

Abstract
Sorafenib is a novel oral multikinase inhibitor that targets Raf serine/threonine and receptor tyrosine kinases, and inhibits tumor cell proliferation and angiogenesis. We have conducted a phase I study of sorafenib to determine the safety, tolerability, pharmacokinetics, and potential efficacy of this agent in 31 Japanese patients with advanced refractory solid tumors. Sorafenib (100–600 mg) was given as a single dose followed by a 7‐day wash‐out period, and then administrated twice daily (bid). The most frequent drug‐related adverse events were rash/desquamation (61%), hand–foot skin reactions (39%), diarrhea (36%), and elevations of serum lipase (36%) and amylase (26%) levels. Dose‐limiting toxicities (DLTs) were grade 3 diarrhea at 200 mg bid and grade 3 fatigue at 600 mg bid. Grade 3 and 4 pancreatic enzyme elevations were observed at 200–600 mg bid, but they were not deemed dose‐limiting because they were asymptomatic and were not associated with pancreatitis or chronic damage to the pancreas. The AUC and Cmax of sorafenib increased linearly with dose up to 400 mg bid. Partial responses were observed in one of 10 patients with non‐small cell lung cancer and one of three patients with renal cell carcinoma. In conclusion, sorafenib 400 mg bid was well tolerated in Japanese patients with advanced refractory solid tumors. The recommended dose for future clinical trials is 400 mg bid. (Cancer Sci 2008; 99: 1492–1498)
Recent research on the molecular mechanisms controlling tumor cell proliferation, invasion, and metastasis has identified several novel targets for cancer therapeutics. The mitogen‐activated protein kinase (MAPK) signaling pathways, which mediate transduction of extracellular signals to the nucleus via a cascade of phosphorylation events through Ras/Raf/MEK/ERK, are often dysregulated in human tumors. Dominant negative mutants of Raf or ERK inhibit both the primary and metastatic growth of human tumor xenografts in vivo. Thus, activation of Raf kinase is considered to be an important mechanism by which human cancer develops. Therefore, the critical components of MAPK signaling pathways, including Raf kinase, represent potential targets for anticancer treatment.( 1 , 2 , 3 , 4 )
Tumor angiogenesis, the proliferation of a vascular network to supply tumors with nutrients and oxygen, is necessary for tumors to maintain growth and to spread. It is supported by angiogenic factors such as vascular endothelial growth factor (VEGF) and platelet‐derived growth factor (PDGF). VEGF and PDGF bind the VEGF receptor (VEGFR) on endothelial cells and the PDGF receptor (PDGFR) on smooth muscle cells, which are both receptor tyrosine kinases, respectively. Thus, the receptors themselves and their signaling pathways are also potential therapeutic targets for cancer.( 5 , 6 )
Sorafenib (BAY 43‐9006) is an orally available small molecule that displays inhibitory activity against multiple kinases including c‐Raf‐1 and B‐Raf. Inhibition of Raf activity is followed by interference with the activation of ERK, thereby inhibiting cell proliferation, differentiation, and transformation.( 7 , 8 ) In addition, sorafenib inhibits receptor tyrosine kinases including VEGFR‐2 and PDGFR, thereby inhibiting angiogenesis. Inhibition of both tumor cell proliferation and angiogenesis is considered to contribute to the potent antitumor activity of sorafenib. In studies of various human tumors, sorafenib exhibited a dose‐dependent inhibition of tumor growth associated with apoptosis in xenograft models.( 7 , 8 )
Various types of clinical development programs for sorafenib are now on‐going worldwide. In the phase III Treatment Approaches in Renal Cancer Global Evaluation Trial (TARGET), sorafenib significantly prolonged progression‐free survival as well as overall survival in patients with advanced renal cell cancer.( 9 ) Sorafenib has recently been approved for advanced renal cell carcinoma and hepatocellular carcinoma in the USA, Europe, and other countries.
The phase I study reported here was planned to determine the safety, dose‐limiting toxicities (DLTs), maximum‐tolerated dose (MTD), and pharmacokinetics of sorafenib in Japanese patients with refractory advanced solid tumors. Pharmacodynamics was also studied using flow cytometric analysis of ERK‐phosphorylation in patients’ peripheral blood mononuclear cells (PBMCs), as well as plasma adrenomedullin levels. Furthermore, disease activity was evaluated by fluorodeoxyglucose‐positron emission tomography (FDG‐PET).
Materials and Methods
Patient selection. Study eligibility criteria included histologically or cytologically confirmed advanced solid cancer, which was refractory to standard therapy or for which no effective therapy was available, patient age ≥ 20 years, Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, estimated life expectancy ≥ 12 weeks, and adequate organ function. Main exclusion criteria were as follows: chronic heart failure (New York Heart Association Grade III or IV), active cardiac diseases, history of HIV infection or chronic hepatitis B or C, active infections, tumor involving the central nervous system, history of seizure, concurrent malignancy, other anticancer therapy within 4 weeks (6 weeks for mitomycin C or nitrosourea, 2 weeks for hormonal therapy, and 3 weeks for radiotherapy), and surgery within 4 weeks prior to this study. Patients treated with CYP3A4 inhibitors or inducers were also excluded because of possible drug interactions with sorafenib and confounding effects on the pharmacokinetics results. The study was approved by the Institutional Review Board of the National Cancer Center and all patients gave written informed consent before entry onto the study.
