

Abstract
Patients with pulmonary adenocarcinoma carrying the epidermal growth factor receptor (EGFR) mutation tend to display dramatic clinical response to treatment with the EGFR tyrosine kinase inhibitor gefitinib. Unfortunately, in many cases the cancer cells eventually acquire resistance, and this limits the duration of efficacy. To gain insight into these acquired resistance mechanisms, we first prepared HEK293T cell line stably transfected with either wild‐type (WT) or mutant (L858R) EGFR, and then expressed oncogenic K‐Ras12V mutant in the latter transfectant. Although 293T cells expressing wild‐type EGFR did not show any growth inhibition by gefitinib treatment similarly to the non‐transfected cells, the cells expressing the EGFR‐L858R were exquisitely sensitive. Consistently, phospho‐Akt levels were decreased in response to gefitinib in cells expressing EGFR‐L858R but not in cells with EGFR‐WT. In contrast, 293T cells expressing both EGFR‐L858R and oncogenic K‐Ras were able to proliferate even in the presence of high concentration of gefitinib probably by inducing Erk1/2 activation. We also expressed K‐Ras12V in the gefitinib‐sensitive pulmonary adenocarcinoma cell line PC‐9, which harbors an in‐frame deletion in the EGFR gene. The activated K‐Ras inhibited the effects of gefitinib treatment on cell growth, cell death induction and levels of phospho‐Akt, as well as phospho‐Erk. These data indicate that activated Ras could substitute most of the upstream EGFR signal, and are consistent with the hypothesis that mutational activation of targets immediately downstream from the EGFR could induce the secondary resistance to gefitinib in patients with lung cancer carrying EGFR mutation. (Cancer Sci 2007; 98: 357–363)
Abbreviations
- ATP
adenosine 5′‐triphosphate
- BP
base pair
- BrdU
5‐bromodeoxyuridine
- DMEM
Dulbecco's Modified Eagle Medium
- FCS
fetal calf serum
- EDTA
ethylene diamine tetra acetate
- EGFR
epidermal growth factor receptor
- FITC
fluorescein isothiocyanate
- HEPES
4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid
- ISEL trial
Iressa Survival Evaluation in Lung Cancer trial
- MEK
methyl ethyl ketone
- MEM
minimum essential medium
- MLB
Mg‐containing lysis buffer
- NSCLC
non‐small cell lung cancer
- PBS
phosphate‐buffered saline
- PDGFR
platelet‐derived growth factor receptor
- RBD
Ras binding domain RT‐PCR, reverse transcription polymerase chain reaction
- SDS‐PAGE
sodium dodecyl sulfate–polyacrylamide gel electrophoresis.
Lung cancer is the leading cause of death due to neoplasia in Japan and elsewhere in the world, and adenocarcinoma is the most prevalent subtype.( 1 ) It has recently been discovered that somatic mutations exist in epidermal growth factor receptor (EGFR) tyrosine kinase in a subset of patients with pulmonary adenocarcinoma.( 2 , 3 , 4 ) Remarkably, these mutations strongly sensitize the cancer cells to growth suppressive effects of the EGFR inhibitors gefitinib (Iressa) and erlotinib (Tarceva),( 2 , 3 , 5 , 6 ) leading to robust clinical responses. Such patients tend to be never‐smokers or females but, most notably, the mutations are more frequent among people of east‐Asian ethnic origin compared to Caucasians.( 6 , 7 ) Indeed, up to 40% of newly diagnosed pulmonary adenocarcinoma cases were found to contain the EGFR mutations in Japan,( 8 , 9 ) while in the USA less than 20% of similar patients have the mutations.( 2 , 10 ) Although in the placebo‐controlled phase III Iressa Survival Evaluation in Lung Cancer (ISEL) trial, gefitinib was not found to be associated with significant improvement in survival in the overall population; preplanned subgroup analyses showed that for patients of Asian origin there was significantly longer survival in the gefitinib group than the placebo group (median survival 9.5 vs 5.5 months, P = 0.01).( 11 )
The EGFR mutations are clustered around the adenosine 5′‐triphosphate (ATP)‐binding pocket of the kinase domain.( 2 , 3 , 4 ) The two most prevalent mutations are L858R missense mutation in exon 21 and 15‐base pair (bp) in‐frame deletion in exon 19, and up to 80% of the patients with EGFR mutation have either of the two.( 8 ) These mutated EGFR, unlike wild‐type EGFR, appear to be constitutively active even without ligand‐induced dimerization, though they can still respond to ligands by increased levels of autophosphorylation.( 12 , 13
Gefitinib is a competitive inhibitor that prevents ATP binding to the ATP‐binding pocket in the kinase domain of EGFR.( 14 ) The robust clinical response to gefitinib treatment indicates two things. First, the growth and survival of the cancer cells are dependent on the signal generated by the mutated EGFR (oncogene addiction).( 15 ) Thus, the response to gefitinib treatment defines a distinct subgroup of lung cancer, in which the mutated EGFR probably promotes an initial phase of carcinogenesis as shown by recent transgenic mice studies.( 16 , 17 ) Second, gefitinib may exert much higher inhibitory effects to the mutated EGFR than to wild‐type EGFR. In vitro studies provided evidence for this,( 2 , 3 , 4 ) although affinity of gefitinib with the EGFR ATP‐binding site was shown to be not significantly different between mutated and wild‐type EGFR.( 18 )
