

Abstract
Peroxisome proliferator‐activated receptor γ (PPARγ) is a ligand‐activated transcription factor that has been implicated in the carcinogenesis and progression of various solid tumors, including pancreatic carcinomas. We aimed to clarify the role of this receptor in pancreatic cell motility in vitro and in metastasis in vivo. Cell motility was examined by assaying transwell migration and wound filling in Capan‐1 and Panc‐1 pancreatic cancer cells, with or without the PPARγ‐specific inhibitor T0070907. A severe combined immunodeficiency xenograft metastasis model was used to examine the in vivo effect of PPARγ inhibition on pancreatic cancer metastasis. In both transwell‐migration and wound‐filling assays, inhibition of PPARγ activity suppressed pancreatic cell motility without affecting in vitro cell proliferation. Inhibition of PPARγ also suppressed liver metastasis in vivo in metastatic mice. In PPARγ‐inhibited cells, p120 catenin accumulation was induced predominantly in cell membranes, and the Ras‐homologous GTPases Rac1 and Cdc42 were inactive. Inhibition of PPARγ in pancreatic cancer cells decreased cell motility by altering p120ctn localization and by suppressing the activity of the Ras‐homologous GTPases Rac1 and Cdc42. Based on these findings, PPARγ could function as a novel target for the therapeutic control of cancer cell invasion or metastasis. (Cancer Sci 2008; 99: 1892–1900)
Abbreviations:
- FITC
fluoresceinisothiocyanate
- GST
glutathione‐S‐transferase
- MTT
3‐[4,5‐dimethylthiazol‐2‐yl]‐2,5‐diphenyltetrazolium bromide
- p120ctn
p120 catenin
- PDAC
pancreatic ductal adenocaricinoma cells
- PPARγ
peroxisome proliferator‐activated receptor γ
- PPRE
PPARγ‐response element
- Rho
Ras‐homologous
- SCID
severe combined immunodeficiency
- siRNA
small interfering RNA
- TZD
thiazolizinedione
Pancreatic ductal adenocarcinoma is associated with one of the highest mortality rates in patients with malignancies.( 1 ) Because of a lack of early symptoms, PDAC is often diagnosed only after a local tumor has disseminated and metastatic disease has already developed in regional lymph nodes or distant organ sites. To overcome this dismal situation, development of novel PDAC therapies involving drugs that target disease‐specific molecules is urgently required. PPARγ, a member of the nuclear receptor family of ligand‐activated transcription factors, is one promising target for such therapies.( 2 )
Activation of PPARγ, which is expressed mainly in adipose tissue, is known to play a central role in adipocyte differentiation and insulin sensitivity.( 3 ) For this reason, synthetic PPARγ‐activating ligands such as TZD are used commonly as oral antihyperglycemic agents to control non‐insulin‐dependent diabetes mellitus. More recently, PPARγ has been investigated as a target for the treatment of a variety of cancers.( 4 , 5 , 6 ) The fact that PPARγ is overexpressed in many tumors, including examples in the esophagus, stomach, breast, lung, and colon, suggests that PPARγ function impacts tumor survival.( 4 , 5 , 6 , 7 , 8 ) Initial efforts to alter PPARγ activity focused on activation with TZD ligands, which have been shown to induce G1 cell‐cycle arrest in a variety of tumor cell lines.( 9 , 10 ) However, the reported benefits of TZD for pancreatic carcinoma patients in clinical trials are modest at best.( 11 , 12 )
Several observations suggest that inhibition of PPARγ function may be beneficial in treating neoplasms.( 13 , 14 ) Although PPARγ is overexpressed in many cancer cell types, loss‐of‐function mutations are rare,( 15 ) which suggests that the receptor is a tumor cell survival factor. Evidence that PPARγ function can contribute to carcinogenesis or cancer cell survival includes reports of a murine colon cancer model in which PPARγ activation leads to increased tumor formation.( 16 , 17 )
Profiles of PPARγ expression in a variety of human malignancies, including pancreatic cancer, have been described. One recent report showed a significant association between high levels of PPARγ expression in pancreatic cancer cells and shorter overall survival time.( 18 ) Prior investigations demonstrating that PPARγ inhibition induces apoptosis in epithelial tumor lines suggest strongly that PPARγ inhibition may also be beneficial in PDAC treatment.( 19 , 20 , 21 ) In hepatocellular carcinoma cell lines, PPARγ inhibitors have been shown to inhibit cell adhesion and induce morphological changes that normally occur prior to the commitment to apoptosis; in contrast, caspase inhibitors do not prevent these changes.( 20 ) We hypothesize that PPARγ inhibition interferes with adhesion‐dependent epithelial cell survival signals, leading to cell death (anoikis). Two additional reports have shown that high doses of PPARγ inhibitors also interfere with Caco‐2 cell survival.( 22 , 23 ) The effect of PPARγ inhibitors (especially at low concentrations) on pancreatic cancer cells has not been investigated.
Ras‐homologous GTPases play a pivotal role in the regulation of numerous cellular functions associated with malignant transformation and metastasis. Members of the Rho family of small GTPases are key regulators of actin reorganization and cell motility, as well as cell–cell and cell–extracellular matrix adhesion. These processes all play critical roles during the development and progression of cancer. Because of their pleiotropic functions, Rho proteins appear to be promising targets for the development of novel anticancer drugs,( 24 , 25 ) including those for PDAC.( 25 ) The ability to modulate pathways regulated by Rho could not only improve the therapeutic efficiency, but also reduce the side effects of conventional antineoplastic therapies.