Study design. A single dose of sorafenib was given orally, followed by a 7‐day wash‐out, and then administration of sorafenib continued twice daily until the occurrence of unacceptable toxicity, withdrawn consent, disease progression, or death.
In this study, the initial dose was 100 mg, which was based on observations in phase I studies performed in foreign countries as well as on preclinical studies. In both dogs and rats, exposures to between 53.5 and 67.1 mg h/L was associated with moderate toxicity. Assuming that oral bioavailability is similar in humans, a single 100 mg dose of sorafenib would be expected to yield a systemic exposure of 5.8 mg h/L. Therefore, 100 mg sorafenib was considered to be a safe starting dose for this phase I study, thereafter escalated to 200, 400, and 600 mg bid.
For each dose level, a cohort of three patients was treated. In the absence of observed DLTs during the first 4 weeks of continuous administration bid, a further cohort of three patients was enrolled to the next higher dose. If one of the first three patients experienced DLTs, three additional patients were treated at that same dose level. The dose was then escalated when no DLTs was observed in the three additional patients.
Definition of dose‐limiting toxicity. Toxicities were evaluated according to the National Cancer Institute Common Toxicity Criteria (NCI‐CTC) version 2.0, with DLTs being defined as grade 3 or 4 non‐hematological toxicity (except anorexia and manageable nausea and vomiting), grade 4 neutropenia lasting for ≥7 days, febrile neutropenia, or thrombocytopenia <25 000/mm3.
Grade 4 elevations of pancreatic enzymes were observed in 200 mg bid cohorts, but ultrasound investigation, magnetic resonance imaging, and computed tomography did not show any evidence of pancreas damage or pancreatitis. Therefore, after the safety of 200 mg bid was confirmed, the definition of DLT was amended to exclude clinically insignificant elevations of pancreatic enzymes and the definition of DLT for serum pancreatic enzymes was amended accordingly. DLTs were deemed dose‐limiting only when they were grade 4 for >4 days, associated with clinical/imaging findings of pancreatitis, or considered to be life‐threatening or result in chronic damage to the pancreas.
Patient evaluation. Physical examination and hematological/biochemical laboratory evaluation were performed weekly for the first 4 weeks of continuous dosing and every 2 weeks thereafter. Laboratory evaluation was also performed on day 4 of the continuous dosing. Tumor measurements were performed at the baseline, and repeated every 8 weeks according to the Response Evaluation Criteria in Solid Tumors (RECIST).( 10 ) Tumor responses were classified as complete response (CR), partial response (PR), stable disease (SD), and progressive disease (PD).
Pharmacokinetics. For the measurement of plasma concentrations of sorafenib and its metabolites, blood samples (5 mL aliquots) were drawn prior to drug administration, as well as 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 24, 36, 48, 72, 96, and 120 h after the single dose administration. For the continuous dosing period, blood was sampled prior to the first dosing on days 1, 4, 7, 10, 14, 21, and 28, along with 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, and 12 h after the first dose on day 14 at 100, 200, 400, and 600 mg bid. The same full sampling was performed on day 28 at 100 and 200 mg bid, while blood was sampled prior to and 12 h after the morning administration at 400 and 600 mg bid. Urine voided up to 48 h after the single administration was collected.
Concentrations of sorafenib and its metabolites in plasma and urine were determined at Bayer HealthCare (Berlin, Germany) using high performance liquid chromatography‐tandem mass spectrometry (HPLC‐MS‐MS) methods.( 11 ) The method was validated within a working range of 0.0100–12.0 mg/L (sorafenib) and 0.0100–2.5 mg/L (metabolite M2; M4; M5; Fig. 1). Mean interassay precision and accuracy for sorafenib quantification ranged from 0.4% to 4.9% and from 91.2% to 96.5%, respectively. Plasma pharmacokinetic parameters, area under the curve (AUC), maximum concentration (Cmax), and elimination half‐life (t1/2) for sorafenib were calculated by non‐compartment analysis using the KINCALC program (Bayer HealthCare).
Figure 1.
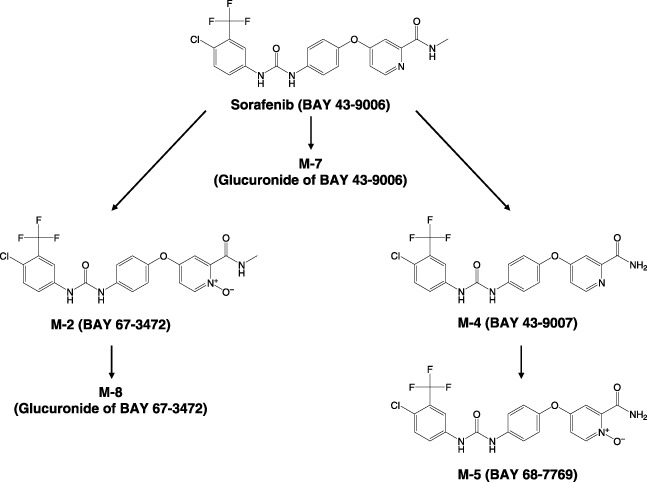
Metabolism map of sorafenib and its metabolites.