Unfortunately, as with other therapies, the majority of patients receiving gefitinib ultimately progressed, and median response durations ranged 8–13 months.( 19 , 20 ) The mechanisms of this acquired resistance have been under intensive investigation.( 21 ) In treatment of chronic myeloid leukemia by Bcr‐Abl tyrosine kinase inhibitor imatinib, similar acquired resistance emerges. More than half of these cases have been explained by an additional mutation in Bcr‐Abl that inhibits imatinib binding.( 22 ) Several groups have looked at the possibility of a similar mechanism in gefitinib resistance, and indeed found one such mutation, T790M, close to the ATP binding site.( 23 , 24 ) However, this mutation has been detected only in a subset of patients with acquired resistance,( 24 ) and sometimes only in a fraction of the cancer cells.( 25 , 26 ) Thus, the presence of T790M does not provide a universal explanation to the acquired resistance phenomenon, and the mechanism could be heterogenous. A number of factors have been reported to influence gefitinib efficacy in patients or tumor models, including Ras mutation,( 10 , 27 ) ErbB‐family receptors/ligands,( 28 , 29 , 30 , 31 , 32 , 33 , 34 ) platelet‐derived growth factor receptor‐alpha (PDGFRA)‐β activation,( 35 ) epithelial–mesenchymal transition/E‐cadherin expression,( 36 , 37 ) PTEN expression( 38 , 39 ) and epithelial membrane protein‐1.( 40 ) However, it is currently unclear whether these mechanisms give a satisfactory explanation for the acquired resistance that emerges during gefitinib treatment.
We hypothesized that an activating mutation on any molecule immediately downstream of the EGFR signaling pathway might be sufficient to deliver a proliferative or survival signal even if EGFR activity is shut down by gefitinib treatment. To investigate this possibility, we utilized two cell systems, and introduced oncogenic K‐Ras12V into these cells. One is the HEK293T cell line transfected with EGFR carrying L858R mutation, and the other is the adenocarcinoma cell line PC‐9 that carries 15‐bp in‐frame deletion in the EGFR gene. Our data suggest that, even in EGFR‐addicted cells, activation of Ras promotes cell growth and survival during gefitinib treatment. Although Ras mutation has been documented only as a mechanism for the primary resistance to gefitinib,( 10 ) a systematic search for activating mutations in signaling molecules downstream of EGFR might be warranted in cancer cells that acquire gefitinib‐resistance, and could lead to the identification of new therapeutic targets.
Materials and Methods
Cell culture and reagents. HEK293T cells were cultured at 37.0°C with 5% CO2 by using Dulbecco's Modified Eagle Medium (DMEM) medium supplemented with 10% heat‐inactivated fetal bovine serum, 1% minimum essential medium (MEM) non‐essential amino acid solution, and 2 mM L‐glutamine. PC‐9 cells were cultured at 37.0°C with 5% CO2 by using RPMI‐1640 medium supplemented with 20% heat‐inactivated fetal bovine serum. Cell growth was assessed by counting viable cells. Gefitinib was provided by AstraZeneca (Waltham, MA, USA).
Expression constructs and transfections. The EGFR expression vectors were constructed by inserting full‐length reverse transcription polymerase chain reaction (RT–PCR) product of human EGFR into pcDNA3.1‐Zeo vector (Invitrogen, Carlsbad, CA, USA). The L858R mutation was generated by site‐directed mutagenesis using a QuikChange kit (Stratagene, La Jolla, CA, USA). The mutation was confirmed by sequencing. The oncogenic Ras expression vectors were constructed by inserting full‐length K‐Ras12V cDNA into either the pApuroII vector( 41 ) or pcDNA3.1‐Zeo. Transfection into 293T cells or PC‐9 cells was done using Lipofectamine 2000 (Invitrogen). Selection of the cells was started 48 h later in 96‐well plates with appropriate antibiotics.
Cell cycle analysis. Cells exposed to gefitinib for 72 h were pulse‐labeled for 30 min with 20 µM 5‐bromodeoxyuridine (BrdU). Cells were then harvested and fixed at 4°C overnight with 70% ethanol, and successively treated in the following conditions: (i) in 4 N HCl/0.5% Triton X‐100 for 30 min at room temperature; (ii) in fluorescein isothiocyanate (FITC)‐conjugated anti‐BrdU antibody (Pharmingen, San Diego, CA, USA) for 1 h at room temperature; (iii) in 5 µg/mL PI in phosphate‐buffered saline (PBS). Between incubations, cells were extensively washed with PBS containing 2% fetal calf serum (FCS) and 0.1% sodium azide. Subsequent flow cytometric analysis was performed on a FACSCalibur (Becton Dickinson, Mountain View, CA, USA). The data were analyzed and displayed as dot plots using the Cell Quest software (Becton Dickinson).