The protein p120ctn is the prototypic member of a subfamily of armadillo repeat‐domain proteins involved in intercellular adhesion. A recent report demonstrated clearly that p120 regulates, at least in part, the activity of Rho GTPases, and that p120 association with classical cadherins regulates their stability.( 26 ) Ectopic expression of p120ctn has been shown to promote cell migration and to induce a wide variety of morphological changes.( 26 )
In the present study, we investigated the effects of PPARγ inhibitors on pancreatic cell lines and xenograft metastatic tissues that function as models for PDAC. Our data demonstrate that inhibition of PPARγ in pancreatic cancer cells decreases cell motility by altering p120ctn localization and suppressing the activity of the Rho GTPases Rac1 and Cdc42. These findings suggest that PPARγ inhibitors may improve the benefit of current PDAC therapeutics.
Materials and Methods
Cell lines and reagents. The PDAC cell line Panc‐1 was purchased from the American Type Culture Collection (Rockville, MD, USA). Other cell lines were provided by the Cell Resource Center for Biomedical Research, Tohoku University (Sendai, Japan). All cell lines were grown in RPMI‐1640 (Sigma‐Aldrich, St Louis, MO, USA) supplemented with 10% fetal bovine serum. Cells were maintained at 37°C in an atmosphere of humidified air with 5% CO2. The PPARγ‐specific inhibitor T0070907 and PPARγ ligand rosiglitazone were purchased from Cayman Chemical (Ann Arbor, MI, USA).
Western blot analysis. Adherent cells were washed in phosphate‐buffered saline, and cell extracts were prepared in Laemmli lysis buffer. Protein concentrations were measured using Bio‐Rad Protein Assay Reagent (Bio‐Rad, Richmond, CA, USA) following the manufacturer's suggested procedure. After electrophoresis of extract aliquots (20 µg protein) on 10% sodium dodecylsulfate–polyacrylamide gels, proteins were transferred to nitrocellulose membranes (Millipore, Bedford, MA, USA), blocked at room temperature for 1 h in Tris‐buffered saline with 5% bovine serum albumin, and then incubated with primary monoclonal antibody for 1 h. Anti‐PPARγ antibody (E‐8) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA); monoclonal antibodies against p120ctn and Rac1 were obtained from BD Transduction Laboratories (Palo Alto, CA, USA). After three washes the membranes were incubated for 1 h at room temperature with secondary antibody, and immune complexes were visualized using the enhanced chemiluminescence detection kit (Amersham, London, UK) following the manufacturer's procedure. Images were captured and analyzed using a LAS‐3000 imaging system (Fujifilm, Tokyo, Japan). The ProteoExtract Subcellular Proteome Extraction Kit (EMD Biosciences, Darmstadt, Germany) was used for the preparation of cytosolic protein extracts.
Cell proliferation and apoptosis assays. Cell proliferation was measured using MTT assays.( 27 ) Approximately 5 × 103 cells in 100 µL medium were plated per well in a 96‐well plate. After 24 h incubation, the medium was changed and supplemented with various concentrations of T0070907 in dimethylsulfoxide, and the cells were incubated for another 24–72 h. After incubating the plates for an additional 4 h with MTT solution (0.5%), sodium dodecylsulfate was added to a final concentration of 20% and absorbance at 595 nm was determined for each well using a microplate reader (Model 550; Bio‐Rad). Control wells were treated with dimethylsulfoxide alone. Three independent experiments were carried out for each cell line. Annexin V staining with the annexin V–FITC apoptosis detection kit (Becton Dickinson, San Jose, CA, USA) followed by FACScan flow cytometry (Becton Dickinson) was used to identify apoptotic cells. Apoptosis measures were carried out in triplicate.
Cell‐motility assays. Motility was assessed by migration of cells in porous‐membrane culture inserts (8.0‐µm pore size; Becton Dickinson). After 24 h of incubation, cells that did not migrate were removed from the upper surface of the membrane with a cotton swab, and migrating cells on the lower surface of the membrane were fixed and stained with toluidine blue. Migrating cell counts were estimated from counts of three independent microscopic visual fields (×100). To estimate cell‐migration activity during wound healing, cells were grown for 2 days (to confluency), after which a scrape in the form of a cross was made through the confluent monolayers with a plastic pipette tip. To measure migration, several wounded areas within each plate were marked for orientation and then photographed periodically by phase‐contact microscopy for 24 h after wounding.
Inhibition of PPARγ function using siRNA. PPARγ siRNA was purchased from Santa Cruz Biotechnology. Panc‐1 and Capan‐1 cells at 70% confluence were transfected with PPARγ siRNA using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) in accordance with the manufacturer's protocol. The cells were treated with 10 nmol/L PPARγ siRNA for 24 h. Stealth RNAi Negative Control Medium GC (Invitrogen) was used for control specimens. Using real‐time reverse transcription–polymerase chain reaction to measure steady‐state mRNA levels in cells, PPARγ‐specific siRNA was found to inhibit PPARγ expression to levels less than 30% of those in control cells (data not shown).
Measuring the effect of T0070907 on PPARγ‐dependent transcription. Capan‐1 cells transfected with plasmid encoding a PPARγ‐response element fused to a luciferase reporter (pHD[×3]PPRE‐Luc) were stimulated as described previously with 1 µmol/L rosiglitazone and various concentrations of T0070907.( 20 ) Luciferase activity was measured 16 h after transfection. Because Renilla luciferase control plasmids are sensitive to steroid/thyroid/retinoid nuclear–receptor stimulation, variability in transfection efficiencies (<20%) were assessed in parallel experiments using the pRL‐TK plasmid (Promega, Madison, WI, USA).