Pharmacodynamics. As a specific marker for the Ras signaling pathway, phosphorylated ERK (pERK) in peripheral blood mononuclear cells (PBMC) was quantified. Peripheral blood samples with EDTA anticoagulant were taken at the baseline and on day 28 of the continuous treatments. PBMCs were prepared from blood, stimulated by phorbol myristate acetate (PMA), and fixed in 4% formaldehyde. pERK in PBMCs was stained using an antipERK and fluorescein isothiocyanate‐conjugated secondary antibody. The cells were resuspended in phosphate‐buffered saline and flow cytometry was performed.( 12 ) The plasma concentration of adrenomedullin was measured by immunoradiometric assay at the baseline and on day 28 of the continuous dosing. FDG‐PET was performed before treatment, 1, 2, and 3 months after the initiation of treatment, and every 2 months thereafter. The maximum standardized uptake values (SUVmax) were recorded. The relationship between trough concentrations of sorafenib on day 28 versus SUVmax 1 month after the start of continuous dosing was investigated by using an inhibitory Emax model:
| E = Emax × (1 – C/[C + EC50]) |
where E is the percentage of SUVmax relative to the baseline, Emax is the maximum effect expressed as a percentage of baseline, C is trough concentration, and EC50 is the concentration yielding 50% of Emax.
Results
Patient characteristics. A total of 31 patients were enrolled in this study: 10 males and 21 females. The median age was 63 years with a range of 32–72 years. The baseline demographics for all patients are shown in Table 1. The commonest cancers were non‐small cell lung (10 patients) and colorectal (six patients) cancers. Six of 10 patients with lung cancer had adenocarcinoma. All patients had an ECOG performance status of 0 or 1. Thirty patients had been pretreated with chemotherapy, 29 had had surgery, and 11 radiotherapy. Four patients discontinued treatment during the initial 4‐week continuous dosing period (cycle 1) because of disease progression in three and withdrawal of consent in one case. All 31 patients were assessable for safety.
Table 1.
Patient characteristics
| Number of patients (female/male) | 31 (10/21) |
| Median age (range) | 63 (32–72) |
| ECOG performance status | |
| 0 | 8 |
| 1 | 23 |
| Cancer type | |
| Non‐small cell lung | 10 |
| Colorectal | 6 |
| Renal | 3 |
| Gastric | 2 |
| Others | 10 |
| Prior therapy | |
| Chemotherapy | 30 |
| Radiotherapy | 11 |
| Surgery | 29 |
EOCG, Eastern Cooperative Oncology Group.
Dose escalations and dose‐limiting toxicity. DLTs were not observed in any of the cohort of three patients at 100 mg bid. A total of 15 patients were enrolled at the 200 mg bid dose level, 12 of whom could be evaluated for DLTs (two patients did not complete cycle 1 due to progressive disease and withdrawal of consent in the other). One of these 12 patients presented with grade 3 diarrhea, classified as a DLT. In addition, two patients had grade 3/4 elevations of pancreatic enzymes including grade 4 lipase and grade 3/4 amylase. However, examination of these patients with pancreatic enzyme elevations using ultrasound, magnetic resonance imaging, and computed tomography did not show any evidence of pancreatitis, and the lipase level began to decrease before sorafenib was stopped. After the safety of 200 mg bid had been thus confirmed, the next dose of 400 mg bid was investigated. Six patients in the 400 mg bid cohorts experienced no DLTs, although two had grade 4 lipase elevations which were not associated with pancreatitis. Next, seven patients at 600 mg bid were studied. One patient was taken off the study because of early disease progression. One of the remaining six patients experienced dose‐limiting grade 3 fatigue. In addition to this observation, hand–foot skin reactions were observed in five patients at 600 mg bid. Therefore, 400 mg bid sorafenib was established as the MTD and is recommended for future clinical studies.
Safety. Thirty patients experienced drug‐related adverse events (2, 3), the most frequent of which were dermatological (77%), gastrointestinal (58%), or elevations of lipase (36%) or amylase (26%). The most common dermatological adverse events were rash/desquamation (61%), hand–foot skin reaction (39%), alopecia (26%), dry skin (23%), and pruritus (16%; Table 2). However, these were mild, beginning mostly 2–8 weeks after the start of sorafenib treatment and resolving with the application of local therapies without requiring a change of sorafenib dosing of any patients. No grade 3/4 dermatological toxicities were observed. The incidence of hand–foot skin reaction tended to be dose‐dependent (Table 3).
Table 2.
Incidence of drug‐related adverse events by worst grade
| Adverse event | All grades n (%) | Grade 3 n (%) | Grede 4 n (%) |
|---|---|---|---|
| Hypertension | 4 (13%) | 1 (3.2%) | 0 |
| Fatigue | 3 (10%) | 1 (3.2%) | 0 |
| Fever | 3 (10%) | 0 | 0 |
| Alopecia | 8 (26%) | 0 | 0 |
| Dry skin | 7 (23%) | 0 | 0 |
| Hand–foot skin reaction | 12 (39%) | 0 | – |
| Rash/desquamation | 19 (61%) | 0 | – |
| Pruritus | 5 (16%) | 0 | 0 |
| Anorexia | 8 (26%) | 0 | 0 |
| Diarrhea | 11 (36%) | 1 (3.2%) | 0 |
| Nausea | 3 (10%) | 0 | 0 |
| Vomiting | 3 (10%) | 0 | 0 |
| Lipase | 11 (36%) | 2 (6.5%) | 5 (16%) |
| Amylase | 8 (26%) | 2 (6.5%) | 1 (3.2%) |
| Alkaline phosphatase (ALP) | 3 (10%) | 1 (3.2%) | 0 |
| Alanine amino transferase (ALT) | 3 (10%) | 1 (3.2%) | 1 (3.2%) |
| Aspartic aminotransferase (AST) | 3 (10%) | 1 (3.2%) | 2 (6.5%) |
| Abdominal pain | 5 (16%) | 0 | 0 |
| Leukocytopenia | 4 (13%) | 4 (13%) | 0 |
Table 3.
