γ‐producing activities were abrogated (Fig. 2a,b) by the depletion of CD8+ T cells by FACS (Fig. 2a, lower panel). In vivo cross‐priming ability was caused by fusion of hsc70 but not by other self proteins, because a proteasome activator, PA28α fused with OVA257‐264 showed only marginal CTL generation as we previously demonstrated,( 14 ) and induced only partial proliferation of CFSE‐labeled CD8+ T cells purified from OTI TCR transgenic mice (Fig. 10b‐3).
Figure 2.
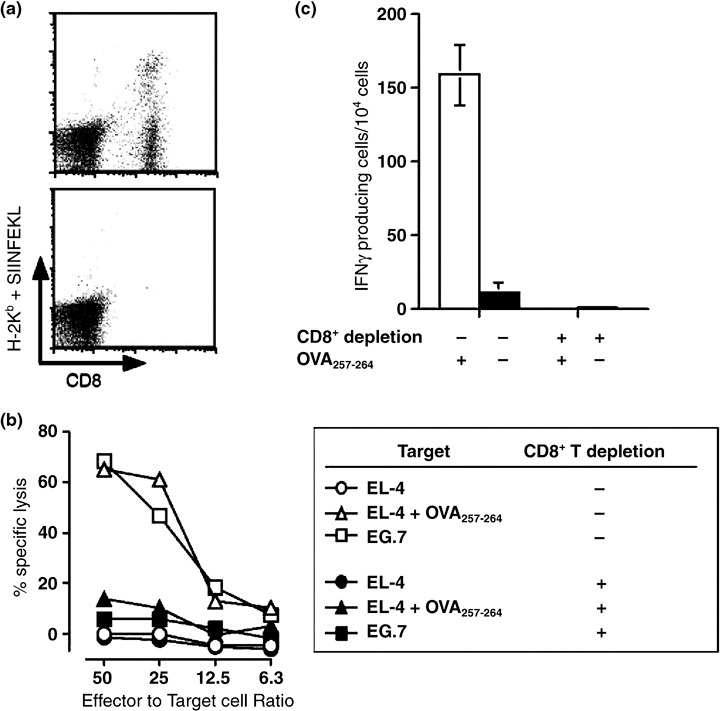
Generation of CD8+ CTL specific to OVA257‐264 by vaccination with hsc70 fusion protein. C57BL/6 mice were immunized intraperitoneally with 10 µg OVA265‐280‐hsc70‐OVA257‐264, twice in a 1‐week interval. Two weeks after the second immunization, the spleen cells were stimulated in vitro with 10−6 M OVA257‐264 for 5 days. (a, upper panel) The resulting cells were stained with tetramer consisting of H‐2Kb and OVA257‐264 (MBL, Nagoya, Japan). (a, lower panel) CD8+ cells were sorted out by fluorescence‐activated cell sorting (FACS) Vantage. Cells in (a) were used for 51Cr‐release assay (b) and cytokine ELISPOT assay (c), as indicated.
Figure 10.
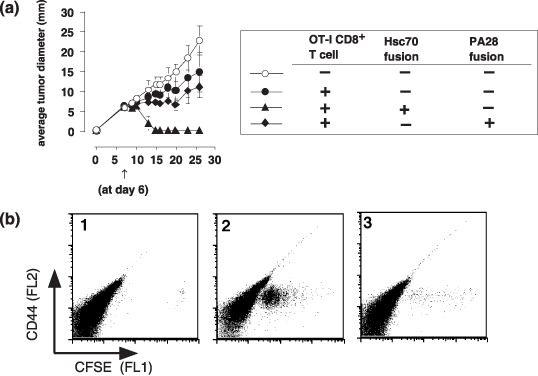
An immune therapy by hsc70 fusion protein for established tumor. (a) On day 0, mice were challenged with a 1 × 106 E.G7 tumor. At day 6, 2 × 106 OTI CD8+T cells were injected via tail vein, and simultaneously, 10 µg hsc70‐OVA257‐264 or equal molar PA28α‐OVA257‐264 was once peritoneally injected. Tumor diameter was monitored every 2–3 days. (n = 4). (b) Carboxyfluorescein succinimidyl ester (CFSE)–labeled 2 × 106 OTI CD8+T cells were adoptively transferred into C57BL/6 mice and simultaneously vaccinated with 10 µg hsc70‐OVA257‐264 (panel 2) or equal molar PA28α‐OVA257‐264 (panel 3). Control mice were not injected with any proteins (column 1). Three days later, proliferating OTI CD8+ T cells in the spleen were investigated by carboxyfluorescein succinimidyl ester dilution (CFSE) and CD44.