Immunofluorescence staining. Cells (5 × 105 per well) were grown on collagen‐1‐coated glass coverslips in six‐well flat‐bottom plates for 24 h. After 24 h incubation, T0070907 was added to a final concentration of 0.1 µmol/L and the cells were grown for an additional 24 h. The cells were then fixed in 4% paraformaldehyde followed by 100% ethanol at –20°C. After permeabilization with 0.1% Triton‐X, non‐specific binding of antibody to the cells was blocked with 2% normal swine serum. Cells were incubated subsequently with anti‐p120 catenin antibody followed by FITC‐labeled secondary antibody. Samples were then mounted using Vectashield (Vector Laboratories, Burlingame, CA, USA) and examined using confocal laser‐scanning microscopy (Carl Zeiss, Oberkochen, Germany). All experiments were repeated in triplicate.
Measurement of Rac‐1 and Cdc42 activities. GST pull‐down assays using a Rac‐1/Cdc42 activation kit were used to evaluate Rac1/Cdc42 activities according to the manufacturer's protocol (Stressgen, Ann Arbor, MI, USA). Briefly, we used a GST fusion polypeptide composed of GST fused to the interactive domain of human p21‐activated kinase‐1, which interacts specifically with GTP‐bound Cdc42 and Rac1 GTPases.( 25 ) The GST fusion target was incubated with cell lysates and then applied to GST‐specific beads to estimate the relative abundance of active Cdc42 and Rac1. Bound Rac1 and Cdc42 proteins were resolved on 12% denaturing polyacrylamide gels and distinguished by western blotting using antibodies specific to each protein. The amount of active GTP‐bound enzyme was quantified relative to the total amount of each GTPase present in whole unprecipitated cell lysates. The experiments were carried out six times.
In vivo metastasis study. Five‐week‐old male SCID mice were obtained from CLEA Japan (Tokyo, Japan) and maintained in a specific pathogen‐free environment. Experiments were carried out according to the guidelines of Yokohama City University. Six‐week‐old mice were used in this experiment. To assay metastatic capability, viable cancer cells were suspended in serum‐free medium, and 20‐µL aliquots of cell suspension containing 2 × 106 cells were inoculated into the spleens of SCID mice under anesthesia. After inoculation, the mice were randomized into two treatment groups (n = 6) and one control group (n = 6). Administration of T0070907 (5 mg/kg/day) to each treatment group began 1 day after cell inoculation and continued daily for 4 weeks. Four weeks after inoculation, the mice were killed and autopsied immediately. Liver metastasis was measured by counting macroscopic lesions, and measuring them to calculate tumor volume:
| length/2 × width/2 × height/2 × 4/3 × π.( 28 ) |
Examination of hematoxylin–eosin‐stained sections of each lesion resulted in assessments of histopathological alterations in liver metastases.
Results
Expression of PPARγ in pancreatic ductal adenocarcinoma cells. Western blotting of PPARγ with the E8 antibody revealed a specific band between 50 and 60 kDa present in all PDAC cell lines examined (Fig. 1a). Among these lines, steady‐state levels of PPARγ protein were highest in Capan‐1 and HPAK cells, and lowest in Panc‐1 and HS766T cells. Immunohistochemical staining with a PPARγ–specific antibody demonstrated that PPARγ expression in PDAC tissues (Fig. 1c) was similar to normal pancreatic ductal epithelium (Fig. 1b).
Figure 1.
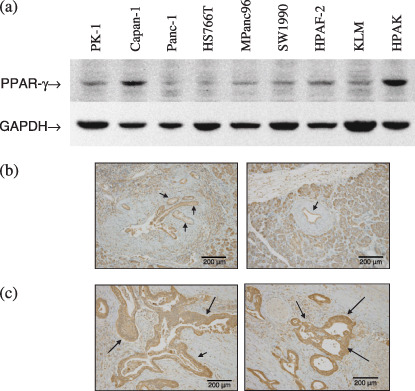
Expression of peroxisome proliferator‐activated receptor γ (PPARγ) in pancreatic cancer cells. (a) Western blots showing PPARγ expression in various pancreatic ductal adenocarcinoma cell lines, as well as a glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) internal control. Immunohistochemical staining of (b) normal ductal epithelium (left and right panels) and (c) pancreatic ductal adenocarcinoma (left and right panels) with anti‐PPARγ antibody. Arrows mark staining of normal pancreatic ductal epithelium in (b) and pancreatic ductal adenocarcinoma in (c).
Peroxisome proliferator‐activated receptor γ inhibitors reduce migration of PDAC cells. The effect of the PPARγ antagonist T0070907 on PDAC cell migration was measured using in vitro wound‐filling assays. The migration of wounded cells treated with T0070907 was inhibited significantly (Fig. 2a,b) relative to untreated, wounded Panc‐1 and Capan‐1 cells. Among cells transfected with PPARγ siRNA to reduce PPARγ expression levels, Capan‐1 cell migration was more severely inhibited than Panc‐1 (Fig. 2c,d). These results indicate that chemical inhibitors or inhibitory siRNA molecules that reduce PPARγ activity lead to inhibition of wound filling. Migration of Capan‐1 and Panc‐1 cells in the absence or presence of several concentrations of T0070907 were also measured in 24‐h transwell migration assays (Fig. 3). In both cell lines, the presence of T0070907 reduced cell migration significantly and in a dose‐dependent manner (Fig. 3a,c). Reduced migration of cells with PPARγ siRNA relative to untreated controls (Fig. 3b,d) demonstrates that transwell migration is inhibited specifically by a reduction in PPARγ activity.