Incidence of common drug‐related adverse events by dose levels
| Adverse event | 100 mg n = 3 | 200 mg n = 15 | 400 mg n = 6 | 600 mg n = 7 |
|---|---|---|---|---|
| Hypertension | 0 | 2 (13%) | 1 (17%) | 1 (14%) |
| Fatigue | 0 | 1 (6.7%) | 1 (17%) | 1 (14%) |
| Alopecia | 0 | 3 (20%) | 3 (50%) | 2 (29%) |
| Dry skin | 0 | 4 (27%) | 3 (50%) | 0 |
| Hand–foot skin reaction | 0 | 3 (20%) | 3 (50%) | 6 (86%) |
| Rash/desquamation | 2 (67%) | 8 (53%) | 6 (100%) | 3 (43%) |
| Pruritus | 0 | 1 (13%) | 2 (33%) | 2 (29%) |
| Anorexia | 1 (33%) | 4 (27%) | 1 (17%) | 2 (29%) |
| Diarrhea | 0 | 6 (40%) | 3 (50%) | 2 (29%) |
| Lipase | 0 | 4 (27%) | 3 (50%) | 4 (57%) |
| Amylase | 0 | 3 (20%) | 3 (50%) | 2 (29%) |
The most common gastrointestinal adverse event was diarrhea (35%). It was mostly mild to moderate and easily managed with oral loperamide. However, grade 3 diarrhea (a DLT) occurred in one patient at the 200 mg bid dose level.
Elevation of lipase or amylase was not observed at the 100 mg bid dose level (Table 3). Of the 15 patients treated with 200 mg bid, four showed elevated lipase (27%) and three elevated amylase (20%). Two of these patients had grade 4 elevated lipase, but no indications of pancreatitis were observed by diagnostic imaging. Three of six patients (50%) in the 400 mg bid group and four of seven (57%) in the 600 mg bid group had elevated levels of pancreatic enzymes, which returned to normal without requiring interruption of sorafenib administration. Serum levels of amylase and lipase began increasing on days 4–7, and then decreased again back to normal levels within 3–10 days with/without stopping administration of sorafenib. No patients had symptoms of pancreatitis. Ultrasound, computed tomography, and magnetic resonance imaging of the pancreas showed no evidence of acute pancreatitis.
Hypertension was observed in four patients, with one occurrence of grade 3 at the 600 mg bid dose level. A causal relationship with the use of the study drug could not be ruled out. These events mostly began 1–7 weeks after the initial sorafenib treatment and returned to normal during continuous treatment thereafter. Treatment‐related abnormalities in hepatic parameters, such as ALT and AST elevations, were reported in two patients as serious adverse events, and drug administration had to be discontinued. Fatigue was reported in three patients including one case of dose‐limiting grade 3.
Pharmacokinetics. Pharmacokinetics data sets after the initial single dosing were obtained in a total of 31 patients. Thereafter, 25 patients were eligible for pharmacokinetics analysis on day 14 during the continuous dosing; six were excluded because of discontinuation of drug administration. The pharmacokinetic parameters of sorafenib are shown in Table 4. Drug absorption was moderate after the single administration, with time to maximum plasma concentration (Tmax) 3–24 h (mean, 8 h) after administration. Plasma half‐life (T1/2) was found to be 24–30 h (mean, 25.5 h). Although considerable interpatient variability was observed, the geometric means of AUC, AUC0‐12 as well as the maximum and trough concentrations increased dose dependently from 100 mg to 400 mg after administration of a single dose and at steady state (day 14). At 600 mg bid, drug exposure seemed to be increased less than proportionally to the dose escalation. Plasma trough concentrations at 400 mg bid (3.75 mg/L) exceeded the IC50 for inhibition of tumor cell proliferation in vitro (ranging between 0.057 and 2.5 mg/L).( 8 )
Table 4.