Next, we investigated CTL generation by immunization with hsc70 fusion proteins and synthetic peptides, OVA257‐264 and OVA265‐280, in an emulsion of IFA as described in Figure 2. Mice were immunized twice in a 1‐week interval with 1 µg of synthetic OVA257‐264 peptide plus IFA, or with 10 µg of the fusion proteins. Two weeks after the second immunization, spleen cells were stimulated with 10−6 M OVA257‐264 peptide for 5 days, and the resulting CTL activity was examined with a standard 51Cr‐release assay. Peptide immunization generated CTL that killed E.G7 and EL4 pulsed with OVA257‐264 but not EL4. Simultaneous immunization with graded doses of OVA265‐280 slightly enhanced the CTL activity in a dose‐dependent manner (Fig. 3, top row). Immunization with 10 µg hsc70 fusion proteins, corresponding to only one‐tenth the molar ratio used for peptide vaccination, generated even higher cytolytic activity toward E.G7 and EL4 pulsed with OVA257‐264, and marginal enhancement was observed with OVA265‐280‐hsc70‐OVA257‐264 compared to hsc70‐OVA257‐264 (Fig. 3, middle row). These results indicate that hsc70, although a ubiquitously expressed self‐antigen in the host, has a powerful adjuvant effect over that of IFA. Not only hsc70‐OVA257‐264 but also hsc70‐TRP2180‐188 induced peptide‐specific CTL. It was noted that hsc70‐TRP2180‐188‐induced CTL could kill MO5 melanoma cells spontaneously expressing TRP2 at the higher E/T ratio. But we observed no enhancement of cytolysis to TRP2180‐188 by immunization with OVA265‐280‐hsc70‐TRP2180‐188, compared to hsc70‐TRP2180‐188 (Fig. 3, bottom row).
Figure 3.
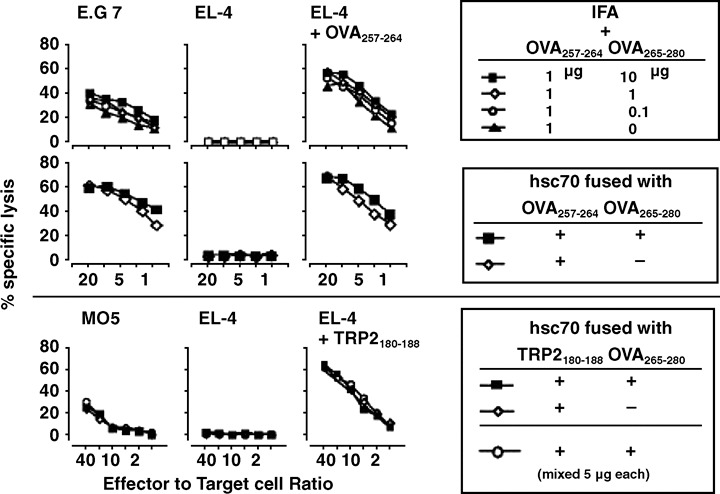
Comparison of the efficiency of cytotoxic T lymphocyte (CTL) generation by vaccination with hsc70 fusion proteins or synthetic peptides. C57BL/6 mice were immunized intraperitoneally with the indicated doses of synthetic peptides plus incomplete Freund's adjuvant (IFA) or with 10 µg (or the indicated volume) hsc70 fusion proteins, twice in a 1‐week interval. Two weeks after the second immunization, the spleen cells were stimulated in vitro with 10−6 M OVA257‐264 or TRP2180‐188 for 5 days. CTL activity against E.G7, EL4, and EL4 pulsed with OVA257‐264 was examined using a standard 51Cr‐release assay.
ELISPOT assay for CTL generated by hsc70 fusion proteins. The effect of a helper epitope, OVA265‐280 in stimulating the generation of peptide specific CTL was difficult to evaluate using a standard 51Cr‐release assay. For this reason, we next carried cytokine ELISPOT assays to examine whether a more sensitive technique would reveal differences in the immunization strategies. The same protocol for in vivo immunization and in vitro peptide stimulation was used as in Figure 3. ELISPOT assays showed a clear effect of the OVA265‐280 helper epitope in a dose‐dependent manner with both IFA‐peptide and hsc70 fusion protein immunization (Fig. 4b,c). The number of IFN‐γ producing cells was much higher in hsc70 fusion proteins, which was consistent with the results obtained in the 51Cr‐release assay. We could still observe the enhanced helper‐effect of OVA265‐280 by immunization with the heterologous combination of OVA265‐280‐hsc70‐TRP2180‐188 (Fig. 4c). Intriguingly, administration of a mixture of hsc70‐TRP2180‐188 and OVA265‐280‐hsc70 gave rise to a higher number of spots than hsc70‐TRP2180‐188 alone (Fig. 4c), suggesting that these hsc70 fusion proteins were targeted to the same specialized antigen presenting cells in vivo through putative cell surface receptors like CD91 or LOX‐1.( 7 , 8 ) Taken together, these data suggest that addition of a helper epitope to the hsc70‐CTL epitope complex increased the population of peptide‐specific CTL.
