

Abstract
Although certain inhibitors of histone deacetylases have been shown to induce cytotoxicity alone or in combination with chemotherapeutic agents in cancer cells, the molecular mechanism is not clear. The goal of the present study was to determine whether the antiseizure drug valproic acid (2‐propylpentanoic acid; VPA), which is also able to inhibit histone deacetylase, exhibits synergistic cytotoxicity with cisplatin, and the possible pathways for this. Our results clearly show that VPA not only exhibits synergistic cytotoxicity with cisplatin in all of the ovarian carcinoma cells tested, but also can resensitize the cells that have acquired resistance to cisplatin. Consistent with the increased cytotoxicity, cotreatment with VPA was shown to upregulate the cisplatin‐mediated DNA damage revealed by phosphorylation of ataxia telangiectasia mutation and histone H2AX. Reactive oxygen species accumulation and tumor suppressor phosphatase and tensin homolog (PTEN) overexpression, which could contribute to the enhanced cytotoxicity, were also observed to be upregulated by VPA. Because PTEN knockdown by small interference RNA or antioxidant treatment can reduce cisplatin‐mediated cytotoxicity, it is suggested that upregulation of PTEN and reactive oxygen species by VPA contributes to the enhancement of cisplatin‐mediated cytotoxicity. These results with resensitization of cisplatin‐resistant cells particularly may provide benefits in the treatment of ovarian cancer patients. (Cancer Sci 2008; 99: 1218–1226)
Platinum‐based chemotherapy is a standard, widely used treatment for ovarian cancer.( 1 ) Despite its great efficacy with an initial response rate of near 80%,( 1 ) the 5‐year survival of advanced‐stage ovarian cancers remains at a low rate, primarily due to the rapid development of resistance. Multiple mechanisms have been described for the ability of cancer cells to become resistant to cisplatin, including reduced accumulation of the drug,( 2 ) increased levels of glutathione( 3 ) and metallothionein,( 4 ) enhanced DNA repair,( 5 ) and alterations in apoptosis.( 6 ) In addition, loss of expression of DNA mismatch repair elements (e.g. the MutLa‐mismatch repair complex with hMLH1 and hPMS2 subunits) has been reported to lead to acquired resistance to cisplatin in ovarian and breast cancer cells.( 7 , 8 ) Until now, no specific treatment has been reported to reduce the resistance of cisplatin during cancer therapy.
Gene silencing associated with tumorigenesis and drug resistance has recently been an area of intense investigation because of the advanced understanding in the correlation between chromatin structure and gene expression.( 9 ) Studies have shown that gene silencing is often correlated with hypoacetylation of histone proteins through the aberrant actions of histone deacetylase (HDAC) enzymes. Given the close correlation between oncogenesis and the transcriptional silencing of certain genes due to the hypoacetylation of histones, inhibitors of HDAC (HDACi) that can modulate chromatin dynamics were tested as a potentially new class of anticancer agent.( 10 ) Recently, a number of studies have shown that the action of HDACi on the reexpression of certain silenced genes can result in cellular changes, including cell cycle arrest,( 11 ) DNA repair and damage assessment,( 12 ) and apoptosis.( 13 )
It was previously shown that hyperacetylation of histones by the HDACi suberoylanilide hydroxamic acid leads to chromatin decondensation, and the chromatin decondensation was shown to associate with a higher degree of DNA damage mediated by Topo II poison in a sequence‐specific manner.( 14 ) Similar observations and conclusions have also been reported by another study.( 15 ) Although these studies highlight important benefits for the clinical development of HDACi, the mechanism of the enhancement of cytotoxicity with certain DNA‐damaging agents is still not quite clear. In addition to the suggestion of the reexpression of certain apoptotic genes due to HDACi‐induced chromatin decondensation mediated by the hyperacetylation of histone proteins, it has been shown that reactive oxygen species (ROS) accumulation and glutathione reduction are critical to the cytotoxicity induced by cisplatin( 3 , 16 ) and HDACi.( 17 , 18 ) Moreover, it has not been shown that HADCi are able to resensitize the cancer cells that have acquired resistance to chemotherapeutic agents.
It has been shown that activation of phosphoinositol 3‐kinase (PI‐3K)–Akt machinery by the reduction of tumor suppressor phosphatase and tensin homolog (PTEN) contributes to cisplatin resistance in OVCAR‐3 ovarian cancer cells, and inhibition of PI‐3K sensitizes cells to cisplatin treatment.( 19 ) PTEN is a tumor suppressor gene encoding a phosphatase that negatively regulates cell survival mediated by the PI‐3K–Akt pathway.( 20 ) Therefore, upregulation of PTEN levels could decrease the survival signals and augment the cytotoxicity induced by DNA‐damaging agents. Moreover, inactivation of Akt and Erk1/2 is suggested to play a primary role in the synergistic apoptosis induced by coadministration of HDACi and perifosine or estradiol metabolite 2‐medroxyestradiol.( 21 , 22 )
Here, we expanded the studies of HDACi to evaluate the effects of the antiseizure drug valproic acid (2‐propylpentanoic acid; VPA) on cisplatin‐mediated cytotoxicity. VPA has been used clinically for a long time to treat seizures and was recently shown to possess HDACi activity. The effects of VPA on synergistic killing with cisplatin were evaluated in various ovarian cancer cell lines, especially cells that were acquired resistance to cisplatin. Our results show that upregulation of ROS production and PTEN protein levels caused by the action of VPA may contribute to the synergistic killing induced by cisplatin‐mediated DNA damage.
Materials and Methods
Cell lines. The human ovarian cancer cell lines SK‐OV‐3, OVCAR‐3, and TOV‐21G were obtained from American Type Culture Collection (Manassas, VA, USA). Cell lines A2780 and A2780/cp70 were obtained from Dr Tim Huang's lab (Ohio State University, Columbus, OH, USA). Cells were maintained at 37°C (5% CO2/air atmosphere) in RPMI‐1640 (Gibco, Rockville, MD, USA) containing 10% heat‐inactivated fetal bovine serum. A2780‐hMLH1+ and A2780‐hMLH1− were subcloned from A2780 cells by limiting dilution.
Cytotoxic agents and chemical reagents. Cisplatin, calboplatin, paclitaxel, topotecan, the HDACi trichostatin A (TSA) and VPA, and N‐acetyl‐cysteine (NAC) were purchased from Sigma (St Louis, MO, USA).