Plasma pharmacokinetic parameters of sorafenib
| Dose (mg bid) | day 1 | day 14 | day 28 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| AUC (mg h/L) | AUC0‐12 (mg h/L) | Cmax (mg/L) | Tmax (h) | T1/2 (h) | CL/f (L/h) | AUC0–12 (mg h/L) | Cmax (mg/L) | Ctrough (mg/L) | AUC0–12 (mg h/L) | Cmax (mg/L) | |
| 100 (n = 3) | 9.4 | 3.3 | 0.43 | 4 | 27.1 | 10.6 | 9.4 | 1.04 | 0.70 | 12.3 | 1.42 |
| (39) | (42) | (41) | (3–8) † | (39) | (39) | (21) | (30) | (43) | (27) | (35) | |
| 200 (n = 15) | 24.3 | 5.1 | 0.74 | 4 | 24.4 | 8.2 | 20.2 ‡ | 2.64 ‡ | 1.38 § | 21.1 ¶ | 2.43 †† |
| (100) | (110) | (107) | (3–24) † | (58) | (100) | (37) | (49) | (588) | (49) | (52) | |
| 400 (n = 6) | 35.4 | 7.0 | 1.21 | 8 | 25.5 | 11.3 | 36.7 | 4.91 | 3.75 | n/a | n/a |
| (195) | (173) | (201) | (3–24)† | (40) | (195) | (73) | (76) | (104) | |||
| 600 (n = 7) | 40.5** | 9.7 | 1.41 | 6 | 30.4 ‡‡ | 14.8 ‡‡ | 33.8 ‡‡ | 4.42 ‡‡ | 4.29 ‡‡ | n/a | n/a |
| (67) | (81) | (70) | (4–23) | (34) | (67) | (43) | (55) | (62) | |||
Data are expressed as geometric mean or median, and percent coefficient of variance is expressed in parentheses.
range;
n = 10,
n = 11,
n = 8,
n = 9,
n = 6 (Calculated by using the half of lower limit of quantification for one patient with Ctrough lower than the lower limit of quantification)
AUC, area under the curve; n/a, not available.
Major metabolites of sorafenib M‐2, M‐4, and M‐5 were detected in plasma, but the AUC0‐12 of each metabolite was less than 13% of the sum of all measured compounds (Table 5). Similar to sorafenib, the AUC0‐12 and Cmax of these metabolites were increased by dose escalations from 100 to 400 mg bid, but were not further increased at 600 mg bid. Sorafenib and M‐2 were not detectable in urine, while the glucuronidated metabolites, M‐7 and M‐8, were excreted in the urine at up to 4% of the administered dose of sorafenib (Table 6).
Table 5.
Plasma pharmacokinetic parameters of metabolites
| Dose (mg bid) | M‐2 (BAY 67‐3472) | M‐4 (BAY 43‐9007) | M‐5 (BAY 68‐7769) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| AUC0–12 (mg h/L) | Ratio (%) | Cmax (mg/L) | AUC0–12 (mg h/L) | Ratio (%) | Cmax (mg/L) | AUC0–12 (mg h/L) | Ratio (%) | Cmax (mg/L) | |
| 100 (n = 3) | 0.63 | 6.07 | 0.07 | 0.16 | 1.54 | 0.02 | 0.21 † | 2.04 † | 0.02 † |
| (57) | (74) | (45) | (40) | (25) | (23) | (54) | (78) | (71) | |
| 200 (n = 10) | 2.47 | 10.01 | 0.31 | 0.70 | 2.83 | 0.11 | 0.83 ‡ | 3.13 ‡ | 0.10 ‡ |
| (79) | (55) | (71) | (179) | (124) | (95) | (50) | (63) | (55) | |
| 400 (n = 6) | 5.84 | 11.7 | 0.73 | 1.89 | 3.81 | 0.24 | 1.79 | 3.60 | 0.22 |
| (269) | (63) | (285) | (324) | (81) | (353) | (563) | (144) | (573) | |
| 600 (n = 6) | 5.44 | 12.2 | 0.66 | 1.81 | 4.09 | 0.23 | 1.48 | 3.34 | 0.18 |
| (140) | (58) | (150) | (139) | (61) | (153) | (185) | (84) | (205) | |
Data are expressed as geometric mean, and percent coefficient of variance is expressed in parentheses.
Ratio of each metabolite to the sum of AUC0–12 of sorafenib, M‐2, M‐4, and M‐5.
n = 2;
n = 9.
AUC, area under the curve.
Table 6.
Urinary excretion of sorafenib and metabolites 48 h after single administration of sorafenib
| Dose (mg bid) | Sorafaneib (BAY 43‐9006) (%) | M‐2 (BAY 67‐3472) (%) | M‐7 (BAY 43‐9006G) (%) | M‐8 (BAY 67‐3472G) (%) |
|---|---|---|---|---|
| 100 | ND | ND | 4.15 (34) † | 0.09 (0) ‡ |
| 200 | ND | ND | 1.97 (55) § | 0.08 (99) ¶ |
| 400 | ND | ND | 1.66 (64) †† | 0.11 (99) ‡‡ |
| 600 | ND | ND | 1.70 (66) †† | 0.09 (120) †† |
Percent coefficient of variance is expressed in parentheses.
BAY 43‐9006G: BAY 43‐9006 glucuronide, BAY 67‐3472G: BAY 67‐3472 glucuronide.
n = 3,
n = 2,
n = 2,
n = 9,
n = 5,
n = 4.
ND, not detected.
Pharmacodynamics. ERK is an essential component of MAPK signaling pathways and a downstream factor of Raf kinase, which is a target molecule of sorafenib.( 7 , 8 ) Adrenomedullin is a bioactive peptide and known to be expressed/secreted by human tumors.( 13 , 14 ) In preclinical studies, expression of adrenomedullin decreased in tumors as the plasma concentration of sorafenib increased. Thus, phosphorylation of ERK and plasma adrenomedullin levels may be a candidate biomarker of sorafenib efficacy. Nevertheless, in the present study, large interindividual variations were observed in changes of pERK‐positive cells in PBMCs and also in plasma adrenomedullin levels, and no obvious trend was recognizable for these parameters (Table 7).