Figure 2.
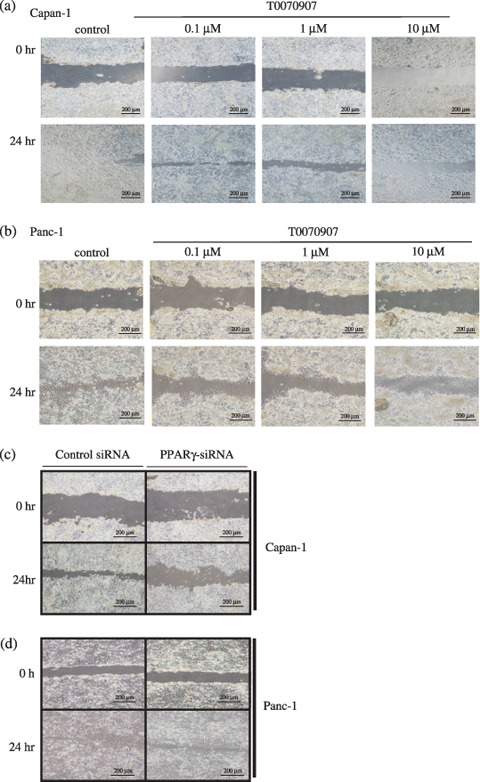
Peroxisome proliferator‐activated receptor γ (PPARγ) inhibition reduces the wound‐filling ability of PDAC cells. Wound‐filling assays in (a) Capan‐1 and (b) Panc‐1 cells treated with various concentrations of T0070907 for 24 h. In both lines, all concentrations of PPARγ inhibitor result in slower cell migration into wound areas. (c) Capan‐1 and (d) Panc‐1 cells transfected with PPARγ small interfering RNA (siRNA) also migrated more slowly into wound areas than cells transfected with control siRNA.
Figure 3.
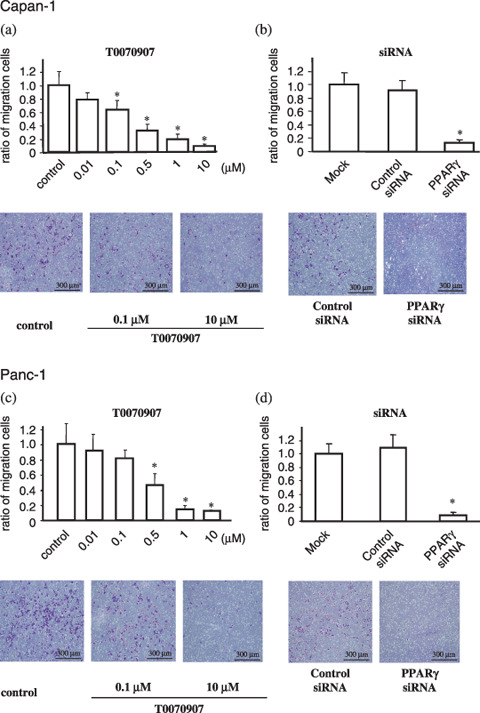
Peroxisome proliferator‐activated receptor γ (PPARγ) inhibition reduces migration of PDAC cells. Cell migrations were estimated from transwell migration assays, in which migrated cells are stained violet and membrane pores can be seen as white dots. Relative to control‐treated cells, (a) Capan‐1 and (c) Panc‐1 cell migration decreased significantly and in a dose‐dependent manner in response to increasing concentrations of PPARγ inhibitor T0070907 (*P < 0.05). (b,d) Significant decreases in cell migration were also observed in PPARγ small interfering RNA (siRNA)‐transfected cells relative to control siRNA‐transfected cells.
Effect of PPARγ inhibitor on cell proliferation and apoptosis. To investigate whether chemical inhibition of PPARγ affects cancer cell proliferation and apoptosis, we used MTT assays to measure cell proliferation and apoptosis in cultured Panc‐1 and Capan‐1 PDAC cell lines. No significant changes in cell proliferation (Fig. 4a,b) or apoptosis (Fig. 4c,d) were observed in T0070907‐treated versus untreated PDAC cells. These results demonstrate that suppression of cell proliferation or apoptosis is not necessarily consequent to T0070907‐mediated suppression of PDAC cell motility.
Figure 4.
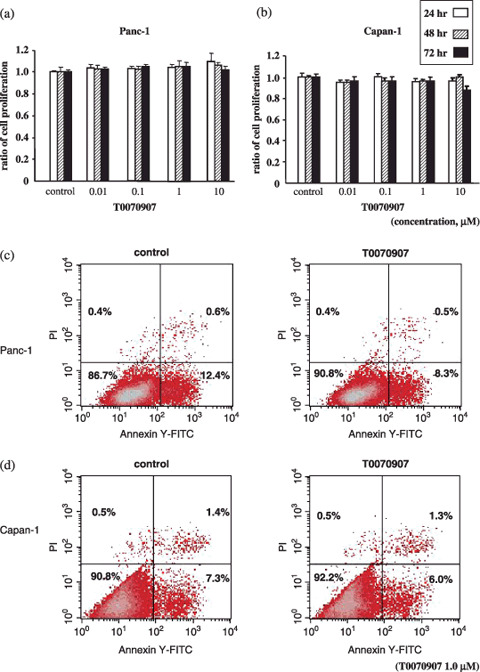
Effect of a peroxisome proliferator‐activated receptor γ (PPARγ) inhibitor on cell proliferation and apoptosis in PDAC cells. Cell proliferation was calculated from 3‐[4,5‐dimethylthiazol‐2‐yl]‐2,5‐diphenyltetrazolium bromide (MTT) assays. (a) Panc‐1 and (b) Capan‐1 cells treated with 1.0 µmol/L T0070907 for 24, 48, and 72 h showed no significant change in MTT values relative to control cells (y‐axis values represent the ratio of MTT optical density readings from treated and untreated cells; columns represent ratio mean ± SD). Apoptotic cell counts were measured by fluorescence activated cell sorting (FACS) after treatment of (c) Panc‐1 or (d) Capan‐1 cells without or with 1.0 µmol/L T0070907 for 24 h. T0070907 treatments did not result in any significant changes in the percentage of apoptotic cells.