Measurement of growth inhibition and cytotoxicity. The cells were plated at 5 × 103 cells/96‐well plate, 1 day before drug treatment, and then cotreated with cisplatin, paclitaxel, or topotecan and TSA or VPA for 24 h. After washing, the cells were incubated with fresh medium for 72 h and then stained with CellTiter 96 Aqueous One Solution Reagent (Promega), which contains a novel tetrazolium compound (3‐[4,5‐dimethylthiazol‐2‐yl]‐5‐[3‐carboxymethoxyphenyl]‐2‐[4‐sulfophenyl]‐2H‐tetrazolium, inner salt; MTS) and an electron‐coupling reagent (phenazine ethosulfate). To measure the amount of soluble formazan produced by the cellular reduction of MTS, the absorbance was recorded at 490 nM using a 96‐well plate reader. Each reaction was assayed at least three times. The results were expressed as the ratio of cytotoxicity to the concentration of cisplatin, paclitaxel, or topotecan as a percentage of one minus MTS reduction in treated samples compared with untreated samples for the drug alone, or compared with TSA‐ or VPA‐treated samples for TSA or VPA plus cisplatin‐treated samples.
To measure the cytotoxicity directly, the cells grown on six‐well tissue culture plates were cotreated with cisplatin and TSA or VPA for 24 h in 2 mL medium supplemented with 10% fetal bovine serum. The cells were then trypsinized and replated to grow for 10 days to observe the surviving colonies.
Cytological immunostaining of hMLH1, hMSH2, hMSH6, and phosphorylated ataxia telangiectasia mutation (ATM) and histone H2AX proteins. Cells were grown on glass coverslips overnight and then fixed with the 50% acetone–50% methanol solution and washed with phosphate‐buffered saline (PBS). Immunostaining was carried out in accordance with the manufacturer's instructions (K0672, LSABII System‐HRP, Dako, Carpinteria, CA, USA). The fixed cells were incubated for 30 min at room temperature with antibodies against human hMLH1 (monoclonal antibody [mAb] G168‐15; PharMingen, USA), hMSH2 (mAb clone 27; Transduction Laboratories, USA), hMSH6/GTBP (mAb clone 44; Transduction Laboratories), phosphorylated‐ATM (mAb 10H11.E12; Calbiochem, USA), or γH2AX (Calbiochem) diluted 1:50. Immunoreactivity was detected using a two‐step peroxidase detection system (Dako). For immunofluorescence staining analysis, rhodamine red‐ or cy3‐conjugated antimouse, antirabbit, or antigoat IgG (Jackson ImmunoResearch Laboratory, USA) was used. Staining was evaluated under a microscope after hematoxylin counterstaining.
Western blotting. Cell extracts were prepared in lysis buffer (50 mM HEPES [pH 7.6], 0.5% sodium dodecylsulfate, 1% sodium deoxycholate, and 5 mM ethylenediaminetetraacetic acid) containing a cocktail of protease inhibitors (Sigma). Protein samples before and after immunoprecipitation were separated by 7% sodium dodecylsulfate–polyacrylamide gel electrophoresis and electroblotted to polyvinylidene fluoride membranes (Amersham). Antibodies against acetylated histone H4 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), PTEN (Cell Signaling, USA) and actin (l‐19; Santa Cruz Biotechnology) were used for western blotting. Secondary antibodies conjugated with horseradish peroxidase (Sigma) and enhanced chemiluminescence agents (Amersham) were used for detection.
Measurement of ROS production. Cells were incubated with 20 µM 2′,7′‐dichlorofluorescein diacetate (Sigma) for 15 min at 37°C,( 23 ) after treatment with cisplatin, VPA, or cisplatin plus VPA for 24 h. The cells were then washed once with 0.1% azide in PBS, tripsinized, and fixed with 2% paraformaldehyde (Sigma) in PBS. The green fluorescence of oxidized 2′,7′‐dichlorofluorescein was observed under a fluorescence microscope or measured at an excitation wavelength of 480 nm and an emission wavelength of 525 nm using a FACS Vantage cell sorter (Becton Dickinson, Mountain View, CA, USA).
To test whether ROS production was responsible for cell death, the cells were pretreated with a scavenger of ROS, 30 mM NAC, for 30 min prior to the addition of cisplatin or cisplatin plus VPA for 24 h. After washing, the cells were cultured for another 72 h before testing the cytotoxicity by MTS assay.
RNA interference. Cells were seeded in 100 × 20 mm culture dishes with complete RPMI‐1640 medium supplemented with 10% fetal bovine serum 24 h prior to transfection. Small interference double‐strand RNA (siRNA) against the PTEN gene was purchased from Cell Signaling and Ambion, and siRNA constructs were introduced into cells using Lipofectomine (Cell Signaling) according to the manufacturer's instructions. Transfected cells were cultured for 48 h prior to undergoing the experiments detecting PTEN protein level and cisplatin‐ and cisplatin plus VPA‐induced cell killing.
Results
Synergistic killing effects of cotreatment with VPA and cisplatin in ovarian cancer cells. To test whether VPA, which was shown to inhibit HDAC,( 24 , 25 ) could result in synergistic cytotoxicity with cisplatin in ovarian cancer cells, cell lines with various sensitivities to cisplatin (Fig. 1a) were cotreated with VPA and cisplatin. Surprisingly, significant populations (more than 40%) of A2780 cells were observed to lose hMLH1 expression by immunostaining (Fig. 1a). The lost gene expression in the parental A2780 cells was restricted to hMLH1 as hMSH2 and hMSH6 were expressed normally (data not shown). For obtaining homogeneous population hMLH1 expression, parental A2780 cells were subcloned by limiting dilution. The expression of hMLH1 in a total of 60 sub‐cloned cells was determined by immunostaining (data not shown), and two clones named A2780‐4 (hMLH1+) and A2780‐12 (hMLH1−) were chosen for further study (Fig. 1b,c). The concentrations of cisplatin that lead to 50% inhibition of cell proliferation (i.e. the IC50) in the ovarian cancer cells shown in Figure 1a were A2780‐4 (hMLH1+; 0.25 µM), A2780‐12 (hMLH1−; 1.25 µM), SKOV‐3 (1.3 µM), OVCAR‐3 (2.8 µM), TOV‐21G (1 µM), SKOV‐3R (2.5 µM), and A2780/cp70 (5 µM). Consistent with results shown in other studies,( 15 , 26 ) the cisplatin‐mediated growth inhibition and cell killing measured by clonogenic assays (data not shown) were enhanced by cotreatment with the near‐non‐toxic dose of VPA (0.6 mM) in all of the tested cells, including A2780‐4 (hMLH1+) (Fig. 1b), which were the most sensitive to cisplatin (Fig. 1a), A2780‐12 (hMLH1−) (Fig. 1c), and SKOV‐3 (hMLH1−) (Fig. 1d), which were more resistant to cisplatin (Fig. 1a). The lost expression of hMLH1 in A2780‐12 and SKOV‐3 cells was not restored by VPA treatment (data not shown). Similarly, as shown in Figure 1e,f, the cisplatin‐mediated cytotoxicity enhanced by VPA was also observed in the other ovarian cancer cells, which were also more resistant to cisplatin than the A2780‐4 cells. These results clearly suggest that VPA can enhance the cisplatin‐mediated cell killing in ovarian cancer cells, including in cells that are intrinsically resistant to cisplatin due to lost expression of the mismatch repair gene hMLH1 (Fig. 1b–f).
Figure 1.