Table 7.
Plasma pharmacodynamics of sorafenib on day 28 of cycle 1
| 100 mg (n = 3) | 200 mg (n = 12) | 400 mg (n = 6) | 600 mg (n = 5) | |
|---|---|---|---|---|
| pERK+ (%) | 44.8 (10.3) | 43.6 (15.4) | 64.1 (29.6) | 57.5 (12.4%) |
| Adrenomedullin (fmol/mL) | 2.18 (0.62) † | 1.90 (0.67) | 2.97 (1.67) | 2.23 (0.61) |
Standard deviation is in parentheses.
pERK+ (phosphorylated ERK+) is expressed as percentage of positive cells in peripheral blood mononuclear cells.
n = 2.
Twenty‐three patients underwent repeated examination by FDG‐PET, with the median value of SUVmax decreasing significantly from 16.2 (range, 3.0–80.3) at the baseline to 11.2 (3.0–57.8) at the first examination after the start of treatment (P = 0.0007 by Wilcoxon signed‐rank test). The median percent change from baseline for each patient was –25% (–54% to 25%). SUVmax was decreased from baseline in 19 patients, with a 25% or greater decrease being observed in 11 patients. A higher trough concentration of sorafenib on day 28 was associated with larger reduction in SUVmax (Fig. 2). This relationship could be described by an Emax model with Emax = 130.1 (SE, 21.0)% and EC50 = 4.8 (2.4) mg/L.
Figure 2.
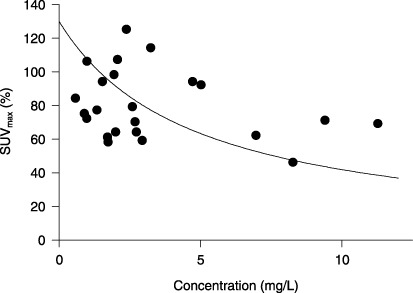
Relationship between the trough concentration of sorafenib and the maximum standardized uptake value (SUVmax) relative to the baseline.
Antitumor activity. Twenty‐nine patients were evaluated for tumor response according to RECIST criteria. Overall duration of treatment was prolonged as the dose was increased. PR was observed in two patients (total, 7%). In a 69‐year‐old patient with renal cell carcinoma previously treated with interferon‐α2b, PR was achieved 1 month after the start of continuous dosing (600 mg bid) and was maintained over 8 months. In another patient with non‐small cell lung cancer (NSCLC) who had been treated with cisplatin, vinorelbine, docetaxel, and gefitinib, tumor size gradually decreased and PR was achieved 11 months after the start of continuous dosing (200 mg bid), and was maintained for more than 20 months. Treatment was discontinued when a second cancer (small cell lung cancer) developed, which was surgically resected and treated with cisplatin and etoposide. The original NSCLC did not grow during the treatment course for a period exceeding 30 months. In addition to the PR, a total of 14 patients (48%) experienced SD. Four of 10 patients with non‐small cell lung cancer achieved SD for more than 6 months.
Discussion
The results of this study showed a favorable safety profile of sorafenib in Japanese patients with advanced refractory solid tumors. The most common drug‐related toxicities including rash/desquamation, hand–foot skin reactions, and diarrhea, and elevations of serum pancreatic enzyme levels were mostly mild to moderate. Dose‐limiting toxicities in this study were diarrhea and fatigue.
Dermatological adverse events were frequently observed. The most common drug‐related events were rash/desquamation (61%) and hand–foot skin reactions (39%), which were grade 2 or milder although their incidence was increased with dose escalation from 400 to 600 mg bid (Table 3). Another type of common toxicity was gastrointestinal, such as diarrhea and anorexia. Diarrhea was reported in 11 patients (36%) and one of them experienced a grade 3 dose‐limiting event. Previous phase I studies in Europe and the United States in patients with advanced refractory solid tumors (100–800 mg bid) showed similar drug‐related adverse events.( 15 , 16 , 17 , 18 , 19 ) The most frequently reported adverse events in four studies were fatigue (40%), anorexia (35%), diarrhea (34%), rash/desquamation (27%), and hand–foot skin reactions (25%). Similarly, the incidence rates of these drug‐related adverse events were higher in the 600 mg group. Diarrhea and fatigue were also dose‐limiting toxicities in those studies, and the most common drug‐related events were dermatological and gastrointestinal toxicities, which were comparable between Japanese and non‐Japanese patients.( 15 , 16 , 17 , 18 , 19 ) Similar to the previous phase I studies, the results of this study suggests that it is appropriate to recommend 400 mg bid for phase II studies in Japan.
