

Abstract
The exchange coupling of electron spins can strongly influence the properties of chemical species. The regulation of this type of electronic coupling has been explored within complexes that have multiple metal ions but to a lesser extent in complexes that pair a redox-active ligand with a single metal ion. To bridge this gap, we investigated the interplay among the structural and magnetic properties of mononuclear Cu complexes and exchange coupling between a Cu center and a redox-active ligand over three oxidation states. The computational analysis of the structural properties established a relationship between the complexes’ magnetic properties and a bonding interaction involving a orbital of the Cu ion and orbital of the redox-active ligand that are close in energy. The additional bonding interaction affects the geometry around the Cu center and was found to be influenced by intramolecular H-bonds introduced by the external ligands. The ability to synthetically tune the d- interactions using H-bonds illustrates a new type of control over the structural and magnetic properties of metal complexes.
Graphical Abstract
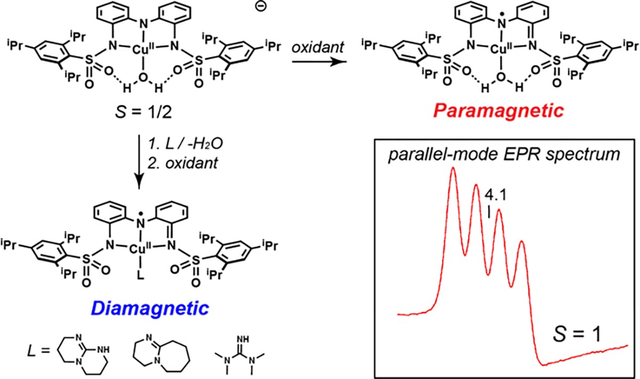
Introduction
Multi-electron processes are prevalent in thermodynamically challenging chemical transformations.1–3 Synthetically, these processes commonly use complexes containing second- and third-row metal ions to supply the necessary redox equivalents. The thermodynamic limitations of first-row metal ions hinder their usage in these types of reactions; however, their natural abundance makes them more accessible than precious metals, and efforts to prepare complexes containing first-row metal ions that can provide reactivity comparable to that of precious metal complexes have been underway for several decades. To overcome the constraints of first-row metal ions, two routes are often utilized: multiple metal ions can be included in the catalytically competent species, or an alternative source of redox equivalents can be supplied through the coordination of a redox-active ligand.4,5 This class of ligands has been employed with notable successes in a variety of metal complexes and is recognized to produce systems with unusual electronic and functional properties (Figure 1A–D).6–19 A key feature of these systems is the occurrence of electronic coupling between the metal center and the redox-active ligand, which potentially provides a means of regulating the physical and functional properties of the complexes.