Western blotting analysis and GST‐RBD pull down. Cells were lyzed with RIPA lysis solution as previously described.( 5 ) Whole cell lysates were separated with sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS‐PAGE), and then transferred to membrane, and detected by antibodies using ECL Plus Western blotting detection reagents (GE Healthcare Biosciences, Piscataway, NJ, USA). The antibodies against EGFR, phospho‐EGFR (Tyr1068), Akt kinase, phospho‐Akt (Ser473), p44/42 Erk1/2 and phospho‐p44/42 Erk1/2 (Thr202/Tyr204) were purchased from Cell Signaling (Beverly, MA, USA). The antipan‐Ras (sc‐30) antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
To detect expression of the activated oncogenic K‐Ras12V, GST‐RBD pull‐down was carried out as described.( 42 ) The GST‐RBD expression plasmid (pGEX‐RBD) was kindly provided by Dr Shalloway (Cornell University, Ithaca, NY, USA). Briefly, Escherichia coli harboring pGEX‐RBD was sonicated on ice three times for 20 sec in lysis buffer (20 mM 4‐[2‐hydroxyethyl]‐1‐piperazineethanesulfonic acid [HEPES], pH 7.5, 120 mM NaCl, 10% Glycerol, 2 mM ethylene diamine tetra acetate [EDTA], 10 µg/mL leupeptin, and 10 µg/mL aprotinin). GST‐RBD beads were prepared by incubating Glutathione Sepharose beads (GE HealthScience) with the lysate, then incubated with lysates prepared from transfected cells using Mg‐containing lysis buffer (MLB): 25 mM HEPES, pH 7.5, 150 mM NaCl, 1% NP‐40, 0.25% Na deoxycholate, 10% glycerol, 25 mM NaF, 10 mM MgCl2, 1 mM EDTA, 1 mM Na ortho‐vanadate, 10 µg/mL leupeptin, and 10 µg/mL aprotinin. The GST‐RBD beads were washed, and bound proteins were analyzed by Western blotting using antipan‐Ras antibody.
Results
Distinct gefitinib sensitivity of 293T cells expressing wild‐type or L858R mutant EGFR. To generate a cellular system by which we could conveniently examine cellular response to wild‐type or mutated EGFR inhibition by gefitinib, we chose human embryonic kidney cell line 293T, and stably transfected with either wild‐type or L858R mutant EGFR (hereafter referred to as EGFR‐WT or EGFR‐L858R). Western blotting analysis showed that EGFR expression levels were approximately equal between the EGFR‐WT and EGFR‐L858R 293T transfectants (Fig. 1a), while non‐transfected 293T cells grew at a comparable rate in the normal culture conditions, although their growth rate was approximately 2‐fold slower compared to non‐transfected 293T cells (data not shown).
Figure 1.
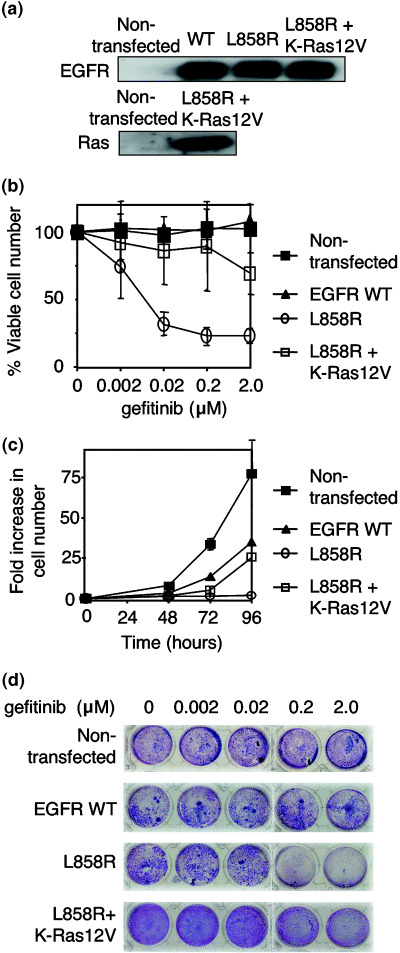
Characterization of 293T cells expressing wild‐type or mutated epidermal growth factor receptor (EGFR). (a) Expression of EGFR and K‐Ras protein in 293T cells. Either wild‐type or EGFR carrying L858R mutation were stably expressed in 293T cells. Cells that express EGFR‐L858R were further transfected with pApuroII‐K‐Ras12V expression vector. Total cell lysates were prepared and blotted with anti‐EGFR antibody. Activated oncogenic K‐Ras12V was detected with the glutathione S‐transferase (GST)‐Ras binding domain pull down followed by immunoblot with antipan‐Ras antibody. (b) Dose‐response curves of the indicated 293T transfectants in the medium containing various concentrations of gefitinib. The percentage of viable cell numbers was calculated at 72 h after treatment relative to cell number without gefitinib treatment. Data shown are mean and standard deviation of the three independent experiments. (c) Growth curves of the indicated 293T transfectants. Cells were treated with gefitinib at a concentration of 2.0 µM, and were counted at the indicated time points. Data shown are mean and standard deviation of the three independent experiments. (d) Cell growth of the indicated 293T transfectants. Cells were cultured for 72 h in the medium containing gefitinib, and stained with crystal violet.
We cultured these cell lines in the presence of various doses of gefitinib, and assessed cell growth by counting viable cells (Fig. 1b) or crystal violet staining (Fig. 1d). Interestingly, 293T cells expressing EGFR‐WT as well as the non‐transfected cells did not show any growth inhibition following gefitinib treatment (Fig. 1b,d), indicating that they grow independently of EGFR signaling. However, 293T cells expressing EGFR‐L858R were exquisitely sensitive to gefitinib, and the cell growth was markedly suppressed even with low doses of gefitinib (Fig. 1b,d). Growth curve experiments also revealed that cells expressing EGFR‐L858R were not able to grow in the presence of 2.0 µM gefitinib (Fig. 1c).