Figure 4.
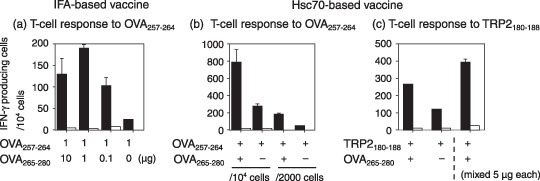
Enzyme‐linked immunospot assay for cytotoxic T lymphocyte (CTL) generated by immunization with peptides or hsc70 fusion proteins. The number of γ‐interferon (IFN‐γ)–producing cells in spleens of mice immunized with peptides or fusion proteins was determined. The same protocol for in vivo immunization and in vitro peptide stimulation was used as for Figure 2. Stimulated spleen cells (1 × 104 or 2 × 103 cells/well, 96‐well plate) were incubated with 5 × 105 naïve spleen cells/well pulsed with (black bar) or without (white bar) 1 µg/mL OVA257‐264 or TRP2 180–188 in triplicate form for 19 h. IFA, incomplete Freund's adjuvant.
In vivo protection effect of hsc70 fusion proteins against intradermal tumor growth. To gain insight into in vivo protection effect of the hsc70 fusion proteins, we vaccinated syngeneic mice at days –14 and –7, days 0 and 7, or days 2 and 9 and monitored the growth of E.G7 inoculated intradermally on day 0. On vaccination at days –14 and –7 or days 0 and 7, hsc70‐OVA257‐264 showed apparent protection, regardless of the presence of the helper epitope (Fig. 5a,b). However, the days 2 and 9 vaccination regimine did not show any protective effect (Fig. 5c), indicating that the vaccine is not strong enough to cure tumors once they have been established in the hosts.
Figure 5.
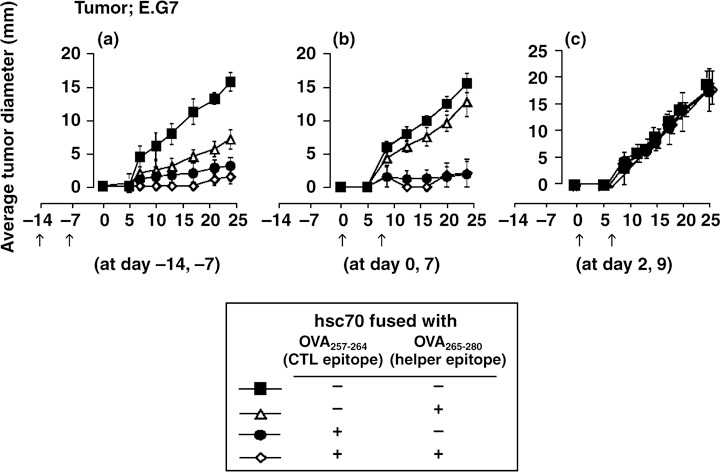
In vivo antitumor effect of hsc70 fusion proteins against E.G7. C57BL/6 mice were vaccinated intraperitoneally with 10 µg of each hsc70 fusion protein as indicated on (a) days –14 and –7, (b) days 0 and 7, or (c) days 2 and 9. The mice were challenged with E.G7 at day 0 and then tumor growth was monitored. (n = 5). CTL, cytotoxic T lymphocyte.
Next, we inoculated the OVA expressing B16 melanoma MO5 to examine the effect of hsc70 fusion proteins in another tumor model. Although Kb expression is difficult to detect by FACS analysis (data not shown), vaccination with hsc70‐OVA257‐264 on days 0 and 7 significantly delayed the appearance of the tumor, and inclusion of the OVA265‐280 helper epitope had a marginally additive effect (Fig. 6a). By contrast, hsc70‐TRP2180‐188 did not show significant protection, demonstrating that the vaccine effect of hsc70 depends on the antigenic peptides used in the fusion protein.
Figure 6.