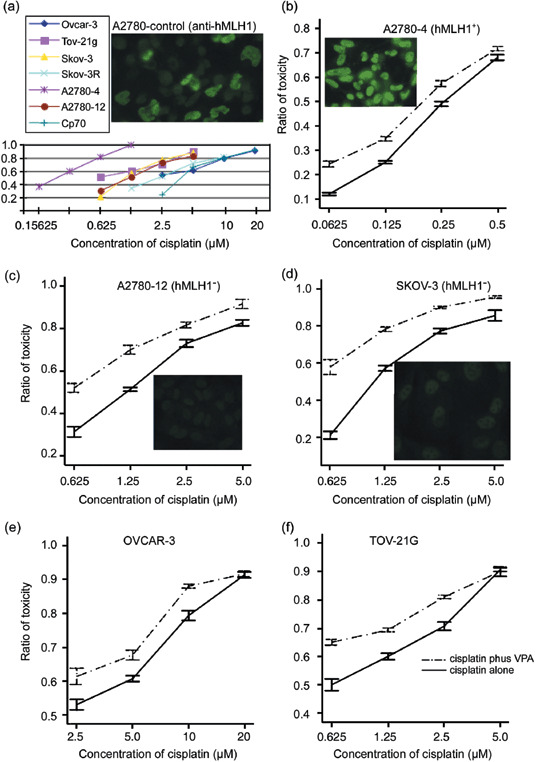
Treatment of valproic acid (VPA) enhances cisplatin‐mediated cytotoxicity. (a) Concentrations of cisplatin that lead to 50% inhibition of cell proliferation (i.e. the IC50) in ovarian cancer cells used for studies are A2780‐4 (hMLH1+; 0.25 µM), A2780‐12 (hMLH1−; 1.25 µM), SKOV‐3 (hMLH1−; 1.3 µM), OVCAR‐3 (2.8 µM), TOV‐21G (1 µM), SKOV‐3R (2.5 µM), and A2780/cp70 (5 µM). (b–f) Evaluation of the cytotoxicity by 3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfophenyl)‐2H‐tetrazolium (MTS) assays in all of the ovarian cancer cells was carried out under 24 h treatment with VPA and different concentrations of cisplatin as indicated. The solid lines indicate treatment with cisplatin alone, and the dotted lines show cotreatment with VPA and cisplatin. The results presented are the average and standard error of three independent experiments.
To further determine whether VPA could also enhance cisplatin‐mediated cytotoxicity in cancer cells that had acquired resistance to cisplatin, SKOV‐3R cells derived from human ovarian cancer SKOV‐3 cells, which were treated with 2.5 µM cisplatin for more than 6 months, and cisplatin‐resistant A2780/cp70 cells derived from A2780 cells that were obtained from Dr Tim Huang were used for the studies. The relative resistance ratios of SKOV‐3R to SKOV‐3 and A2780/cp70 to A2780‐4 cells were 2 and 20, respectively (Fig. 1a). As shown in Figure 2a, cisplatin‐mediated cell killing in both SKOV‐3R and A2780/cp70 cells was also shown to be enhanced by the near‐non‐toxic dose (0.6 mM) of VPA (Fig. 2b). In summary, these data show that the near‐non‐toxic dose of VPA can enhance cisplatin‐mediated cytotoxicity in ovarian cancer cells no matter whether they have intrinsic (Fig. 1) or acquired (Fig. 2) resistance to cisplatin.
Figure 2.
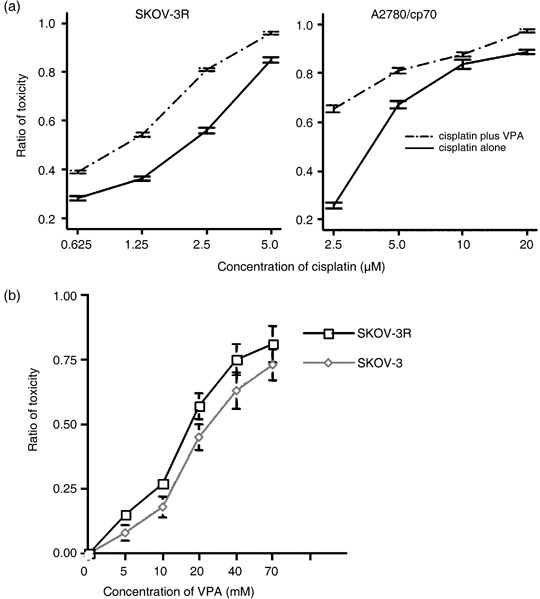
Valproic acid (VPA) exhibits synergistic cytotoxicity with cisplatin in ovarian cancer cells that have acquired resiatance to cisplatin. (a) Evaluation of cytotoxicity by 3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfophenyl)‐2H‐tetrazolium (MTS) assays was carried out for 24 h in A2780/cp70 cells or 72 h in SKOV‐3R cells by treatment with VPA and various concentrations of cisplatin. The solid lines indicate treatment with cisplatin alone, and the dotted lines indicate cotreatment with VPA and cisplatin. (b) Concentrations of VPA that lead to 50% inhibition of cell proliferation (i.e. the IC50) in ovarian cancer cells used for studies are SKOV‐3 (21 mM) and SKOV‐3R (17.5 mM). The results presented are the average and standard error of three independent experiments.
To further demonstrate the effects of combining cisplatin and VPA, the combination index (CI) was determined using the simplified index‐isobologram method based on the median principle developed by Chou and Talalay.( 27 ) The CI were calculated as:
| CI = CA,50/IC50,A + CB,50/IC50,B, |
CA,50 and CB,50 were the concentrations of drugs A and B used in combination to achieve 50% of the drug effect, and IC50,A and IC50,B were the IC50 of the single agents A and B. A CI equal to 1 indicates an additive effect; CI < 1 indicates synergy by the combination of two drugs; and CI > 1 indicates antagonism. For SKOV‐3 cells, the IC50 of cisplatin was 1.25 µM (Fig. 1a) and the IC50 of VPA was 21 mM (Fig. 2b). The combination of 0.625 µM cisplatin and 2 mM VPA resulted in 50% cytotoxicity. Therefore:
| CI = 0.625/1.25 + 2/21 = 0.60. |
For SKOV‐3R cells, the IC50 of cisplatin was 2.5 µM (Fig. 1a) and the IC50 of VPA was 17.5 mM (Fig. 2b). The combination of 1.25 µM cisplatin and 2 mM VPA resulted in 50% cytotoxicity. Therefore:
| CI = 1.25/2.5 + 2/17.5 = 0.61. |
Similar results were observed in three independent experiments with P < 0.03. Therefore, these results clearly show that VPA exhibits synergistic cytotoxicity with cisplatin in ovarian cancer cells that have either intrinsic or acquired resistance to cisplatin.