Inhibition of PPARγ alters the subcellular localization of p120ctn. Association of p120ctn with the intracellular domains of cadherins promotes cell–cell adhesion and cell motility by regulating the activation of Rho GTPases.( 29 , 30 ) Because cytoplasmic p120ctn is the only known activator of Rho GTPases that functions in cell motility, the ratio of cadherin‐bound p120ctn to p120ctn in the cytplasmic pool is an important factor regulating motility. To examine the involvement of p120ctn in T0070907‐mediated suppression of cell motility, we used immunocytochemical analyses to examine the subcellular distribution of p120ctn in PDAC cells. In T0070907‐treated Capan‐1 cells, p120ctn was found predominantly on the plasma membranes (relative to more free p120ctn in the cytoplasm of untreated cells) (Fig. 5a). In contrast, there were no significant changes in the distribution of p120ctn in T0070907‐treated Panc‐1 cells (data not shown). The intracellular distributions of PPARγ and p120ctn did not overlap (merged) (Fig. 5a).
Figure 5.
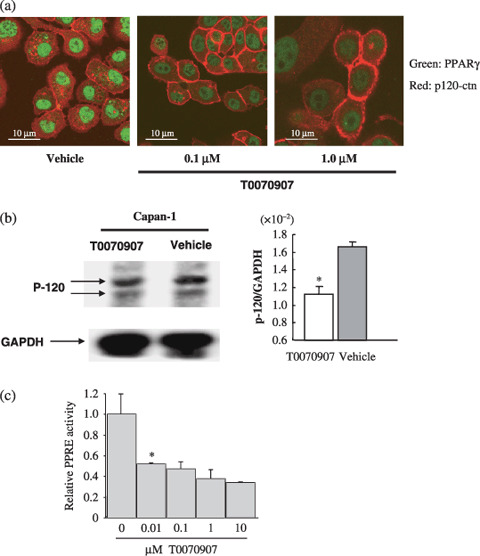
Peroxisome proliferator‐activated receptor γ (PPARγ) inhibitor T0070907 affects the subcellular localization of p120 catenin (p120ctn) in Capan‐1 cells. (a) The subcellular distribution of p120ctn in Capan‐1 control cells or those treated with 0.1 or 1.0 µmol/L T0070907 were compared following immunostaining with antip120ctn (red) and anti‐PPARγ (green). Although p120ctn accumulated in the cytoplasm of vehicle‐treated cells, it localized predominantly to cell membranes in T0070907‐treated Capan‐1 cells. Merged images of PPARγ and p120ctn immunohistochemical staining patterns show no colocalization. (b) Western‐blot analysis of cytosolic protein extracts with p120ctn antibody and glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) control antibody. T0070907 treatment resulted in reduced signal strength from the cytosolic p120ctn band. Changes in the cytosolic p120ctn expression can be seen by the reduction in the ratio of p120ctn : GAPDH signal. Bars represents the mean ratio of p120 : GAPDH signal strength ± SD. *P < 0.05. (c) Capan‐1 cells transfected with a PPARγ‐response element (PPRE)‐luciferase reporter plasmid were stimulated with 1 mmol/L rosiglitazone (synthetic ligand of PPARγ) in the presence of concentrations of T0070907 shown. Luciferase activity was measured after 16 h of treatment. Bars represent relative PPRE activity as measured by the ratio of PPARγ‐dependent luciferase activity in treated cells relative to untreated cells.
Although western blots of fractionated cells revealed that cytoplasmic p120ctn levels decreased in T0070907‐treated Capan‐1 cells (Fig. 5b), no significant change in distribution was observed between untreated and treated Panc‐1 cells (data not shown). These results indicate that in Capan‐1 cells, PPARγ inhibition increases the relative amount of cadherin‐bound p120ctn. We speculate that relatively low levels of PPARγ expression in Panc‐1 cells may confound our ability to measure any similar change in p120ctn subcellular localization following T0070907 treatment. To investigate whether PPARγ activity in Capan‐1 cells is inhibited by low concentrations of T0070907, the effect of a range of T0070907 concentrations on PPRE‐dependent transcription was measured (Fig. 5c). With 0.1 µmol/L T0070907, PPRE‐dependent transcription in Capan‐1 cells was inhibited to approximately half maximum.
Peroxisome proliferator‐activated receptor γ inhibitor suppresses the activity of Rac‐1 and Cdc42. Previous reports suggest that p120ctn affects cell motility in association with Rac1 and Cdc42 Rho GTPases.( 30 , 31 , 32 , 33 ) The activities of Rac1 and Cdc42 GTPases were measured in lysates of T0070907‐treated and ‐untreated Capan‐1 cells using a GST pull‐down target that interacts specifically with active GTPases. In T0070907‐treated cells, we observed a significant decrease in the percent‐active fractions of Rac1 and Cdc42 GTPases (Fig. 6).