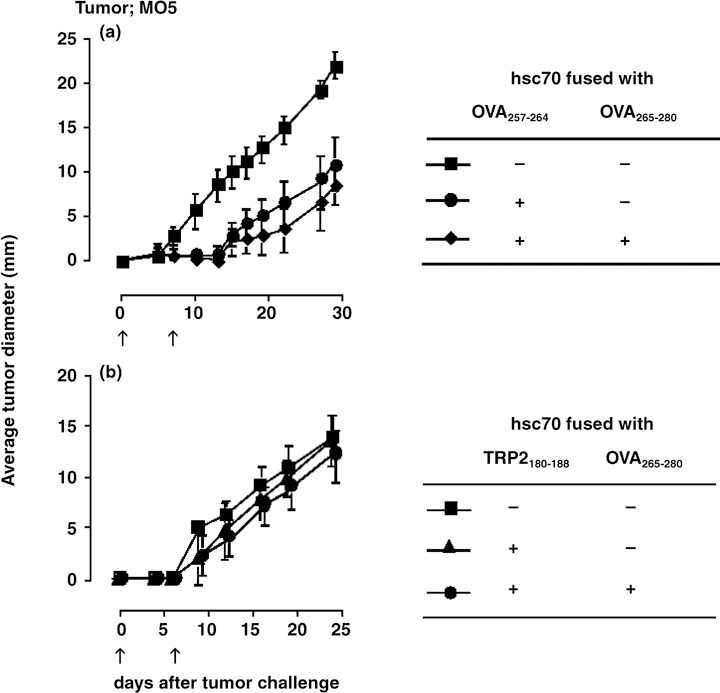
In vivo antitumor effect of hsc70 fusion proteins against MO5. C57BL/6 mice were vaccinated intraperitoneally with 10 µg of each hsc70 fusion protein as indicated on days 0 and 7. Mice were challenged with MO5 at day 0 and tumor growth was monitored (n = 5).
In vivo protection effect of hsc70 fusion proteins against melanoma metastasis. The induction of tumor immunity by any vaccination strategy has two aspects, (i) the remission of the original tumor mass; and (ii) the prevention/elimination of metastatic lesions. To examine the effect of hsc70 fusion proteins on the later, we injected MO5 melanoma cells intravenously in a standard metastasis model and vaccinated with hsc70‐OVA257‐264 at days 0 and 7 or at days 2 and 9. The numbers of pigmented metastatic spots on the lung surface were counted on day 15 after sacrificing the mice. The average spot number without vaccination was 70.6. In contrast, vaccination at days 0 and 7 and days 2 and 9 resulted in 17 and 61.2 spots, respectively (Fig. 7a), indicating that hsc70‐OVA257‐264 suppressed the MO5 metastasis. Next, we immunized mice with hsc70‐OVA257‐264 and OVA265‐280‐hsc70‐OVA257‐264 on days 0, 2, 5, and 7, and the number of lung spots were counted at day 21. OVA265‐280‐hsc70‐OVA257‐264 significantly suppressed metastasis, while on the other hand, hsc70‐OVA257‐264 showed no protection (Fig. 7b), indicating that addition of the OVA265‐280‐helper epitope to hsc70‐OVA257‐264 significantly enhanced the protection against MO5 lung metastasis. Representative pictures of the lung metastasis are shown in Figure 8a,b, in which the figure designations correspond to those of Figure 7. We confirmed that hsc70 alone gave no protection against MO5 metastasis, whereas OVA265‐280‐hsc70‐OVA257‐264 reproducibly suppressed the metastasis as shown in Figure 8c.
Figure 7.
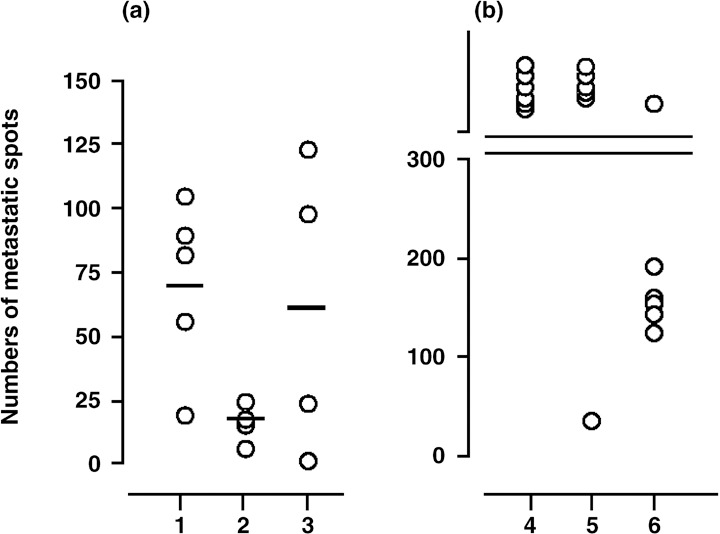
In vivo antitumor effect of hsc70 fusion proteins against MO5 lung metastasis. (a) On day 0, mice were challenged with MO5 via the tail vein, and vaccinated with hsc70‐OVA257‐264 at days 0 and 7 (column 2) or at days 2 and 9 (column 3). The numbers of metastatic spots of the lung surface were counted on day 15 after sacrificing the mice. Control mice were not vaccinated (column 1) (n = 4 or 5). (b) Mice were immunized with hsc70‐OVA257‐264 (column 5) and OVA265‐280‐hsc70‐OVA257‐264 (column 6) at days 0, 2, 5, and 7, and the spot numbers of lungs, were counted at day 21. Control mice were not vaccinated (column 4) (n = 6).