We also evaluated cell cycle progression of these cells by BrdU pulse‐labeling with or without gefitinib treatment (Fig. 2). Only 293T cells expressing EGFR‐L858R displayed a significant change in cell cycle distribution following gefitinib treatment. In those cells treated with gefitinib, the proportion of the cells in S phase was significantly decreased compared to non‐treated cells (46% to 10%), and viable cells seemed to be accumulated mostly in the G1 phase. Furthermore, the percentage of dead cells with sub‐G1 DNA content was increased (12% to 45%), indicating that cell death likely due to apoptosis was induced following gefitinib treatment. Taken together, our data indicate that expression of the EGFR‐L858R, but not EGFR‐WT, converted 293T cells to proliferate depending on cell survival/proliferation signal via EGFR. These results also support the view that these cell lines may serve as a model system to investigate biological responses of cells carrying EGFR mutation to gefitinib treatment.
Figure 2.

Analysis of cell cycle distribution. Cells treated with gefitinib (2.0 µM) for 72 h were pulse‐labeled with 5‐bromodeoxyuridine (BrdU) and stained with anti‐BrdU antibody and PI (left panel). Numbers in lower middle, upper, lower right, and left regions indicate the percentages of cells in the G1, S, G2/M phases, and with sub‐G1 DNA content, respectively. In the right panel, these data are shown as a bar graph.
Activated Akt levels were reduced by gefitinib treatment in cells expressing L858R mutant but not in cells with wild‐type EGFR. We next evaluated the phosphorylation status of EGFR in 293T cells expressing EGFR‐WT versus EGFR‐L858R. In the normal cell culture conditions without gefitinib treatment, both EGFR‐L858R and EGFR‐WT proteins were highly phosphorylated in 293T stable transfectants as shown by Western blotting using antiphospho‐EGFR Y1068 antibody (Fig. 3). In keeping with the previous reports,( 2 , 3 , 4 ) treatment with 2.0 µM gefitinib abrogated the Y1068 phosphorylation levels of EGFR‐L858R protein, while the same treatment inhibited Y1068 phosphorylation of EGFR‐WT only partially and there still remained a significant amount of the phosphorylation. These results are consistent with the previous reports that gefitinib is more effective in suppressing mutant EGFR than wild‐type EGFR.( 2 , 3 , 4 )
Figure 3.

Analysis of the EGFR signaling in the 293T cells expressing wild‐type EGFR, EGFR‐L858R, and both EGFR‐L858R and K‐Ras12V. Cells were treated with gefitinib at a concentration of 2.0 µM for 6 h or left untreated. Total cell lysates were prepared and blotted with indicated antibodies.
Activated EGFR transmits signals downstream by recruiting adapter molecules to phosphorylation sites in the kinase domain and the C‐terminal tail, or by directly phosphorylating specific substrates. Among such downstream effectors, we examined activation/phosphorylation status of Erk1/2 and Akt kinase in the transfectants using respective phospho‐specific antibodies. In both cell lines not treated with gefitinib or exogenous EGFR ligands, we could detect constitutive phosphorylation in Erk1/2 and Akt. Gefitinib treatment in cells expressing EGFR‐L858R mostly abrogated this phosphorylation, hence activation, of Erk1/2 and Akt, consistent with the growth suppressive effect of the treatment. Of note, compared to cells expressing EGFR‐L858R, phospho‐Akt levels in 293T cells expressing EGFR‐WT as well as non‐transfected 293T cells (Fig. 3) were more intense, and the phosphorylation levels of Akt or Erk1/2 in these cells were not decreased in response to gefitinib (Fig. 3). These results may support a notion that, in cells expressing EGFR‐L858R or EGFR‐WT, the levels of Akt and Erk phosphorylation are regulated by EGFR or a growth factor receptor other than EGFR, respectively. Identity of the latter receptor is currently unknown.
Transfected oncogenic Ras reduced gefitinib sensitivity of 293T cells expressing mutant EGFR. To test whether Ras activation by an oncogenic mutation 12 V could compensate EGFR suppression by gefitinib, 293T cells that express EGFR‐L858R were further transfected with the oncogenic K‐Ras12V mutant. Expression of K‐Ras12V was detected with the GST‐RBD pull down followed by immunoblotting with antipan‐Ras antibody (Fig. 1a). We found that all of the four transfectants expressing K‐Ras12V were able to proliferate even in the presence of a high concentration of gefitinib (data not shown), suggesting that a single K‐Ras mutation could induce gefitinib resistance. One clone of the K‐Ras12V/EGFR‐L858R double transfectant was examined further in terms of survival, cell growth and cell cycle distribution. Cell survival and growth of the double transfectant following gefitinib treatment was significantly increased compared to cells expressing only EGFR‐L858R (Fig. 1b–d). Interestingly, the percentage of dead cells with sub‐G1 DNA content was still mildly increased in response to gefitinib treatment in the double transfectant (Fig. 2). These data suggest that K‐Ras12V expression provides a growth‐promoting signal in 293T cells but cannot completely suppress gefitinib‐induced cell death. Thus, it seems likely that when the EGFR signal is shut off by gefitinib, constitutively active K‐Ras can substitute part of the signal generated through EGFR.