Figure 6.
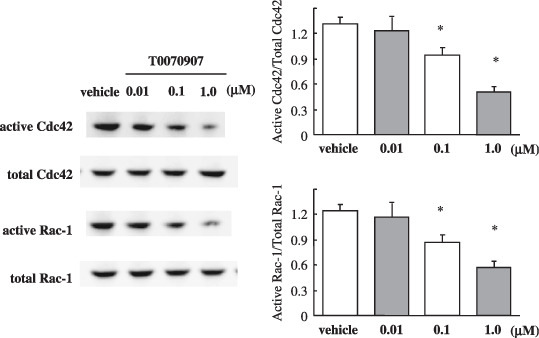
Effect of the peroxisome proliferator‐activated receptor γ (PPARγ) inhibitor T0070907 on the activity of Rac‐1 and Cdc42 GTPases in Capan‐1 cells. GTPase activity for Rac‐1 and Cdc42 was measured using GST pull‐down assays that are capable of distinguishing the percentage of the active fraction (relative to total) of Rac‐1 and Cdc42 in lysates of Capan‐1 cells. Treatment with T0070907 (0.1 and 1.0 µmol/L) resulted in a significant decrease in the percentage of active Rac‐1 and Cdc42. Each column represents the mean ± SD. The experiments were carried out six times. *P < 0.05.
Peroxisome proliferator‐activated receptor γ inhibitor reduces liver metastasis in a mouse xenograft model. To investigate whether PPARγ inhibitors affect metastatic cell spreading, we tested the ability of T0070907 to reduce metastatic tumor formation in a Capan‐1/SCID mouse xenograft model. Capan‐1 cells were injected into the spleens of SCID mice, and the number and size of metastatic lesions in livers were measured after 4 weeks (Fig. 7). Mice treated orally with 5 mg/kg/day of T0070907 contained two‐thirds fewer metastatic foci (P < 0.05), with an average tumor volume of only 12% of tumors in control mice (P < 0.05) (Table 1). Serum l‐alanine aminotransferase (ALT) levels were within the normal range in the all mice (Suppl. Fig. S1).
Figure 7.
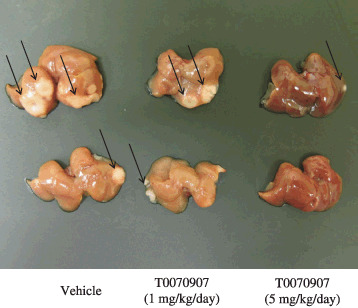
Peroxisome proliferator‐activated receptor γ (PPARγ) inhibitor T0070907 reduces the number and volume of metastases in a murine xenograft model. Numbers and areas of metastases (arrows) in the liver decreased markedly in mice treated with T0070907 relative to control mice.
Table 1.
Effects of peroxisome proliferator‐activated receptor γ inhibitor (T0070907) on liver metastasis of Capan‐1 cells
| Incidence | Number of metastatic colonies (mean ± SD) | Total tumor volume (mm3) (mean ± SD) |
|---|---|---|
| Vehicle | 3.50 ± 1.05 | 738.6 ± 415.7 |
| T0070907 (5 mg/kg/day) | 1.00 ± 1.27 | 86.8 ± 173.2 |
| P < 0.05 | P < 0.05 |
Capan‐1 cells were injected into the spleen of male severe combined immunodeficiency mice. One day after injection, three groups (n = 6) were randomized into vehicle or 5 mg/kg/day T0070907. After 4 weeks, livers were harvested, and the number of metastases and total tumor volume of all metastatic lesions was determined.
Discussion
We demonstrated that levels of PPARγ expression vary among pancreatic adenocarinoma cell lines (Fig. 1) and tested the effect of the PPARγ–specific inhibitor T0070907 on PDAC cells. In Capan‐1 and Panc‐1 cells, both T0070907 and PPARγ siRNA suppressed cell motility, migration, and invasion, but did not inhibit cell proliferation (2, 3, 4). These results suggest strongly that PPARγ plays a crucial role in PDAC cell motility, migration, and invasion. Elucidating the mechanism that underlies cell motility is of clinical importance as a means for controlling tumor cell invasion, dissemination, and metastasis in patients with pancreatic cancer.
Following treatment with T0070907 PPARγ inhibitor, p120ctn was found predominantly in Capan‐1 cell membranes. Recent reports demonstrate that p120ctn associates with all classic cadherin subtypes, and is involved in the regulation of cell motility and cell adhesion.( 26 ) p120ctn is known to also regulate actin cytoskeleton configuration. We did not observe colocalization of PPARγ and p120ctn expression in PPARγ inhibitor‐treated or ‐untreated cells, indicating that PPARγ may not interact directly with p120ctn.