To determine whether this enhancement of cytotoxicity by cotreatment with VPA was specific to cisplatin, ovarian cancer cells were cotreated with VPA and topotecan (Hycamtin) or paclitaxel (Phyxol) that are also common chemotherapeutic drugs for ovarian cancer. Whereas VPA exhibits synergistic cytotoxicity with cisplatin, VPA does not show the enhanced effect of cell killing in SKOV‐3 and SKOV‐3R cells with topotecan and paclitaxel (Fig. 3),
Figure 3.
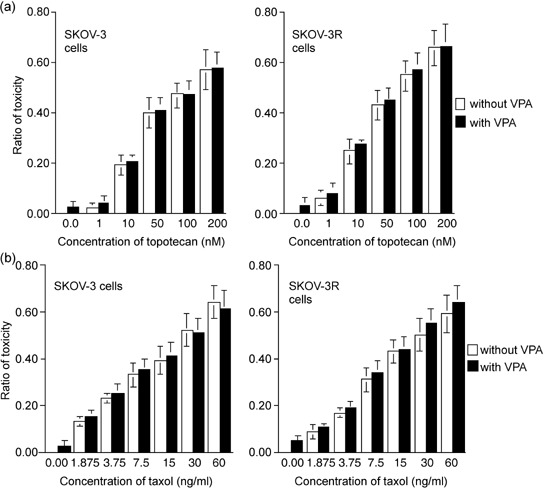
Valproic acid (VPA) does not exhibit synergistic cytotoxicity with paclitaxel and topotecan in the ovarian cancer cells. Evaluation of cytotoxicity by 3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfophenyl)‐2H‐tetrazolium (MTS) assays was carried out in SKOV‐3 and SKOV‐3R cells by treatment with VPA and various concentrations of (a) topotecan and (b) paclitaxel. The results presented are the average and standard error of three independent experiments.
Valproic acid upregulates the reduced expression of PTEN in cisplatin‐resistant cells. To determine the possible pathways of synergistic killing induced by VPA, we chose to measure the expression level of PTEN in the beginning. As shown in Figure 4a, the protein level of PTEN in SKOV‐3R and A2780/cp70 cells that had acquired resistance to cisplatin was observed to be lower than that in the parental SKOV‐3 and A2780‐4 cells. Furthermore, treatment with VPA alone or cotreatment with VPA and cisplatin was found to increase the protein levels of PTEN in both SKOV‐3 and SKOV‐3R cells (Fig. 4a), suggesting that upregulation of PTEN might sensitize or resensitize cancer cells to cisplatin due to the effect of reducing the survival signals mediated by PI‐3K–Akt machinery. To further determine whether the modulation of PTEN could indeed play a role in the synergistic cytotoxicity during the cotreatment with VPA and cisplatin, we examined the levels of PTEN and cytotoxicity under treatment with two siRNA constructs that were shown to knockdown the expression of PTEN. Similar results were observed with the two constructs. As shown in Figure 4b, knockdown of PTEN expression by the related siRNA but not by the control siRNA was demonstrated by the analysis of flow cytometry and western blotting with anti‐PTEN antibody in SKOV‐3 cells. Consistent with the results that protein level of PTEN in SKOV‐3R cells was observed to be lower than that in SKOV‐3 cells as shown in Figure 4a, cells that were transfected with PTEN siRNA were more resistant to cisplatin‐mediated cell killing either alone or with VPA cotreatment as shown in Figure 4c. These data show that upregulation of PTEN may contribute to VPA‐mediated synergistic cytotoxicity with cisplatin.
Figure 4.
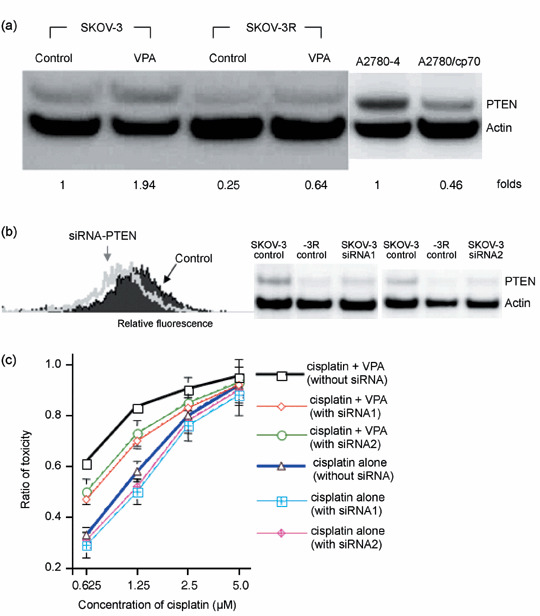
Knockdown of phosphatase and tensin homolog (PTEN) results in resistance to cisplatin‐ or cisplatin plus valproic acid (VPA)‐induced cytotoxicity. (a) Downregulation of PTEN expression in both cisplatin‐resistant SKOV‐3R and A2780/cp70 cells was observed by Western blotting analysis. Upregulation of PTEN expression in both SKOV‐3 and SKOV‐3R cells upon VPA treatment was observed by Western blotting analysis. The fold increase in PTEN expression was calculated after normalizing to the level of actin. (b) Knockdown of PTEN by specific small interfering RNA (siRNA) in SKOV‐3 cells was demonstrated with control siRNA by flow cytometry and Western blotting. (c) Knockdown of PTEN by specific siRNA resulted in resistance to cisplatin‐ or cisplatin plus VPA‐induced cytotoxicity. SKOV‐3 cells with or without the transfected siRNA that showed knockdown of PTEN expression were treated with cisplatin alone or cisplatin plus VPA for 24 h and assayed for cytotoxicity after incubation in fresh medium for 72 h. The results presented are the average and standard error of three independent experiments.
Valproic acid further upregulates cisplatin‐mediated ROS accumulation. To demonstrate whether VPA could further upregulate the accumulation of ROS, the levels of ROS were analyzed by flow cytometry under the treatment with VPA and cisplatin. As shown in Figure 5a, ROS were upregulated by either VPA or cisplatin alone, and ROS were further accumulated by cotreatment with cisplatin and VPA in both SKOV‐3 and SKOV‐3R cells. To determine whether upregulation of ROS could indeed play a role in the synergistic cytoxicity mediated by VPA, the cells were cotreated with the antioxidant NAC, a precursor of glutathione and scavenger of ROS,( 28 ) upon treatment with VPA and cisplatin. Pretreatment for 15 min with 30 mM NAC reduced both ROS production (data not shown) and the cytotoxicity induced by cotreatment with cisplatin and VPA (Fig. 5b). However, it has been suggested that inhibition of drug uptake by NAC contributes to the abrogation of cisplatin‐mediated cytotoxicity in renal tubule epithelial cells.( 29 ) To prevent the inhibition of cisplatin uptake, A2780‐12 (hMLH1−) and SKOV‐3 cells were pretreated with cisplatin for 6 h followed by cotreatment with NAC and VPA without removing the cisplatin. As shown in Figure 5c, the effect of synergistic cytotoxicity mediated by 0.6 mM VPA was still observed with 0.5 µM cisplatin pretreatment for 6 h without NAC. As expected, although 30 mM NAC did not inhibit the cytotoxicity mediated by pretreatment with 0.5 µM cisplatin alone for 6 h, it significantly reduced the synergistic cell killing effect mediated by cotreatment with VPA and cisplatin (Fig. 5c). These results show that an antioxidant (NAC) that can reduce the accumulation of ROS decreases the synergistic cytotoxicity mediated by VPA, suggesting that further upregulation of ROS by VPA contributes to the synergistic cytotoxicity with cisplatin.