Figure 8.
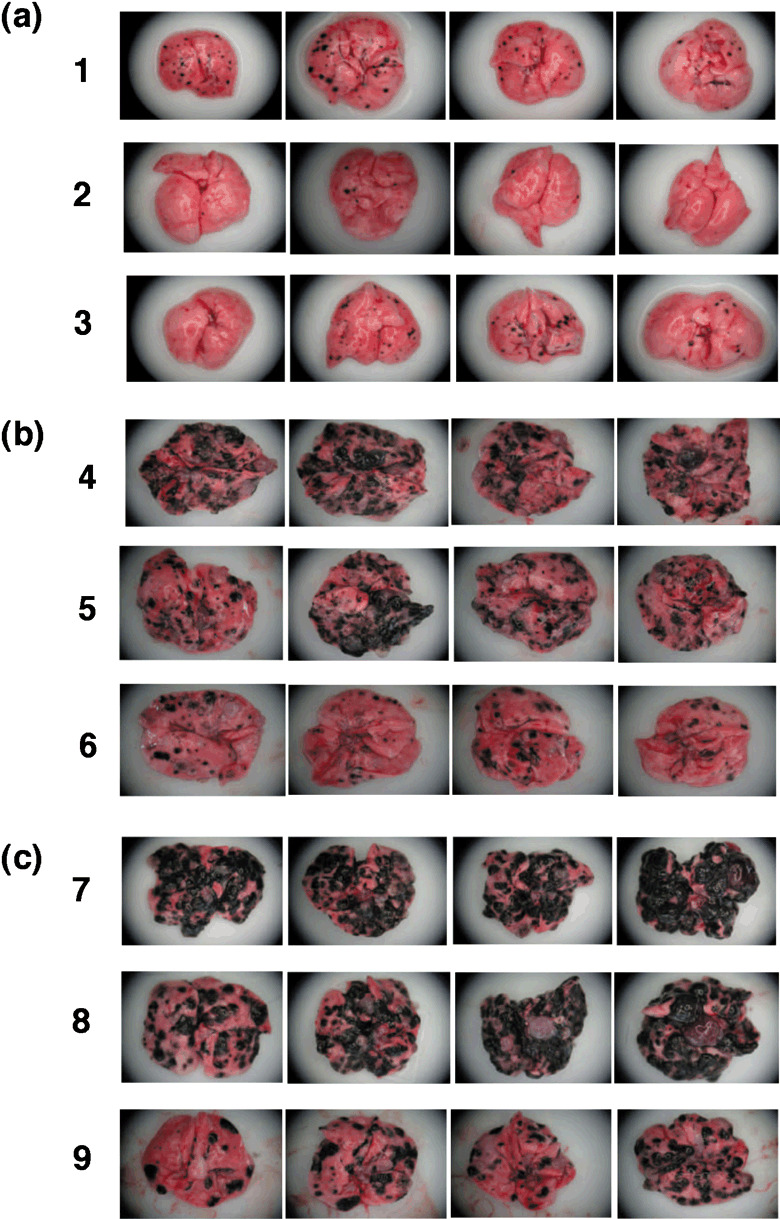
Macroscopic visualization of lung metastases. Metastatic spots on lung surfaces were observed with zoom stereo microscope at ×10 magnification. (a,b) The designations correspond to those in Figure 7. (c) On day 0, mice were challenged with MO5 via the tail vein, and vaccinated with hsc70 (column 8) or OVA265‐280‐hsc70‐OVA257‐264 (column 9) at days 0, 2, 5, and 7. At day 21, mice were sacrificed, and the spots were quantified. Control mice were not vaccinated (column 7).
In order to analyze effect of CD4+ T cells, we injected anti‐CD4 mAb to eliminate in vivo before vaccination with OVA265‐280‐hsc70‐OVA257‐264, and monitored metastatic spots. CD4+ T cell depletion slightly reduced the vaccination effect of OVA265‐280‐hsc70‐OVA257‐264 (Fig. 9a 2,3,b2,3) and the effect was nearly comparable for that of hsc70‐OVA257‐264 (Fig. 9a 3,4,b3,4).
Figure 9.