Ras‐homologous GTPases, which localize to membranes in a GDP‐bound state, are activated to a GTP‐bound state upon stimulation of cell‐surface receptors. Upon activation, Rho GTPases bind effectors that trigger specific cellular responses. As Rho proteins are known to play essential roles in signaling events that regulate cadherin‐dependent motility, specific inhibitors of individual Rho functions (notably RhoA‐, RhoB‐, Rac1‐, or Cdc42‐related functions) could provide therapeutic benefits in controlling cancer metastasis. Indeed, compounds developed as specific inhibitors of the RhoA‐effector molecule Rho‐kinase have been demonstrated to exert antimetastatic activity in vivo. ( 24 )
The inactivation of Rac1 and Cdc42 that we observed in response to PPARγ inhibition indicates that these molecules are involved in PPARγ‐mediated PDAC cell motility. We also demonstrated liver metastasis inhibition in response to PPARγ inhibition in an in vivo metastatic model. Previous reports have demonstrated an induction in apoptosis in response to PPARγ inhibition in other epithelial tumor lines.( 19 , 20 , 21 ) Anoikis, which is a loss of adhesion‐induced apoptosis, was also reported in response to PPARγ inhibition by T0070907; however, concentrations greater than 10 µmol/L T0070907 have been shown to be required to induce anoikis in a variety of carcinoma cell lines. In the present study, we observed a significant inhibitory effect of T0070907 on cell migration at much lower T0070907 concentrations (0.01–1 µmol/L; Fig. 5c) that had no effect on cell proliferation or cell death as measured in MTT and apoptosis assays (Fig. 4). Our findings at low concentrations of T0070907 suggest that inhibition of cancer cell migration is due to the specificity of T0070907's pharmacological effect on PPARγ, and not by anoikis, which is induced at higher concentrations of PPARγ inhibitor. The relatively low concentrations of T0070907 required for inhibition and the dose‐dependent effect of the inhibitor on cell migration make it unlikely that inhibition was non‐specific.
Transwell migration and wound‐filling assays in both Capan‐1 and Panc‐1 pancreatic cancer cell lines demonstrated the inhibitory effects of PPARγ on cell motility in vitro. In contrast, the effects of PPARγ inhibitor on p120ctn subcellular localization, and on Rac1 and Cdc42 GTPase activities, were exclusive to Capan‐1 cells, and not seen in Panc‐1. We speculate that relative to Panc‐1 cells, steady‐state levels of PPARγ in Capan‐1 cells are much higher (Fig. 1a), which may contribute to these discrepancies. This hypothesis is supported by a recent clinical report in which a significant positive association was measured between high levels of PPARγ expression in pancreatic cancer cells and shorter overall survival time.( 18 ) Further investigation will be required to better understand the mechanism of PPARγ inhibition of pancreatic cancer cell motility, invasion, and metastasis.
In conclusion, we have demonstrated that inhibition of PPARγ in pancreatic cancer cells decreases cell motility by altering p120ctn localization and suppressing Rac1 and Cdc42 Rho GTPase activities. PPARγ could function as a novel therapeutic target for controlling cancer cell dissemination or metastasis.
Supporting information
Fig. S1. Serum levels of alanine aminotransferase (ALT) in the T0070907‐treated metastatic model mice. There was no liver injury at this dose of T0070907.
Please note: Blackwell Publishing are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Acknowledgments
We thank Machiko Hiraga and Yuko Satoh for their technical assistance. The present work was supported in part by a Grant‐in‐Aid for research on the Third Term Comprehensive Control Research for Cancer from the Ministry of Health, Labour and Welfare, Japan to A.N., a grant from the National Institute of Biomedical Innovation to A.N., a grant from the Ministry of Education, Culture, Sports, Science and Technology, Japan (KIBAN‐B) to A.N., a grant from the Princess Takamatsu Cancer Research Foundation to A.N., and a grant from the Japanese Human Science Research Foundation to A.N.
References
- 1. Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin 2007; 57: 43–66. [DOI] [PubMed] [Google Scholar]
- 2. Kersten S, Desvergne B, Wahli W. Roles of PPARs in health and disease. Nature 2000; 405: 421–4. [DOI] [PubMed] [Google Scholar]
- 3. Desvergne B, Wahli W. Peroxisome proliferator‐activated receptors: nuclear control of metabolism. Endocr Rev 1999; 20: 649–88. [DOI] [PubMed] [Google Scholar]
- 4. Takashima T, Fujiwara Y, Higuchi K et al . PPAR‐γ ligands inhibit growth of human esophageal adenocarcinoma cells through induction of apoptosis, cell cycle arrest and reduction of ornithine decarboxylase activity. Int J Oncol 2001; 19: 465–71. [PubMed] [Google Scholar]
- 5. Chang TH, Szabo E. Induction of differentiation and apoptosis by ligands of peroxisome proliferator‐activated receptor gamma in non‐small cell lung cancer. Cancer Res 2000; 60: 1129–38. [PubMed] [Google Scholar]
- 6. DuBois RN, Gupta R, Brockman J, Reddy BS, Krakow SL, Lazar MA. The nuclear eicosanoid receptor, PPARγ, is aberrantly expressed in colonic cancers. Carcinogenesis 1998; 19: 49–53. [DOI] [PubMed] [Google Scholar]
- 7. Mueller E, Sarraf P, Tontonoz P et al . Terminal differentiation of human breast cancer through PPAR gamma. Mol Cell 1998; 1: 465–70. [DOI] [PubMed] [Google Scholar]