We also found that the phosphorylation status of EGFR and Akt in the double transfectant was remarkably suppressed by gefitinib treatment similar to cells singly expressing EGFR‐L858R (Fig. 3). However, phospho‐Erk1/2 levels were an exception. They were constitutively elevated in the double transfectant, and still detectable following gefitinib treatment. This finding is not unexpected, because Ras can activate Raf kinase, and Raf activates methyl ethyl ketone (MEK), leading to Erk activation.( 43 )
Pulmonary adenocarcinoma cell line PC‐9 became resistant to gefitinib treatment following expression of oncogenic Ras. To test whether the above observation could be reproducible in a lung cancer cell line carrying EGFR mutation, we decided to use PC‐9, a gefitinib‐sensitive lung cancer cell line derived from a female Japanese patient with pulmonary adenocarcinoma,( 44 ) which harbors an in‐frame deletion in exon 19 of the EGFR gene.( 45 ) We transfected K‐Ras12V expression vector into PC‐9 cells, and confirmed expression of activated Ras using GST‐RBD pull down followed by immunoblot with antipan‐Ras antibody (Fig. 4a). Expression levels of EGFR were not altered after the transfection as examined by Western blotting (Fig. 4a) or by flow cytometric analysis (data not shown).
Figure 4.

Characterization of PC‐9 cells with or without K‐Ras12V transfection. (a) Expression of EGFR and K‐Ras12V in PC‐9 cells. PC‐9 cells were stably transfected with pcDNA3.1/Zeo‐K‐Ras12V expression vector. Expression of oncogenic K‐Ras12V was detected using GST‐Ras binding domain pull down followed by immunoblot with antipan‐Ras antibody. The level of EGFR protein expression was measured by immunoblot with anti‐EGFR antibody. (b) PC‐9 cells with or without K‐Ras12V expression treated with dimethylsulfoxide (DMSO) or gefitinib (2.0 µM) for 72 h. (c) Dose‐response curves of PC‐9 cells with or without K‐Ras12V expression in the presence of indicated concentrations of gefitinib. The percentage of viable cell number was calculated at 72 h after the treatment. (d) Cell cycle analysis. Cells treated with gefitinib (2.0 µM) for 72 h were pulse‐labeled with BrdU, and stained with anti‐BrdU antibody and PI (left panel). Numbers in lower middle, upper, lower right, and left regions indicate the percentages of cells in the G1, S, G2/M phases, and with sub‐G1 DNA content, respectively. In the right panel, these data are shown as a bar graph.
While parental PC‐9 cells were highly sensitive to gefitinib, expression of the activated K‐Ras converted PC‐9 cells being clearly less sensitive to gefitinib treatment in cell growth assay (Fig. 4b). We also assessed the dose‐response by culturing cells in gefitinib‐containing medium. In this assay, IC50 of PC‐9 cells or PC‐9 expressing K‐Ras12V was approximately 0.06 µM or >2 µM, respectively (Fig. 4c). Cell cycle analysis showed that fewer dead cells with sub‐G1 DNA content were present in PC‐9 expressing K‐Ras12V in response to gefitinib treatment relative to parental PC‐9 cells (Fig. 4d), indicating that cell death induction by gefitinib was somehow cancelled by Ras activation in PC‐9 cells.
Then, we characterized EGFR signal transduction by phospho‐specific antibodies in PC‐9 and the transfectant expressing K‐Ras12V in response to gefitinib treatment. Phospho‐EGFR levels were similarly decreased in both parental and Ras12V expressing PC‐9 cells (Fig. 5a). On the other hand, levels of phosphorylation on Erk1/2 or Akt were significantly suppressed only in parental PC‐9 cells, and PC‐9 expressing K‐Ras12V had increased amount of phospho‐Erk1/2 and phospho‐Akt compared to parental PC‐9 cells even at highest gefitinib dose (2.0 µM) (Fig. 5a). Thus, in PC‐9 cells, oncogenic Ras expression delivered an activation signal to both Erk1/2 and Akt.
Figure 5.

Analysis of the EGFR signaling in PC‐9 cells expressing K‐Ras12V (a) Cells were treated with the indicated concentrations of gefitinib for 6 h. Total cell lysates were prepared and blotted with indicated antibodies. (b) A schematic of EGFR signaling generated by the mutated EGFR in the absence (PC‐9) or in the presence of oncogenic K‐Ras (transfectant) with or without gefitinib treatment. In PC‐9 cells, gefitinib treatment shuts down both PI‐3K‐Akt and Ras‐Raf‐MEk‐Erk pathways. Expression of oncogenic K‐Ras abrogated effects of gefitinib treatment on both of these pathways, leading to continuous cell growth and survival.
Discussion
It has been generally accepted that Ras is activated by growth factor receptors like EGFR by the recruitment of adaptors and nucleotide exchange factor Shc/Grb‐2/SOS complex.( 43 , 46 ) Thus, Ras functions downstream of EGFR and is an important part of signal transduction following EGFR activation. In the current study, we have shown that expression of oncogenic Ras can convert cell lines that are dependent on mutated EGFR to be gefitinib‐resistant. By doing so, we have tested a hypothesis that a single hit mutation in a signaling molecule downstream of EGFR is sufficient to compensate for inhibition of EGFR signaling by gefitinib. Another report has recently shown that activated forms of Akt or p110 PI3K can exert similar effects.( 47 ) Thus, it seems possible that the acquired resistance to gefitinib could be induced by mutations in molecules downstream of EGFR.