Figure 5.
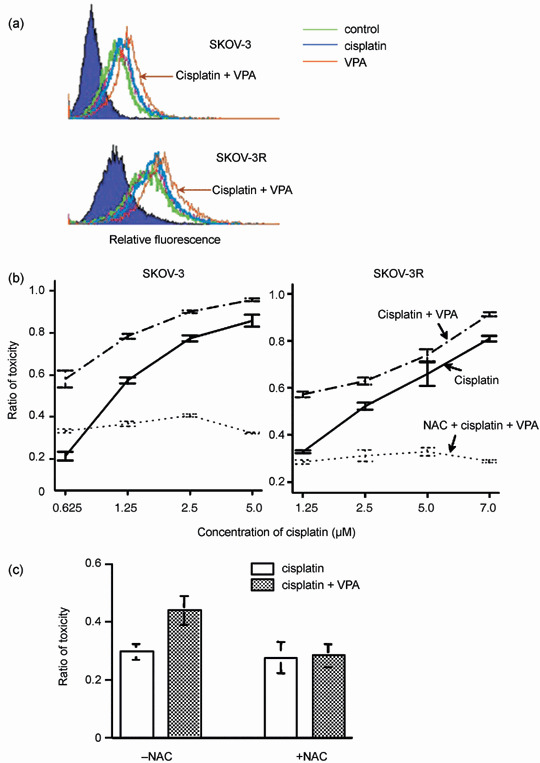
Upregulation of cisplatin‐mediated reactive oxygen species (ROS) production by valproic acid (VPA) and the attenuation of VPA‐mediated synergistic cytotoxicity by the free radical scavenger N‐acetylcysteine (NAC). (a) Production of ROS in SKOV‐3 and SKOV‐3R cells was determined under treatment without (control) or with cisplatin or VPA alone, or with VPA plus cisplatin for 24 h. The intensity of ROS‐mediated oxidation of the fluorochrome was measured by flow cytometry. (b,c) The inhibition of VPA‐mediated synergistic cell killing by a ROS scavenger. (b) SKOV‐3 and SKOV‐3R cells were preincubated with NAC for 20 min and then treated with cisplatin plus VPA for 24 h. Then the cells were cultured in fresh medium for 72 h before assays for the inhibition of cytotoxicity. (c) SKOV‐3 cells were pretreated with cisplatin for 6 h before the addition of VPA and NAC. The results presented are the average and standard error of three independent experiments.
Effects of VPA on cisplatin‐mediated DNA damage and histone acetylation. To determine the correlation between the upregulation of DNA damage and the synergistic cytotoxicity by VPA, we measured the level of phosphorylated ATM, which is a central signal tranducer of DNA double‐strand breaks. Despite the unknown molecular mechanism, cisplatin has been shown to induce DNA double‐strand breaks, which lead to the activation of ATM by phosphorylation, and cell death.( 30 ) Consistent with the VPA‐induced synergistic cytotoxicity shown in 1, 2, the upregulation of ATM activation, shown by immunostaining with antiphosphorylated ATM antibody, was also observed in the nuclei of SKOV‐3 and SKOV‐3R cells as shown in Figure 6a. Consistent with the upregulation of phosphorylated ATM by VPA shown in Figure 6a, the increased level of foci revealed by the phosphorylation of histone H2AX, a protein that becomes evident at the site of each double strand break (DSB) that can be visualized by immunofluorescent staining, was also detected by immunostaining with antiγH2AX antibody upon cotreatment with VPA and cisplatin but not with cisplatin or VPA alone (Fig. 6b).
Figure 6.
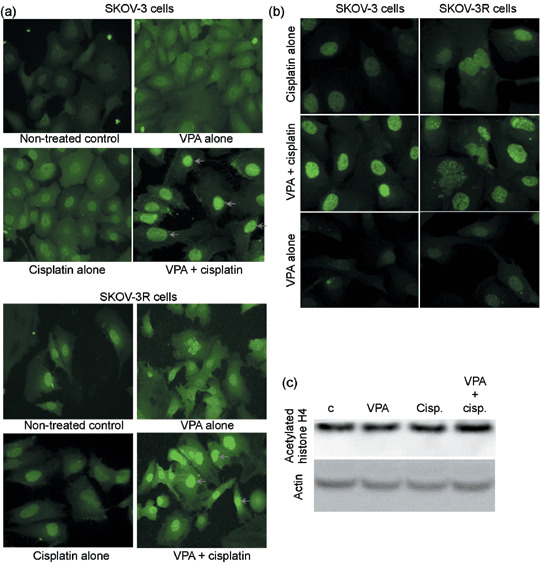
Treatment with valproic acid (VPA) enhances the levels of histone acetylation and DNA damage induced by cisplatin. (a,b) Increased DNA damage upon treatment with VPA and cisplatin was observed by immunostaining with antiphsphorylated ataxia telangiectasia mutation (ATM) and γH2AX antibodies. Increased DNA damage upon cotreatment with cisplatin and VPA for 24 h was observed clearly in both SKOV‐3 and SKOV‐3R cells by comparison of the foci of phosphorylated ATM and γH2AX, indicated with arrows, between treatments with cisplatin or VPA alone and cotreatment with VPA and cisplatin. The results presented here show one of three independent experiments that obtained almost identical results. (c) VPA‐induced hyperacetylation of histones. Acetylation of histone H4 was evaluated by western blotting analysis in SKOV‐3 cells under 24 h treatment with VPA without or with cisplatin.
To determine whether the effect of VPA in augmenting DNA damage induced by cisplatin is due to chromatin relaxation induced by the VPA‐mediated hyperacetylation of histones, we measured the levels of acetylated histone H4 using a specific antibody. As shown in Figure 6c, slightly higher levels of acetylated histone H4 were observed with VPA treatment at the non‐toxic dose of 0.6 mM. These results suggest that the relaxation of chromatin mediated by the hyperacetylation of histone H4 upon VPA treatment may not contribute to the upregulation of cisplatin‐mediated DNA damage as revealed by the increased levels of phosphorylated ATM and H2AX in Figure 6a,b.