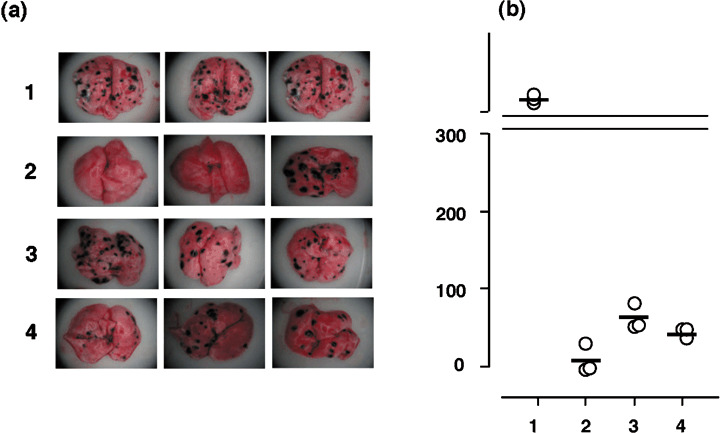
Effect of CD4+ cell depletion on vaccination against MO5 lung metastasis. On day –2, 0, 2, and 5, mice were injected with control mouse ascites (column 1, 2, and 4) or GK1.5 (anti‐CD4) ascites (column 3). On day 0, mice were challenged with MO5 via the tail vein, and vaccinated with OVA265‐280‐hsc70‐OVA257‐264 (column 2,3) and hsc70‐OVA257‐264 (column 4) at days 0, 2, 5, and 7. Control mice were not vaccinated (column 1). (a) Metastatic spots on lung surfaces were observed as in Figure 8. (b) The numbers of metastatic spots were counted on day 21 after sacrificing the mice. (n = 3).
An immune therapy for established tumors by hsc70 fusion protein. One of the goals of hsc70 fusion proteins is an immune therapy for established tumors in vivo. Since vaccination at day 6 after E.G7 inoculation did not show eradication of the tumor (data not shown), we simultaneously injected 2 × 106 OTI‐derived, purified CD8+ T cells, in addition to hsc70‐OVA257‐264, to tumor‐bearing mice. As a control, PA28α‐OVA257‐264 was injected. Adoptively transferred OTI CD8+ T cells alone or with an injection of PA28α‐OVA257‐264 did not confer tumor regression; however, OTI CD8+ T cells plus hsc70‐OVA257‐264 completely rejected once‐established tumor masses (Fig. 10a). We observed vigorous proliferation of adoptively transferred CFSE‐labeled OTI CD8+ T cells in the spleen after vaccination with hsc70‐OVA257‐264 but not by PA28α‐OVA257‐264 (Fig. 10b). We also observed by FACS the proliferating CFSE‐labeled OTI CD8+ T cells in the regional lymph node and tumor tissues (data not shown), suggesting that infiltration of activated OTI CD8+ T cells into the tumor mass is important for tumor rejection.
Discussion
HSP are evolutionally conserved across species and are constitutively expressed in all cells. Their expression level is dramatically increased by heat shock, a phenomenon termed the heat shock response. Heat shock response is caused not only by heat but also by other stresses such as heavy metals, hypoxia, alcohol intoxication, glucose starvation, and even cytokine stimulation.( 16 ) HSP stimulate proper folding of proteins damaged by these stressors and thus constitute a protein salvage system. On the other hand, it was recently suggested that HSP are also linked to the ubiquitin‐proteasome pathway to degrade proteins.
Hsp90 and hsc70 recognize unfolded proteins and peptides in the cytosol, where they recruit the E3 ubiquitin ligase CHIP (carboxyl terminus of hsc70 interacting protein) to polyubiquitinylate these substrates.( 17 , 18 ) The polyubiquitinylated proteins are degraded by the 26S proteasome, and some of the degradation products can potentially serve as peptide ligands for MHC class I molecules.
The proteins and peptides bound to HSP, once released outside cells, are efficiently trapped by antigen‐presenting cells via a number of HSP receptors and can then be cross presented to CD8+ T cells in the context of MHC class I molecules.( 19 ) In this context, mild biochemical procedures for the purification of HSP from tumor cells allow the copurification of unknown tumor antigens bound to HSP, which is the rationale behind the use of antigen‐HSP complexes for cancer vaccine therapy without knowing the identity of the tumor antigens recognized by T cells. One disadvantage of this vaccine approach may be the quantity of tumor antigens bound to HSP. Thus, many HSP isolated from tumor cells seem to lack associated tumor antigens.( 4 ) Fusion of mini genes encoding T cell epitopes to HSP cDNAs to construct a fusion protein may be a solution to this problem. Indeed, we previously found efficient generation of CTL by vaccination with several epitopes fused to hsc70.( 11 ) A question that remained to be addressed was whether this type of vaccine could eradicate tumors in vivo. In the present study, we have demonstrated that vaccination with a CD8+ T cell epitope fused to hsc70 resulted in a significant protective effect against in vivo tumor growth, and that simultaneous fusion of an epitope recognized by CD4+ T cells to HSP increased the population of primed CD8+ T cells, resulting in enhanced eradication of tumors.