There are a group of patients who have pulmonary adenocarcinoma in which Ras is constitutively activated by mutation. They are often smokers and distinct from patients with mutated EGFR.( 4 , 8 ) Activating mutations in EGFR and Ras are considered to be mutually exclusive in the same primary tumors,( 8 , 27 ) in keeping with the concept that EGFR and Ras act in the same biochemical pathway. Alternatively, it even seemed possible that Ras mutation that has an ability to induce cell death or senescence,( 48 ) may eradicate lung cancer cells with EGFR mutation. However, our current data indicate that cells carrying mutations in both EGFR and Ras can survive and proliferate at least in vitro. In addition, there are a few reports that describe lung cancer patients that have both EGFR and Ras mutations, and they all exhibit primary resistance when treated with gefitinib or erlotinib.( 27 , 49 ) Although our experiment seems to be highly artificial, these data indicate that Ras mutation can render cells carrying EGFR mutation to be genitinib resistant.
Epidermal growth factor receptor has several effector pathways including Ras/Erk or PI3K/Akt (Fig. 5b, left upper panel), and we examined by Western blotting whether these pathways could be activated by oncogenic Ras expression in the presence of gefitinib. In 293T cells transfected with EGFR‐L858R, expression of K‐Ras12V activated the Ras/Erk pathway even in the presence of gefitinib, which probably maintains S‐phase progression as observed in Figure 2. In contrast, phosphorylation of Akt, which provides an important anti‐apoptotic signal,( 50 ) by phosphorylating pro‐apoptotic molecules like BAD( 51 ) or PAR‐4,( 52 ) was shut down in those cells by gefitinib. Thus, it seems reasonable that gefitinib‐induced cell death was only mildly suppressed by K‐Ras12V in 293T cells expressing EGFR‐L858R (Fig. 2). Interestingly, the situation was different in PC‐9 cells expressing oncogenic Ras (Fig. 5b, right lower panel). Not only Erk but also Akt were still phosphorylated in the presence of gefitinib, consistent with the remarkable suppression of cell death induction. Thus, depending on cell types, the signal from K‐Ras to PI3K/Akt pathway may or may not be efficiently transduced.
It is perhaps surprising that expression of EGFR‐L858R does sensitize 293T cells to gefitinib treatment, although a similar phenomenon has been reported a using related cell line, HEK293, and the EGFR transgene carrying 15‐bp in‐frame deletion.( 45 ) The sensitization, hence the oncogene addiction to mutated EGFR, does not necessarily happen when mutated EGFR are expressed in cells. For example, lung cancer cell line H1299 could not be rendered dependent on EGFR signal by expression of mutated EGFR.( 32 , 53 ) Interestingly, in cells with EGFR‐L858R, but not in cells with EGFR‐WT, Akt is placed under control of EGFR signal transduction, as seen in antiphospho‐Akt Western blotting (Fig. 3). Elucidation of the mechanism of this change in signal transduction may shed light on carcinogenesis induced by EGFR mutation.
In summary, we have characterized EGFR signal transduction and cellular behavior following gefitinib treatment in cells expressing both mutated EGFR and oncogenic K‐Ras. K‐Ras expression induced gefitinib resistance which was associated with activation of Erk and/or Akt, supporting S‐phase progression and/or apoptosis suppression in the presence of gefitinib. These results support the hypothesis that activation of signaling molecules in pathways downstream from the EGFR could be responsible for the development of resistance to gefitinib. Inhibitors of these signaling molecules may have potential for the treatment of patients who develop resistance to EGFR inhibitors.
Acknowledgments
We would like to thank Ms Masayo Kimura and Emi Uchida for expert technical support; Ms Kazuko Hikasa, Kyoko Takahashi, and Hisayo Saito for secretarial assistance; AstraZeneca for providing gefitinib; and Dr David Shalloway (Cornell University, Ithaca, NY, USA) for providing the pGEX‐RBD construct. This work was supported in part by grants from the Ministry of Education, Culture, Sports, Science and Technology (KK).