Effects of cotreatment with TSA and cisplatin in ovarian cancer cells on cell killing and histone acetylation. To determine whether TSA, which is well known to inhibit the broad spectrum of HDAC, could result in synergistic cytotoxicity with cisplatin in ovarian cancer cells, ovarian cancer cells were cotreated with TSA and cisplatin. As shown in Figure 7a, cisplatin‐mediated cell killing in both SKOV‐3 and SKOV‐3R cells was also shown to be enhanced by a very low toxic dose (50 nM) of TSA. To determine whether the effect of TSA in augmenting the cytotoxicity induced by cisplatin is correlated with chromatin relaxation induced by the TSA‐mediated hyperacetylation of histones, we measured the levels of acetylated histone H4 using a specific antibody. As shown in Figure 7b, significantly higher levels of acetylated histone H4 were observed in the treatment with 50 nM TSA. These results suggest that the relaxation of chromatin mediated by the hyperacetylation of histone H4 upon TSA treatment may contribute to the upregulation of cisplatin‐mediated cell killing.
Figure 7.
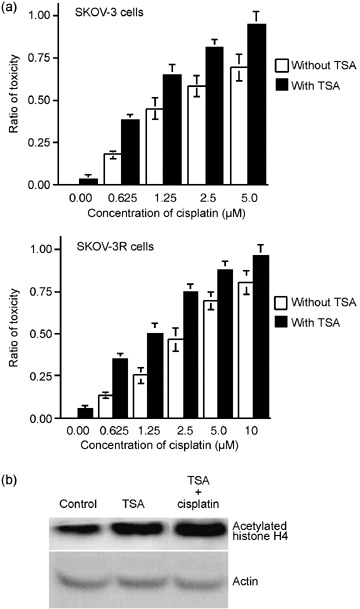
Treatment with trichostatin A (TSA) enhances the cisplatin‐mediated cytotoxicity and the levels of the histone acetylation. (a) The increased cytotoxicity upon treatment with TSA and cisplatin was observed clearly in both SKOV‐3 and SKOV‐3R cells. Results presented here show one of three independent experiments that obtain almost identical results. (b) TSA‐induced hyperacetylation of histone. Acetylation of histone H4 was evaluated by Western blotting analysis in SKOV‐3 cells under 24 h treatment by TSA without or with cisplatin.
Discussion
The present study reports the effects and mechanisms of VPA, which has been reported to inhibit the action of HDAC in enhancing antitumor action with platinated agents in ovarian cancer cells.( 24 , 25 ) VPA has been used to treat seizures for more than 30 years, and recent studies have indicated that VPA itself may possess anticancer activity against a variety of tumors.( 31 ) In fact, the Food and Drug Administration in USA granted VPA orphan drug status to treat familial adenomatous polyposis in July 2005. In addition to their anticancer activity,( 32 ) HDACi have also been shown to increase the cytotoxicity of certain anticancer drugs targeting DNA or enzymes acting on DNA such as etoposide, doxorubicin, and cisplatin.( 15 ) However, the effects of combination therapy mediated by HDACi have not been tested intensively with VPA.
In the current study, we test the synergistic actions of cotreatment with VPA and cisplatin on ovarian cancer cell lines that had either inherent (A2780‐hMLH1−, SKOV‐3, OVCAR‐3, and TOV‐21) or acquired (A2780/cp70 and SKOV‐3R) resistance to cisplatin. Indeed, our data clearly indicate that VPA, at near‐non‐toxic and clinically relevant concentrations,( 33 ) could still exhibit synergistic cytotoxicity with cisplatin in all of the tested cells (1, 2). Unexpectedly, cancer cells that had acquired resistance to cisplatin were slightly more sensitive to VPA (Fig. 2b). As VPA does not show an enhanced cell killing effect in SKOV‐3 and SKOV‐3R cells with topotecan (Hycamtin) or paclitaxel (Phyxol), which are also common chemotherapeutic drugs for ovarian cancer (Fig. 3), this enhancement of cytotoxicity by cotreatment with VPA is specific to cisplatin. Elucidating the mechanisms responsible for this sensitivity may lead to novel strategies for improving cancer therapy with cisplatin‐resistance and defective PTEN (Fig. 4).
It has been shown that activation of the PI‐3K–Akt pathway by PTEN reduction results in resistance to cisplatin in an ovarian cancer cell line.( 19 ) Consistent with this observation, the amount of PTEN is much lower in SKOV‐3R and A2780/cp70 cells, which have been acquired resistance to cisplatin (Fig. 4a). This observation lead us to test the possibility that VPA could upregulate PTEN expression, which contributes to the synergistic cytotoxic effect with cisplatin. It has been shown that PTEN overexpression results in the enhancement of cisplatin‐mediated cytotoxicity in ovarian cancer cells.( 34 ) As shown in Figure 4a, VPA indeed upregulates PTEN in both SKOV‐3 and SKOV‐3R cells. Moreover, as the cells transfected with PTEN‐related siRNA were more resistant to either cisplatin alone or cotreatment with VPA and cisplatin (Fig. 4c), upregulation of PTEN by VPA is suggested to contribute to the synergistic cytotoxicity with cisplatin.
Consistent with the suggestion that the increased accumulation of ROS contributes to cytotoxicity by certain HDACi,( 17 , 18 ) cotreatment with VPA was shown to further upregulate the production of ROS induced by cisplatin in both SKOV‐3 and SKOV‐3R cells (Fig. 5a), and the effect of synergistic cytotoxicity was also shown to be inhibited by cotreatment with the antioxidant NAC (Fig. 5c). These results suggest that further upregulation of ROS by VPA contributes to synergistic cytotoxicity with cisplatin. Consistent with these results, it has recently been shown that NAC not only reduces ROS accumulation substantially but also blocks the induction of cell killing mediated by cotreatment with the multiple receptor tyrosine kinase inhibitor AEE788 and the HDACi LBH589.( 35 )
Further accumulation of ROS mediated by VPA (Fig. 5) may upregulate the levels of DNA damage (Fig. 6a) and the cell killing in concert with the action of PTEN (Fig. 4). Very recently, it has been shown that inhibition of PI‐3K–Akt signaling by upregulation of PTEN or a PI‐3K inhibitor radiosensitizes cancer cells.( 36 , 37 ) Therefore, it is highly possible that upregulation of PTEN by VPA also contributes to enhance the cytotoxicity induced by the increased levels of DNA damage upon cotreatment with VPA and cisplatin. In contrast to these reports, it has been shown that PTEN null cells exhibit accumulation of DNA double‐strand breaks,( 38 ) and it has also been shown that PTEN acts on chromosome centromeres and upregulates the expression of Rad51 that is involved in the repair of DNA double‐strand breaks.( 39 ) Clearly, further studies are required to clarify the roles of PTEN in DNA damage repair and cell killing. Another important question raised in the present study is how VPA can upregulate the expression of PTEN. It has been reported that ROS can downregulate the expression level and inhibit the action of PTEN.( 40 , 41 ) However, both ROS and PTEN are increased by VPA and were shown to contribute to cisplatin‐mediated cytotoxicity in the present study. It is intriguing to know how ROS can inhibit PTEN and enhance its cytotoxicity. One possibility is that ROS inhibit PTEN‐mediated DNA damage repair, and upregulated PTEN inhibits PI‐3K–Akt signaling following cisplatin damage.