Although antigens are readily cross‐presented to CTL in vitro by DC or other antigen presenting cells pulsed with HSP‐epitope complexes, these results do not necessarily imply that administration of these HSP‐epitope complexes in vivo will generate CTL efficiently enough to eradicate tumors. In order to obtain in vivo protection, T cells need to be fully activated, which can be accomplished by suitable activation of DC with HSP. Hsp70 and Gp96 have been shown to interact with toll‐like receptors 4 and 2 (TLR4 and 2),( 5 , 20 ) an encounter that activates DC to fully prime T cells. However, the idea that DC can be activated with HSP is still controversial.( 21 ) Endotoxin contaminating of the recombinant proteins, even though rigorously removed, might, at least in part, participate in activation of DC.( 22 , 23 ) On the other hand, Datta et al. showed that among agonists for various TLR, only those for TLR3,7 and 9 but not for TLR2 and 4, enabled DC to cross‐prime CD8+ T cells.( 24 ) We also observed that cross‐priming by hsp70 fusion proteins occurs equally well in vivo, even in TLR2 and 4 double KO mice (data not shown). These results suggest that the effect of endotoxin contamination is minimum or negligible in cross‐priming. It is possible that interaction between CD40 and hsp70 activates DC, as previously demonstrated.( 25 )
Addition of a CD4+ T cell epitope to the hsc70‐CTL epitope augmented the vaccination effect, especially in the MO5 lung metastasis model (7, 8, 9). This effect apparently resulted from an increased population of IFN‐γ‐producing CD8+ T cells as determined by ELISPOT assay (Fig. 4). Both helper and CTL epitopes should be expressed on the same DC because the two epitopes are within a single hsc70 molecule. Therefore, there may be simultaneous activation of both CD4+ and CD8+ T cells on the surface of a single DC. Activated CD4+ T cells express CD40L, which can interact with CD40 on DC. Thus, conditioning of DCs by CD4+ T cells is a likely possibility, and this might provoke much more efficient activation of CD8+ T cells in vivo. ( 26 , 27 ) Along this line, we attempted to generate OVA265‐280 specific CD4+ T cells in vitro by peptide stimulation but were unsuccessful.
From a clinical viewpoint, it is important to successfully produce a fusion protein containing hsc70 together with an epitope from a human cancer antigen. In this context, we recently demonstrated that ESO p157–165 of NY‐ESO‐1, a cancer/testis antigen, fused to human hsc70, was cross‐presented by DC to specific CTL clone, although the hsc70 preparation did not induce maturation of DC.( 28 ) Moreover, repetitive stimulation of CD8+ T cells with human DC pulsed with the fusion protein generated CTL against ESO p157–165. Taken together, the use of HSP‐peptide complexes for cancer vaccination is a reasonable approach, not only from theoretical and practical perspectives.
Acknowledgments
The authors are grateful to Ms. Hiroiwa for recombinant protein preparations. This work was supported by grants‐in‐aid for Scientific Research on Priority Areas from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
References
- 1. Udono H, Srivastava PK. Heat shock protein 70‐associated peptides elicit specific cancer immunity. J Exp Med 1993; 178: 1391–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Udono H, Srivastava PK. Comparison of tumor‐specific immunogenicities of stress‐induced proteins gp96, hsp90, and hsp70. J Immunol 1994; 152: 5398–403. [PubMed] [Google Scholar]
- 3. Tamura Y, Peng P, Liu K, Daou M, Srivastava PK. Immunotherapy of tumors with autologous tumor‐derived heat shock protein preparation. Science 1997; 278: 117–20. [DOI] [PubMed] [Google Scholar]
- 4. Ishii T, Udono H, Yamano T et al . Isolation of MHC class I‐restricted tumor antigen peptide and its precursors associated with heat shock proteins hsp70, hsp90, and gp96. J Immunol 1999; 162: 1303–9. [PubMed] [Google Scholar]
- 5. Asea A, Kraeft SK, Kurt‐Jones EA et al . HSP70 stimulates cytokine production through a CD14‐dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat Med 2000; 6: 435–42. [DOI] [PubMed] [Google Scholar]
- 6. Asea A, Rehli M, Kabingu E et al . Novel signal transduction pathway utilized by extracellular HSP70: role of toll‐like receptor (TLR) 2 and TLR4. J Biol Chem 2002; 277: 15028–34. [DOI] [PubMed] [Google Scholar]