References
- 1. Stewart BW, Kleihues P. World cancer reported. Lyon: IARC press, 2003. [Google Scholar]
- 2. Paez JG, Janne PA, Lee JC et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004; 304: 1497–500. [DOI] [PubMed] [Google Scholar]
- 3. Lynch TJ, Bell DW, Sordella R et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non‐small‐cell lung cancer to gefitinib. N Engl J Med 2004; 350: 2129–39. [DOI] [PubMed] [Google Scholar]
- 4. Pao W, Miller V, Zakowski M et al. EGF receptor gene mutations are common in lung cancers from ‘never smokers’ and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA 2004; 101: 13 306–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib‐sensitizing EGFR mutations in lung cancer activate anti‐apoptotic pathways. Science 2004; 305: 1163–7. [DOI] [PubMed] [Google Scholar]
- 6. Tsao MS, Sakurada A, Cutz JC et al. Erlotinib in lung cancer – molecular and clinical predictors of outcome. N Engl J Med 2005; 353: 133–44. [DOI] [PubMed] [Google Scholar]
- 7. Shigematsu H, Lin L, Takahashi T et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst 2005; 97: 339–46. [DOI] [PubMed] [Google Scholar]
- 8. Kosaka T, Yatabe Y, Endoh H, Kuwano H, Takahashi T, Mitsudomi T. Mutations of the epidermal growth factor receptor gene in lung cancer: biological and clinical implications. Cancer Res 2004; 64: 8919–23. [DOI] [PubMed] [Google Scholar]
- 9. Tokumo M, Toyooka S, Kiura K et al. The relationship between epidermal growth factor receptor mutations and clinicopathologic features in non‐small cell lung cancers. Clin Cancer Res 2005; 11: 1167–73. [PubMed] [Google Scholar]
- 10. Pao W, Wang TY, Riely GJ et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med 2005; 2: e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thatcher N, Chang A, Parikh P et al. Gefitinib plus best supportive care in previously treated patients with refractory advanced non‐small‐cell lung cancer: results from a randomised, placebo‐controlled, multicentre study (Iressa survival evaluation in lung cancer). Lancet 2005; 366: 1527–137. [DOI] [PubMed] [Google Scholar]
- 12. Greulich H, Chen TH, Feng W et al. Oncogenic transformation by inhibitor‐sensitive and – resistant EGFR mutants. PLoS Med 2005; 2: e313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jiang J, Greulich H, Janne PA, Sellers WR, Meyerson M, Griffin JD. Epidermal growth factor‐independent transformation of Ba/F3 cells with cancer‐derived epidermal growth factor receptor mutants induces gefitinib‐sensitive cell cycle progression. Cancer Res 2005; 65: 8968–74. [DOI] [PubMed] [Google Scholar]
- 14. Herbst RS, Fukuoka M, Baselga J. Gefitinib – a novel targeted approach to treating cancer. Nat Rev Cancer 2004; 4: 956–65. [DOI] [PubMed] [Google Scholar]
- 15. Weinstein IB. Cancer. Addiction to oncogenes – the Achilles heal of cancer. Science 2002; 297: 63–4. [DOI] [PubMed] [Google Scholar]
- 16. Ji H, Li D, Chen L et al. The impact of human EGFR kinase domain mutations on lung tumorigenesis and in vivo sensitivity to EGFR‐targeted therapies. Cancer Cell 2006; 9: 485–95. [DOI] [PubMed] [Google Scholar]
- 17. Politi K, Zakowski MF, Fan PD, Schonfeld EA, Pao W, Varmus HE. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down‐regulation of the receptors. Genes Dev 2006; 20: 1496–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fabian MA, Biggs WH III, Treiber DK et al. A small molecule–kinase interaction map for clinical kinase inhibitors. Nat Biotechnol 2005; 23: 329–36. [DOI] [PubMed] [Google Scholar]
- 19. Hotta K, Kiura K, Ueoka H et al. Effect of gefitinib (‘Iressa’, ZD1839) on brain metastases in patients with advanced non‐small‐cell lung cancer. Lung Cancer 2004; 46: 255–61. [DOI] [PubMed] [Google Scholar]
- 20. Fukuoka M, Yano S, Giaccone G et al. Multi‐institutional randomized phase II trial of gefitinib for previously treated patients with advanced non‐small‐cell lung cancer (The IDEAL 1 Trial) [corrected]. J Clin Oncol 2003; 21: 2237–46. [DOI] [PubMed] [Google Scholar]
- 21. Clark J, Cools J, Gilliland DG. EGFR inhibition in non‐small cell lung cancer: resistance, once again, rears its ugly head. PLoS Med 2005; 2: e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gorre ME, Mohammed M, Ellwood K et al. Clinical resistance to STI‐571 cancer therapy caused by BCR‐ABL gene mutation or amplification. Science 2001; 293: 876–80. [DOI] [PubMed] [Google Scholar]
- 23. Kobayashi S, Boggon TJ, Dayaram T et al. EGFR mutation and resistance of non‐small‐cell lung cancer to gefitinib. N Engl J Med 2005; 352: 786–92. [DOI] [PubMed] [Google Scholar]
- 24. Pao W, Miller VA, Politi KA et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2005; 2: e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kwak EL, Sordella R, Bell DW et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci USA 2005; 102: 7665–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Inukai M, Toyooka S, Ito S et al. Presence of epidermal growth factor receptor gene T790M mutation as a minor clone in non‐small cell lung cancer. Cancer Res 2006; 66: 7854–8. [DOI] [PubMed] [Google Scholar]
- 27. Eberhard DA, Johnson BE, Amler LC et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non‐small‐cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol 2005; 23: 5900–9. [DOI] [PubMed] [Google Scholar]