Although studies have shown that HDACi can selectively kill tumor cells alone or through synergistic cytotoxicity with certain anticancer agents,( 32 , 42 ) whether the effects are indeed caused by direct inhibition of HDAC remains to be elucidated. The modulation of chromatin structure by changes in the acetylation and deacetylation of histones for increasing the accessibility of anticancer agents targeting DNA is commonly suggested for the synergistic cytotoxicity.( 14 , 15 , 26 ) In addition to chromatin remodeling, to clarify the cytotoxic effect specifically for transformed cells but not normal cells, the effect of HDAC inhibition should also be linked with non‐histone proteins that are important for cell growth, differentiation, or signaling of DNA damage, such as p53.( 43 ) Furthermore, enhancement of the cytotoxic effects mediated by oncogenic lesions (e.g. the RB–E2F1 pathway) in breast cancer cells have also been suggested for killing tumor cells with certain HDACi.( 44 ) In the present report, we observed that VPA at a near‐non‐cytotoxic dose can increase the levels of cisplatin‐mediated DNA damage (Fig. 6a) but not histone H4 acetylation (Fig. 6b). These results suggest that chromatin remodeling mediated by the modulation of histone acetylation does not contribute to the synergistic cytotoxicity and upregulation of ROS and PTEN mediated by cotreatment with VPA and cisplatin. Consistent with these observations, pretreatment with VPA for 24 h followed by cisplatin does not result in a higher level of cytotoxicity than that with VPA and cisplatin cotreatment (data not shown). Therefore, although VPA was shown to directly inhibit class I HDAC( 24 , 25 ) and stimulate the degradation of class II HDAC,( 45 ) it is possible that the enhancement of cisplatin‐mediated cytotoxicity does indeed occur through the modulation of other targets. Although the effect of enhancing cisplatin‐mediated cytotoxicity by VPA may not occur through the modulation of histone acetylation, the enhancement of cisplatin‐induced cytotoxicity by the well‐known HDACi TSA was shown to correlate with higher levels of histone H4 acetylation (Fig. 7).
In summary, these findings, in which VPA can significantly enhance cisplatin‐mediated cytotoxicity in all of the tested ovarian cancer cells, especially cells that are resistant to cisplatin, and the pathways revealed that contribute to the synergistic effect, may provide better therapeutic strategies for treating cancers.
Acknowledgments
We would like to thank Dr Tim Huang for providing the ovarian cancer cells A2780 and A2780/cp70. This work was supported by grants NSC96‐2321‐B‐182A‐011 and CMRPG660131 for C.‐T. Lin and NSC96‐2314‐B‐016‐046, TSGH‐C97‐6‐s01 and TSGH‐C96‐2‐s01 for M.‐H. Yu from National Science Council, Chang Gung Memorial Hospital and Tri‐Service General Hospital in Taiwan.
References
- 1. Agarwal R, Kaye SB. Ovarian cancer: strategies for overcoming resistance to chemotherapy. Nat Rev Cancer 2003; 3: 502–16. [DOI] [PubMed] [Google Scholar]
- 2. Andrews PA, Velury S, Mann SC, Howell SB. cis‐Diamminedichloroplatinum (II) accumulation in sensitive and resistant human ovarian carcinoma cells. Cancer Res 1988; 48: 68–73. [PubMed] [Google Scholar]
- 3. Godwin AK, Meister A, O'Dwyer PJ, Huang CS, Hamilton TC, Anderson ME. High resistance to cisplatin in human ovarian cancer cell lines is associated with marked increase of glutathione synthesis. Proc Natl Acad Sci USA 1992; 89: 3070–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kelley SL, Basu A, Teicher BA, Hacker MP, Hamer DH, Lazo JS. Overexpression of metallothionein confers resistance to anticancer drugs. Science 1988; 241: 1813–15. [DOI] [PubMed] [Google Scholar]
- 5. Parker RJ, Eastman A, Bostick‐Bruton F, Reed E. Acquired cisplatin resistance in human ovarian cancer cells is associated with enhanced repair of cisplatin DNA lesions and reduced drug accumulation. J Clin Invest 1991; 87: 772–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu JR, Opipari AW, Tan L et al . Dysfunctional apoptosome activation in ovarian cancer: implications for chemoresistance. Cancer Res 2002; 62: 924–31. [PubMed] [Google Scholar]
- 7. Drummond JT, Anthoney A, Brown R, Modrich P. Cisplatin and adriamycin resistance are associated with MutLα and mismatch repair deficiency in an ovarian tumor cell line. J Biol Chem 1996; 271: 19 645–8. [DOI] [PubMed] [Google Scholar]
- 8. Brown R, Hirst GL, Gallagher WM et al . hMLH1 expression and cellular responses of ovarian tumour cells to treatment with cytotoxic anticancer agents. Oncogene 1997; 15: 45–52. [DOI] [PubMed] [Google Scholar]
- 9. Spencer VA, Davie JR. Role of covalent modifications of histones in regulating gene expression. Gene 1999; 240: 1–12. [DOI] [PubMed] [Google Scholar]
- 10. Vigushin DM, Coombes RC. Histone deacetylase inhibitors in cancer treatment. Anticancer Drugs 2002; 13: 1–13. [DOI] [PubMed] [Google Scholar]
- 11. Ogryzko VV, Hirai TH, Russanova VR, Barbie DA, Howard BH. Human fibroblast commitment to a senescence‐like state in response to histone deacetylase inhibitors is cell cycle dependent. Mol Cell Biol 1996; 16: 5210–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen Z, Clark S, Birkeland M et al . Induction and superinduction of growth arrest and DNA damage gene 45 (GADD45) α and β messenger RNAs by histone deacetylase inhibitors trichostatin A (TSA) and butyrate in SW620 human colon carcinoma cells. Cancer Lett 2002; 188: 127–40. [DOI] [PubMed] [Google Scholar]
- 13. Marks PA, Richon VM, Rifkind RA. Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J Natl Cancer Inst 2000; 92: 1210–16. [DOI] [PubMed] [Google Scholar]
- 14. Marchion DC, Bicaku E, Daud AI, Richon V, Sullivan DM, Munster PN. Sequence‐specific potentiation of topoisomerase II inhibitors by the histone deacetylase inhibitor suberoylanilide hydroxamic acid. J Cell Biochem 2004; 92: 223–37. [DOI] [PubMed] [Google Scholar]
- 15. Kim MS, Blake M, Baek JH, Kohlhagen G, Pommier Y, Carrier F. Inhibition of histone deacetylase increases cytotoxicity to anticancer drugs targeting DNA. Cancer Res 2003; 63: 7291–300. [PubMed] [Google Scholar]