- 7. Binder RJ, Han DK, Srivastava PK. CD91: a receptor for heat shock protein gp96. Nat Immunol 2000; 1: 151–5. [DOI] [PubMed] [Google Scholar]
- 8. Delneste Y, Magistrelli G, Gauchat J et al . Involvement of LOX‐1 in dendritic cell‐mediated antigen cross‐presentation. Immunity 2002; 17: 353–62. [DOI] [PubMed] [Google Scholar]
- 9. Berwin B, Hart JP, Rice S et al . Scavenger receptor‐A mediates gp96/GRP94 and calreticulin internalization by antigen‐presenting cells. Embo J 2003; 22: 6127–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ackerman AL, Giodini A, Cresswell P. A role for the endoplasmic reticulum protein retrotranslocation machinery during crosspresentation by dendritic cells. Immunity 2006; 25: 607–17. [DOI] [PubMed] [Google Scholar]
- 11. Udono H, Yamano T, Kawabata Y, Ueda M, Yui K. Generation of cytotoxic T lymphocytes by MHC class I ligands fused to heat shock cognate protein 70. Int Immunol 2001; 13: 1233–42. [DOI] [PubMed] [Google Scholar]
- 12. Udono H, Levey DL, Srivastava PK. Cellular requirements for tumor‐specific immunity elicited by heat shock proteins: tumor rejection antigen gp96 primes CD8+ T cells in vivo. Proc Natl Acad Sci USA 1994; 91: 3077–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Suzue K, Young RA. Adjuvant‐free hsp70 fusion protein system elicits humoral and cellular immune responses to HIV‐1 p24. J Immunol 1996; 156: 873–9. [PubMed] [Google Scholar]
- 14. Udono H, Saito T, Ogawa M, Yui K. Hsp‐antigen fusion and their use for immunization. Methods 2004; 32: 21–4. [DOI] [PubMed] [Google Scholar]
- 15. Maecker HT, Umetsu DT, DeKruyff RH, Levy S. Cytotoxic T cell responses to DNA vaccination: dependence on antigen presentation via class II MHC. J Immunol 1998; 161: 6532–6. [PubMed] [Google Scholar]
- 16. Lindquist S. The heat‐shock response. Annu Rev Biochem 1986; 55: 1151–91. [DOI] [PubMed] [Google Scholar]
- 17. Connell P, Ballinger CA, Jiang J et al . The co‐chaperone CHIP regulates protein triage decisions mediated by heat‐shock proteins. Nat Cell Biol 2001; 3: 93–6. [DOI] [PubMed] [Google Scholar]
- 18. Murata S, Minami Y, Minami M, Chiba T, Tanaka K. CHIP is a chaperone‐dependent E3 ligase that ubiquitylates unfolded protein. EMBO Rep 2001; 2: 1133–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Binder RJ, Srivastava PK. Peptides chaperoned by heat‐shock proteins are a necessary and sufficient source of antigen in the cross‐priming of CD8+ T cells. Nat Immunol 2005; 6: 593–9. [DOI] [PubMed] [Google Scholar]
- 20. Vabulas R, Braedel S, Hilf N et al . The endoplasmic reticulum‐resident heat shock protein Gp96 activates dendritic cells via the Toll‐like receptor 2/4 pathway. J Biol Chem 2002; 277: 20847–53. [DOI] [PubMed] [Google Scholar]
- 21. Tsan MF, Gao B. Endogenous ligands of Toll‐like receptors. J Leukoc Biol 2004; 76: 514–9. [DOI] [PubMed] [Google Scholar]
- 22. Bausinger H, Lipsker D, Ziylan U et al . Endotoxin‐free heat‐shock protein 70 fails to induce APC activation. Eur J Immunol 2002; 32: 3708–13. [DOI] [PubMed] [Google Scholar]
- 23. Warger T, Hilf N, Rechtsteiner G et al . Interaction of TLR2 and TLR4 ligands with the N‐terminal domain of Gp96 amplifies innate and adaptive immune responses. J Biol Chem 2006; 281: 22545–53. [DOI] [PubMed] [Google Scholar]
- 24. Datta SK, Redecke V, Prilliman KR et al . A subset of Toll‐like receptor ligands induces cross‐presentation by bone marrow‐derived dendritic cells. J Immunol 2003; 170: 4102–10. [DOI] [PubMed] [Google Scholar]
- 25. Wang Y, Kelly CG, Karttunen JT et al . CD40 is a cellular receptor mediating mycobacterial heat shock protein 70 stimulation of CC‐chemokines. Immunity 2001; 15: 971–83. [DOI] [PubMed] [Google Scholar]
- 26. Schoenberger SP, Toes RE, Van Der Voort EI, Offringa R, Melief CJ. T‐cell help for cytotoxic T lymphocytes is mediated by CD40–CD40L interactions. Nature 1998; 393: 480–3. [DOI] [PubMed] [Google Scholar]
- 27. Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T‐helper and a T‐killer cell. Nature 1998; 393: 474–8. [DOI] [PubMed] [Google Scholar]
- 28. Susumu S, Nagata Y, Ito S et al . Cross‐presentation of NY‐ESO‐1 cytotoxic T lymphocyte epitope fused to human heat shock cognate protein 70 by dendritic cells. Cancer Sci 2008; 99: 107–12. [DOI] [PMC free article] [PubMed] [Google Scholar]