- 28. Moulder SL, Yakes FM, Muthuswamy SK, Bianco R, Simpson JF, Arteaga CL. Epidermal growth factor receptor (HER1) tyrosine kinase inhibitor ZD1839 (Iressa) inhibits HER2/neu (erbB2)‐overexpressing breast cancer cells in vitro and in vivo. Cancer Res 2001; 61: 8887–95. [PubMed] [Google Scholar]
- 29. Hirata A, Hosoi F, Miyagawa M et al. HER2 overexpression increases sensitivity to gefitinib, an epidermal growth factor receptor tyrosine kinase inhibitor, through inhibition of HER2/HER3 heterodimer formation in lung cancer cells. Cancer Res 2005; 65: 4253–60. [DOI] [PubMed] [Google Scholar]
- 30. Fujimoto N, Wislez M, Zhang J et al. High expression of ErbB family members and their ligands in lung adenocarcinomas that are sensitive to inhibition of epidermal growth factor receptor. Cancer Res 2005; 65: 11 478–85. [DOI] [PubMed] [Google Scholar]
- 31. Pino MS, Shrader M, Baker CH et al. Transforming growth factor alpha expression drives constitutive epidermal growth factor receptor pathway activation and sensitivity to gefitinib (Iressa) in human pancreatic cancer cell lines. Cancer Res 2006; 66: 3802–12. [DOI] [PubMed] [Google Scholar]
- 32. Engelman JA, Janne PA, Mermel C et al. ErbB‐3 mediates phosphoinositide 3‐kinase activity in gefitinib‐sensitive non‐small cell lung cancer cell lines. Proc Natl Acad Sci USA 2005; 102: 3788–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang SE, Narasanna A, Perez‐Torres M et al. HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell 2006; 10: 25–38. [DOI] [PubMed] [Google Scholar]
- 34. Zhou BB, Peyton M, He B et al. Targeting ADAM‐mediated ligand cleavage to inhibit HER3 and EGFR pathways in non‐small cell lung cancer. Cancer Cell 2006; 10: 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kassouf W, Dinney CP, Brown G et al. Uncoupling between epidermal growth factor receptor and downstream signals defines resistance to the antiproliferative effect of Gefitinib in bladder cancer cells. Cancer Res 2005; 65: 10 524–35. [DOI] [PubMed] [Google Scholar]
- 36. Thomson S, Buck E, Petti F et al. Epithelial to mesenchymal transition is a determinant of sensitivity of non‐small‐cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res 2005; 65: 9455–62. [DOI] [PubMed] [Google Scholar]
- 37. Witta SE, Gemmill RM, Hirsch FR et al. Restoring E‐cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res 2006; 66: 944–50. [DOI] [PubMed] [Google Scholar]
- 38. Kokubo Y, Gemma A, Noro R et al. Reduction of PTEN protein and loss of epidermal growth factor receptor gene mutation in lung cancer with natural resistance to gefitinib (IRESSA). Br J Cancer 2005; 92: 1711–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mellinghoff IK, Wang MY, Vivanco I et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med 2005; 353: 2012–24. [DOI] [PubMed] [Google Scholar]
- 40. Jain A, Tindell CA, Laux I et al. Epithelial membrane protein‐1 is a biomarker of gefitinib resistance. Proc Natl Acad Sci USA 2005; 102: 11 858–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Takata M, Sabe H, Hata A et al. Tyrosine kinases Lyn and Syk regulate B cell receptor‐coupled Ca2+ mobilization through distinct pathways. EMBO J 1994; 13: 1341–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Taylor SJ, Shalloway D. Cell cycle‐dependent activation of Ras. Curr Biol 1996; 6: 1621–7. [DOI] [PubMed] [Google Scholar]
- 43. Kolch W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev Mol Cell Biol 2005; 6: 827–37. [DOI] [PubMed] [Google Scholar]
- 44. Lee YC, Saijo N, Sasaki Y et al. Clonogenic patterns of human pulmonary adenocarcinoma cell lines (PC‐9, PC‐13 and PC‐14) and how they influence the results of test for chemosensitivity to cisplatin in the human tumor clonogenic assay. Jpn J Clin Oncol 1985; 15: 637–44. [PubMed] [Google Scholar]
- 45. Arao T, Fukumoto H, Takeda M, Tamura T, Saijo N, Nishio K. Small in‐frame deletion in the epidermal growth factor receptor as a target for ZD6474. Cancer Res 2004; 64: 9101–4. [DOI] [PubMed] [Google Scholar]
- 46. Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med 2004; 10: 789–99. [DOI] [PubMed] [Google Scholar]
- 47. Engelman JA, Mukohara T, Zejnullahu K et al. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR‐amplified lung cancer. J Clin Invest 2006; 116: 2695–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997; 88: 593–602. [DOI] [PubMed] [Google Scholar]
- 49. Han SW, Kim TY, Jeon YK et al. Optimization of patient selection for gefitinib in non‐small cell lung cancer by combined analysis of epidermal growth factor receptor mutation, K‐ras mutation, and Akt phosphorylation. Clin Cancer Res 2006; 12: 2538–44. [DOI] [PubMed] [Google Scholar]
- 50. Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C. PI3K/Akt and apoptosis: size matters. Oncogene 2003; 22: 8983–98. [DOI] [PubMed] [Google Scholar]
- 51. She QB, Solit DB, Ye Q, O’Reilly KE, Lobo J, Rosen N. The BAD protein integrates survival signaling by EGFR/MAPK and PI3K/Akt kinase pathways in PTEN‐deficient tumor cells. Cancer Cell 2005; 8: 287–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gurumurthy S, Goswami A, Vasudevan KM, Rangnekar VM. Phosphorylation of Par‐4 by protein kinase A is critical for apoptosis. Mol Cell Biol 2005; 25: 1146–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen YR, Fu YN, Lin CH et al. Distinctive activation patterns in constitutively active and gefitinib‐sensitive EGFR mutants. Oncogene 2006; 25: 1205–15. [DOI] [PubMed] [Google Scholar]