- 16. Miyajima A, Nakashima J, Tachibana M, Nakamura K, Hayakawa M, Murai M. N‐Acetylcysteine modifies cis‐dichlorodiammineplatinum‐induced effects in bladder cancer cells. Jpn J Cancer Res 1999; 90: 565–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yu C, Subler M, Rahmani M et al . Induction of apoptosis in BCR/ABL+ cells by histone deacetylase inhibitors involves reciprocal effects on the RAF/MEK/ERK and JNK pathways. Cancer Biol Ther 2003; 2: 544–51. [DOI] [PubMed] [Google Scholar]
- 18. Ungerstedt JS, Sowa Y, Xu WS et al . Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc Natl Acad Sci USA 2005; 102: 673–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee S, Choi EJ, Jin C, Kim DH. Activation of PI3K/Akt pathway by PTEN reduction and PIK3CA mRNA amplification contributes to cisplatin resistance in an ovarian cancer cell line. Gynecol Oncol 2005; 97: 26–34. [DOI] [PubMed] [Google Scholar]
- 20. Stambolic V, Suzuki A, De La Pompa JL et al . Negative regulation of PKB/Akt‐dependent cell survival by the tumor suppressor PTEN. Cell 1998; 95: 29–39. [DOI] [PubMed] [Google Scholar]
- 21. Rahmani M, Reese E, Dai Y et al . Coadministration of histone deacetylase inhibitors and perifosine synergistically induces apoptosis in human leukemia cells through Akt and ERK1/2 inactivation and the generation of ceramide and reactive oxygen species. Cancer Res 2005; 65: 2422–32. [DOI] [PubMed] [Google Scholar]
- 22. Gao N, Rahmani M, Shi X, Dent P, Grant S. Synergistic antileukemic interactions between 2‐medroxyestradiol (2‐ME) and histone deacetylase inhibitors involve Akt down‐regulation and oxidative stress. Blood 2006; 107: 241–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bass DA, Parce JW, Dechatelet LR, Szejda P, Seeds MC, Thomas M. Flow cytometric studies of oxidative product formation by neutrophils: a graded response to membrane stimulation. J Immunol 1983; 130: 1910–17. [PubMed] [Google Scholar]
- 24. Gottlicher M, Minucci S, Zhu P et al . Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J 2001; 20: 6969–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem 2001; 276: 36 734–41. [DOI] [PubMed] [Google Scholar]
- 26. Marchion DC, Bicaku E, Daud AI, Sullivan DM, Munster PN. Valproic acid alters chromatin structure by regulation of chromatin modulation proteins. Cancer Res 2005; 65: 3815–22. [DOI] [PubMed] [Google Scholar]
- 27. Chou TC, Talalay P. Quantitative analysis of dose–effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 1984; 22: 27–55. [DOI] [PubMed] [Google Scholar]
- 28. Datta R, Yoshinaga K, Kaneki M, Pandey P, Kufe D. Phorbol ester‐induced generation of reactive oxygen species is protein kinase Cβ‐dependent and required for SAPK activation. J Biol Chem 2000; 275: 41 000–3. [DOI] [PubMed] [Google Scholar]
- 29. Kroning R, Lichtenstein AK, Nagami GT. Sulfur‐containing amino acids decrease cisplatin cytotoxicity and uptake in renal tubule epithelial cell lines. Cancer Chemother Pharmacol 2000; 45: 43–9. [DOI] [PubMed] [Google Scholar]
- 30. Nowosielska A, Marinus MG. Cisplatin induces DNA double‐strand break formation in Escherichia coli dam mutants. DNA Repair (Amst) 2005; 4: 773–81. [DOI] [PubMed] [Google Scholar]
- 31. Blaheta RA, Michaelis M, Driever PH, Cinatl J Jr. Evolving anticancer drug valproic acid: insights into the mechanism and clinical studies. Med Res Rev 2005; 25: 383–97. [DOI] [PubMed] [Google Scholar]
- 32. Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov 2006; 5: 769–84. [DOI] [PubMed] [Google Scholar]
- 33. Chavez‐Blanco A, Segura‐Pacheco B, Perez‐Cardenas E et al . Histone acetylation and histone deacetylase activity of magnesium valproate in tumor and peripheral blood of patients with cervical cancer. A phase I study. Mol Cancer 2005; 4: 22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yan X, Fraser M, Qiu Q, Tsang BK. Over‐expression of PTEN sensitizes human ovarian cancer cells to cisplatin‐induced apoptosis in a p53‐dependent manner. Gynecol Oncol 2006; 102: 348–55. [DOI] [PubMed] [Google Scholar]
- 35. Yu C, Friday BB, Lai JP et al . Abrogation of MAPK and Akt signaling by AEE788 synergistically potentiates histone deacetylase inhibitor‐induced apoptosis through reactive oxygen species generation. Clin Cancer Res 2007; 13: 1140–8. [DOI] [PubMed] [Google Scholar]
- 36. Pappas G, Zumstein LA, Munshi A, Hobbs M, Meyn RE. Adenoviral‐mediated PTEN expression radiosensitizes non‐small cell lung cancer cells by suppressing DNA repair capacity. Cancer Gene Ther 2007; 14: 543–9. [DOI] [PubMed] [Google Scholar]
- 37. Kao GD, Jiang Z, Fernandes AM, Gupta AK, Maity A. Inhibition of phosphatidylinositol‐3‐OH kinase/Akt signaling impairs DNA repair in glioblastoma cells following ionizing radiation. J Biol Chem 2007; 282: 21 206–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Puc J, Keniry M, Li HS et al . Lack of PTEN sequesters CHK1 and initiates genetic instability. Cancer Cell 2005; 7: 193–204. [DOI] [PubMed] [Google Scholar]
- 39. Shen WH, Balajee AS, Wang J et al . Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 2007; 128: 157–70. [DOI] [PubMed] [Google Scholar]
- 40. Flaherty DM, Monick MM, Hinde SL. Human alveolar macrophages are deficient in PTEN. The role of endogenous oxidants. J Biol Chem 2006; 281: 5058–64. [DOI] [PubMed] [Google Scholar]
- 41. Cruz CM, Rinna A, Forman HJ, Ventura AL, Persechini PM, Ojcius DM. ATP activates a reactive oxygen species‐dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem 2007; 282: 2871–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer 2006; 6: 38–51. [DOI] [PubMed] [Google Scholar]
- 43. Roy S, Packman K, Jeffrey R, Tenniswood M. Histone deacetylase inhibitors differentially stabilize acetylated p53 and induce cell cycle arrest or apoptosis in prostate cancer cells. Cell Death Differ 2005; 12: 482–91. [DOI] [PubMed] [Google Scholar]
- 44. Zhao Y, Tan J, Zhuang L, Jiang X, Liu ET, Yu Q. Inhibitors of histone deacetylases target the Rb‐E2F1 pathway for apoptosis induction through activation of proapoptotic protein Bim. Proc Natl Acad Sci USA 2005; 102: 16 090–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kramer OH, Zhu P, Ostendorff HP et al . The histone deacetylase inhibitor valproic acid selectively induces proteasomal degradation of HDAC2. EMBO J 2003; 22: 3411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]