Elevated lipase (36%) and amylase (26%) levels were also common drug‐related adverse events, and seven patients (23%) experienced grade 3 or worse. The incidences seemed to be dose‐dependent, suggesting that it was related to sorafenib. In a preclinical study, histological changes in the pancreas were observed. Such elevations have been rarely reported in previous clinical studies of sorafenib performed in other countries, where pancreatic enzyme levels were not routinely measured. Lack of symptoms and the transient nature of this toxicity could have led to underestimation in previous studies. The elevation of lipase was also reported in patients treated with sunitinib, a receptor tyrosine kinase inhibitor,( 20 ) which inhibits VEGFR‐2, PDGFR, Flt‐3, and c‐KIT.( 21 , 22 ) The mechanism of the elevation of pancreatic enzymes may be related to kinase inhibition or to some chemical property, rather than to inhibition of angiogenesis, because patients treated with bevacizumab, an anti‐VEGF antibody, did not experience this.( 23 , 24 ) Importantly, elevations of pancreatic enzyme levels did not cause any clinically relevant events. They were transient, and did not interrupt the sorafenib administration schedule in most patients in the present study. However, as pancreatitis was reported in other clinical trials of sorafenib,( 25 ) physicians treating patients with this drug need to recognize the possibility of occurrence of pancreatitis, although the diagnosis of pancreatitis should not be made solely on the basis of elevation of pancreatic enzymes.
The results of pharmacokinetic analysis suggested that the AUCs of sorafenib and metabolites were related to dose within the range of 100–400 mg bid, but with no further increase at 600 mg. Although the N‐oxide of sorafenib (M‐2) is the main drug metabolite in plasma, sorafenib exists in plasma mostly in an unchanged form. The ratio of the metabolite to the sum of the unchanged drug and three metabolites was 6–12%, which was lower than the 17% measured in healthy volunteers who received [14C]‐sorafenib.( 11 ) The difference might be related to variation in subjects, methodology, and the dose.
Preclinical data suggested that sorafenib is metabolized by CYP3A4. However, coadministration of ketoconazole, a CYP3A4 inhibitor, did not change the concentration of sorafenib in healthy volunteers. In this case, the formation of the main metabolite decreased, suggesting other metabolic pathways, such as glucuronidation.( 11 ) This is the first report that has investigated urinary excretion of sorafenib and metabolites in cancer patients. It was found that glucuronidated sorafenib and other glucuronidated metabolites but not sorafenib itself were in fact excreted in the urine. The amount of metabolites excreted in urine was 2–4%. Following oral administration of [14C]‐sorafenib to healthy volunteers, 19% of the dose was excreted as glucuronides in urine, and 77% in feces (50% as unchanged drug).( 11 ) A gain, variation in subjects, methodology, and the dose might explain the difference in the amount of drug excreted in urine. Consistent with the results of previous phase I studies in non‐Japanese patients, considerable interpatient variability in relation to the pharmacokinetics of sorafenib was observed in Japanese patients as well.( 15 ) Although drug exposures in Japanese patients were slightly lower than in non‐Japanese patients,( 15 ) available data suggest that no dosage adjustment due to ethnicity will be necessary.
We assessed pharmacodynamics in patients treated with sorafenib. ERK is a downstream kinase of Raf kinase, and when sorafenib inhibits Raf kinase, the phosphorylation levels of ERK may also be decreased.( 8 ) Previous clinical studies indicated a significant reduction of pERK levels with increasing sorafenib dose.( 18 ) In the present study, pERK‐positive cells within PBMCs were not found to change at any of the dose tested. In addition, adrenomedullin was suggested to be a biomarker of sorafenib in preclinical studies, but no significant changes were observed in our clinical study. In contrast, FDG‐PET analysis, performed one month after the start of continuous dosing, showed that treatment with sorafenib decreased disease activity in 83% of patients. Furthermore, reduction in FDG uptake was associated with drug exposure. These observations imply that FDG‐PET may be used as a surrogate endpoint. Validity of FDG‐PET in evaluating the activity of molecular targeted drugs needs to be further investigated.
Preliminary efficacy data in this study indicated one confirmed PR in a patient with renal cell carcinoma. Angiogenesis is suggested as an essential factor in the progression and metastasis of the disease.( 26 ) The anti‐VEGF antibody bevacizumab inhibits VEGF signalings and has demonstrated antitumor activity against renal cell carcinoma. Sorafenib targets VEGFR‐2 and PDGFR and inhibits angiogenesis. The efficacy of sorafenib for renal cell carcinoma has been demonstrated in a clinical phase III study (TARGET), in which it significantly prolonged progression‐free survival and overall survival.( 9 ) In addition, one PR in a patient with non‐small cell lung cancer was observed in the present study and SD for more than 24 weeks was achieved in four patients. These responses support a clinical benefit of sorafenib and suggest that further clinical studies are warranted in Japanese patients.
In conclusion, sorafenib was generally well tolerated, and continuous administration at a dose of 400 mg bid is recommended for further studies in Japanese patients.