- 8. Sato H, Ishihara S, Kawashima K et al . Expression of peroxisome proliferator‐activated receptor (PPAR) γ in gastric cancer and inhibitory effects of PPARγ agonists. Br J Cancer 2000; 83: 1394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guan YF, Zhang YH, Breyer RM, Davis L, Breyer MD. Expression of peroxisome proliferator‐activated receptor γ (PPARγ) in human transitional bladder cancer and its role in inducing cell death. Neoplasia 1999; 1: 330–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kitamura S, Miyazaki Y, Shinomura Y, Kondo S, Kanayama S, Matsuzawa Y. Peroxisome proliferator‐activated receptor γ induces growth arrest and differentiation markers of human colon cancer cells. Jpn J Cancer Res 1999; 90: 75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Debrock G, Vanhentenrijk V, Sciot R, Debiec‐Rychter M, Oyen R, Van Oosterom A. A phase II trial with rosiglitazone in liposarcoma patients. Br J Cancer 2003; 89: 1409–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kulke MH, Demetri GD, Sharpless NE et al . A phase II study of troglitazone, an activator of the PPARγ receptor, in patients with chemotherapy‐resistant metastatic colorectal cancer. Cancer J 2002; 8: 395–9. [DOI] [PubMed] [Google Scholar]
- 13. Martelli ML, Iuliano R, Le Pera I et al . Inhibitory effects of peroxisome poliferator‐activated receptor γ on thyroid carcinoma cell growth. J Clin Endocrinol Metab 2002; 87: 4728–35. [DOI] [PubMed] [Google Scholar]
- 14. Panigrahy D, Shen LQ, Kieran MW, Kaipainen A. Therapeutic potential of thiazolidinediones as anticancer agents. Expert Opin Invest Drugs 2003; 12: 1925–37. [DOI] [PubMed] [Google Scholar]
- 15. Posch MG, Zang C, Mueller W, Lass U, Von Deimling A, Elstner E. Somatic mutations in peroxisome proliferator‐activated receptor‐γ are rare events in human cancer cells. Med Sci Monit 2004; 10: BR250–4. [PubMed] [Google Scholar]
- 16. Lefebvre AM, Chen I, Desreumaux P et al . Activation of the peroxisome proliferator‐activated receptor γ promotes the development of colon tumors in C57BL/6J‐APCMin/+ mice. Nat Med 1998; 4: 1053–7. [DOI] [PubMed] [Google Scholar]
- 17. Saez E, Tontonoz P, Nelson MC et al . Activators of the nuclear receptor PPARγ enhance colon polyp formation. Nat Med 1998; 4: 1058–61. [DOI] [PubMed] [Google Scholar]
- 18. Kristiansen G, Jacob J, Buckendahl AC et al . Peroxisome proliferator‐activated receptor γ is highly expressed in pancreatic cancer and is associated with shorter overall survival times. Clin Cancer Res 2006; 12: 6444–51. [DOI] [PubMed] [Google Scholar]
- 19. Masuda T, Wada K, Nakajima A et al . Critical role of peroxisome proliferator‐activated receptor γ on anoikis and invasion of squamous cell carcinoma. Clin Cancer Res 2005; 11: 4012–21. [DOI] [PubMed] [Google Scholar]
- 20. Schaefer KL, Wada K, Takahashi H et al . Peroxisome proliferator‐activated receptor γ inhibition prevents adhesion to the extracellular matrix and induces anoikis in hepatocellular carcinoma cells. Cancer Res 2005; 65: 2251–9. [DOI] [PubMed] [Google Scholar]
- 21. Takahashi H, Fujita K, Fujisawa T et al . Inhibition of peroxisome proliferator‐activated receptor γ activity in esophageal carcinoma cells results in a drastic decrease of invasive properties. Cancer Sci 2006; 97: 854–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lea MA, Sura M, Desbordes C. Inhibition of cell proliferation by potential peroxisome proliferator‐activated receptor (PPAR) γ agonists and antagonists. Anticancer Res 2004; 24: 2765–71. [PubMed] [Google Scholar]
- 23. Ramilo G, Valverde I, Lago J, Vieites JM, Cabado AG. Cytotoxic effects of BADGE (bisphenol A diglycidyl ether) and BFDGE (bisphenol F diglycidyl ether) on Caco‐2 cells in vitro . Arch Toxicol 2006; 80: 748–59. [DOI] [PubMed] [Google Scholar]
- 24. Fritz G, Kaina B. Rho GTPases: promising cellular targets for novel anticancer drugs. Curr Cancer Drug Targets 2006; 6: 1–14. [PubMed] [Google Scholar]
- 25. Taniuchi K, Nakagawa H, Hosokawa M et al . Overexpressed P‐cadherin/CDH3 promotes motility of pancreatic cancer cells by interacting with p120ctn and activating Rho‐family GTPases. Cancer Res 2005; 65: 3092–9. [DOI] [PubMed] [Google Scholar]
- 26. Anastasiadis PZ. p120‐ctn: a nexus for contextual signaling via Rho GTPases. Biochim Biophys Acta 2007; 1773: 34–46. [DOI] [PubMed] [Google Scholar]
- 27. Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Meth 1983; 65: 55–63. [DOI] [PubMed] [Google Scholar]
- 28. Yasui N, Sakamoto M, Ochiai A et al . Tumor growth and metastasis of human colorectal cancer cell lines in SCID mice resemble clinical metastatic behaviors. Invasion Metastasis 1997; 17: 259–69. [PubMed] [Google Scholar]
- 29. Nobes CD, Hall A. Rho GTPase control polarity, protrusion, and adhesion during cell movement. J Cell Biol 1999; 144: 1235–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Noren NK, Liu Bp Burridge K, Kreft B. p120 catenin regulates the actin cytoskeleton via Rho family GTPase. J Cell Biol 2000; 150: 567–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nobes CD, Hall A. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J Cell Biol 1999; 144: 1235–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Noren NK, Liu BP, Burridge K, Kreft B. p120 catenin regulates the actin cytoskeleton via Rho family GTPases. J Cell Biol 2000; 150: 567–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Anastasiadis PZ, Reynolds AB. Regulation of Rho GTPases by p120‐catenin. Curr Opin Cell Biol 2001; 13: 604–10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Serum levels of alanine aminotransferase (ALT) in the T0070907‐treated metastatic model mice. There was no liver injury at this dose of T0070907.
Please note: Blackwell Publishing are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item