References
- 1. Bos JL. RAS oncogenes in human cancer: a review. Cancer Res 1989; 49: 4682–9. [PubMed] [Google Scholar]
- 2. Hilger RA, Scheulen ME, Strumberg D. The Ras‐Raf‐MEK‐ERK pathway in the treatment of cancer. Onkologie 2002; 25: 511–18. [DOI] [PubMed] [Google Scholar]
- 3. Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer 2003; 3: 11–22. [DOI] [PubMed] [Google Scholar]
- 4. Kolch W, Kotwaliwale A, Vass K et al . The role of Raf kinases in malignant transformation. Expert Rev Mol Med 2002: 1–18. [DOI] [PubMed] [Google Scholar]
- 5. Perona R. Cell signalling: growth factors and tyrosine kinase receptors. Clin Transl Oncol 2006; 8 (2): 77–82. [DOI] [PubMed] [Google Scholar]
- 6. Homsi J, Daud AI. Spectrum of activity and mechanism of action of VEGF/PDGF inhibitors. Cancer Control 2007; 14 (3): 285–94. [DOI] [PubMed] [Google Scholar]
- 7. Wilhelm S, Chien DS. BAY 43‐9006: preclinical data. Curr Pharm Des 2002; 8: 2255–7. [DOI] [PubMed] [Google Scholar]
- 8. Wilhelm SM, Carter C, Tang L et al . BAY 43‐9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res 2004; 64: 7099–109. [DOI] [PubMed] [Google Scholar]
- 9. McKeage K, Wagstaff AJ. Sorafenib: in advanced renal cancer. Drugs 2007; 67 (3): 475–83. [DOI] [PubMed] [Google Scholar]
- 10. Therasse P, Arbuck SG, Eisenhauer EA et al . New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 2000; 92: 205–16. [DOI] [PubMed] [Google Scholar]
- 11. Lathia C, Lettieri J, Cihon F et al . Lack of effect of ketoconazole‐mediated CYP3A inhibition on sorafenib clinical pharmacokinetics. Cancer Chemother Pharmacol 2006; 57: 685–92. [DOI] [PubMed] [Google Scholar]
- 12. Chow S, Patel H, Hedley DW. Measurement of MAP kinase activation by flow cytometry using phospho‐specific antibodies to MEK and ERK. potential for pharmacodynamic monitoring of signal transduction inhibitors. Cytometry 2001; 46: 72–8. [DOI] [PubMed] [Google Scholar]
- 13. Hinson JP, Kapas S, Smith DM. Adrenomedullin, a multifunctional regulatory peptide. Endocr Rev 2000; 21: 138–67. [DOI] [PubMed] [Google Scholar]
- 14. Nikitenko LL, Fox SB, Kehoe S et al . Adrenomedullin and tumour angiogenesis. Br J Cancer 2006; 94: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Strumberg D, Clark JW, Awada A et al . Safety, pharmacokinetics, and preliminary antitumor activity of sorafenib: a review of four phase I trials in patients with advanced refractory solid tumors. Oncologist 2007; 12 (4): 426–37. [DOI] [PubMed] [Google Scholar]
- 16. Awada A, Hendlisz A, Gil T et al . Phase I safety and pharmacokinetics of BAY 43‐9006 administered for 21 days on/7 days off in patients with advanced, refractory solid tumours. Br J Cancer 2005; 92: 1855–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Moore M, Hirte HW, Siu L et al . Phase I study to determine the safety and pharmacokinetics of the novel Raf kinase and VEGFR inhibitor BAY 43‐9006, administered for 28 days on/7 days off in patients with advanced, refractory solid tumors. Ann Oncol 2005; 16: 1688–94. [DOI] [PubMed] [Google Scholar]
- 18. Clark JW, Eder JP, Ryan D et al . Safety and pharmacokinetics of the dual action Raf kinase and vascular endothelial growth factor receptor inhibitor, BAY 43‐9006, in patients with advanced, refractory solid tumors. Clin Cancer Res 2005; 11: 5472–80. [DOI] [PubMed] [Google Scholar]
- 19. Strumberg D, Awada A, Hirte H et al . Pooled safety analysis of BAY 43‐9006 (sorafenib) monotherapy in patients with advanced solid tumours: Is rash associated with treatment outcome? Eur J Cancer 2006; 42: 548–56. [DOI] [PubMed] [Google Scholar]
- 20. Motzer RJ, Rini BI, Bukowski RM et al . Sunitinib in patients with metastatic renal cell carcinoma. JAMA 2006; 295: 2516–24. [DOI] [PubMed] [Google Scholar]
- 21. O’Farrell AM, Foran JM, Fiedler W et al . An innovative phase I clinical study demonstrates inhibition of FLT3 phosphorylation by SU11248 in acute myeloid leukemia patients. Clin Cancer Res 2003; 9: 5465–76. [PubMed] [Google Scholar]
- 22. Mendel DB, Laird AD, Xin X et al . In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet‐derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res 2003; 9: 327–37. [PubMed] [Google Scholar]
- 23. Caprioni F, Fornarini G. Bevacizumab in the treatment of metastatic colorectal cancer. Future Oncol 2007; 3: 141–8. [DOI] [PubMed] [Google Scholar]
- 24. Sanborn RE, Sandler AB. The safety of bevacizumab. Expert Opin Drug Saf 2006; 5: 289–301. [DOI] [PubMed] [Google Scholar]
- 25. Strumberg D, Richly H, Hilger RA et al . Phase I clinical and pharmacokinetic study of the Novel Raf kinase and vascular endothelial growth factor receptor inhibitor BAY 43‐9006 in patients with advanced refractory solid tumors. J Clin Oncol 2005; 23: 965–72. [DOI] [PubMed] [Google Scholar]
- 26. Larkin JM, Chowdhury S, Gore ME. Drug insight: advances in renal cell carcinoma and the role of targeted therapies. Nat Clin Pract Oncol 2007; 4: 470–9. [DOI] [PubMed] [Google Scholar]