Figure 1.
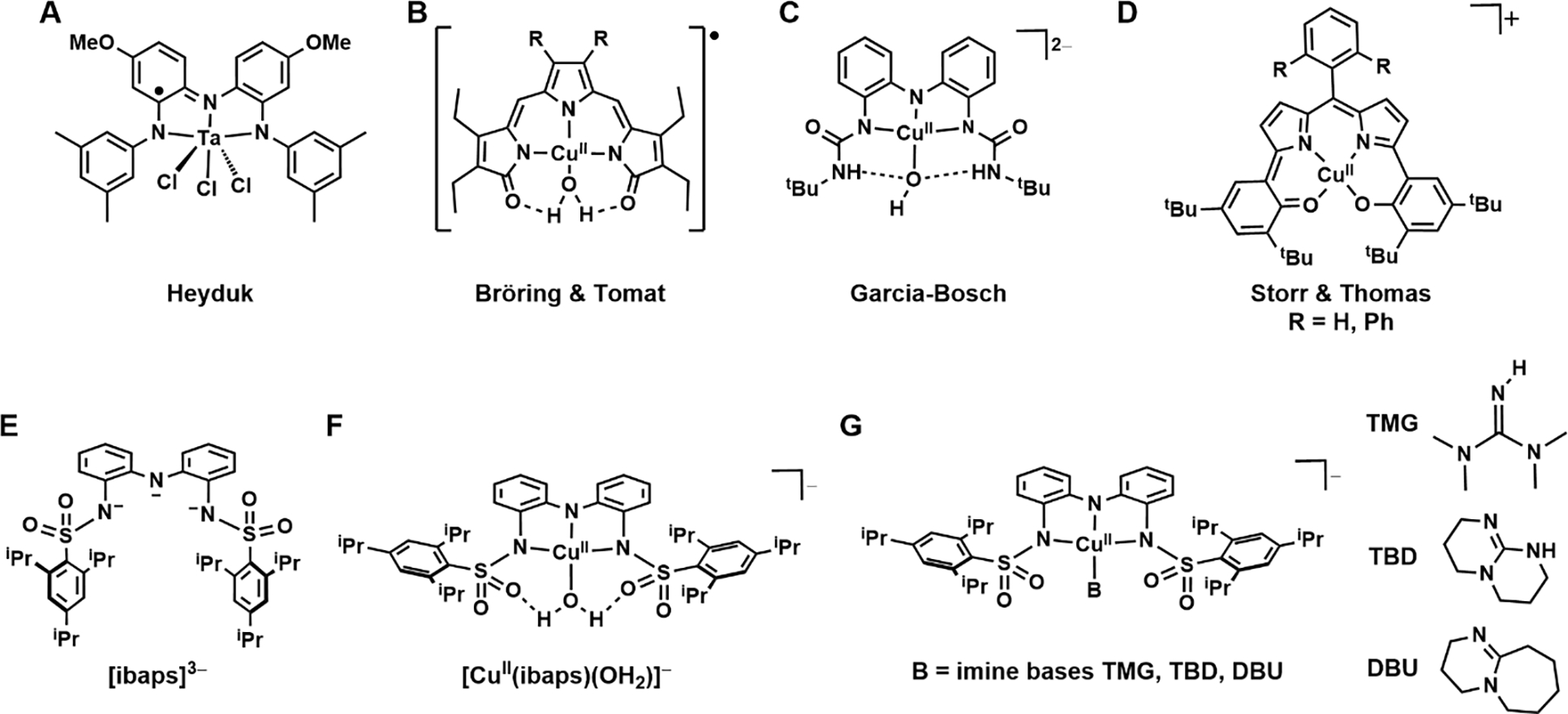
Synthetic metal complexes from the literature (A-D); the redox-active ligand (E) and complexes used in this study (F,G).
Pincer-type ligands, including those with multiple N-atom donors (denoted [NNN]), are often used as redox sources.20 Heyduk has shown the utility of redox-active ligands in establishing reactivity towards small molecules. This utility is demonstrated by a Ta(V) complex that, despite being d0, exhibits reactivity that is attributed solely to the [NNN] redox-active ligand (Figure 1A).6,21 Work by Bröring and Tomat shows that the tripyrrindione ligand precursor hexaethyltripyrrin-1,14-dione (H3TD1) coordinates to CuII centers as a radical dianion in the one-electron-oxidized form, which can supply an additional electron (Figure 1B).7,8 The primary coordination sphere of the CuII center is completed by an aqua ligand that forms two intramolecular hydrogen bonds (H-bonds) with the [TD1·]2− ligand. Despite H-bonds being noncovalent interactions, they have been shown to impact the structure and function of many systems.22–27 This secondary coordination sphere motif demonstrated by the [TD1·]2− ligand is also applied in related systems by Garcia-Bosch, where a diurea pincer ligand interacts with either a hydroxido or aqua ligand within a Cu–OHx system (Figure 1C); reactivity toward phenols was found for the oxidized Cu–OH species in which the oxidative equivalents were stored within the redox-active ligand framework.9 Similarly, a four-coordinate redox-active ligand from Storr and Thomas that can store two oxidizing equivalents has been used to prepare Cu complexes that exhibit reactivity toward organic substrates (Figure 1D).10
The presence of multiple sites capable of redox chemistry within a single molecule affords the possibility of intramolecular exchange coupling of the electron spins. This coupling can influence the properties of the molecule, including its overall spin state, in ways unattainable by molecules containing a single redox site. Previous investigations have compared the magnetic properties of related compounds containing redox-active ligands,28–32 but the modulation of exchange coupling by varying an ancillary ligand has been understudied in synthetic systems. This type of modulation is observed in heme proteins, which contain an axially coordinated ligand that directs the mode of electronic coupling between the metal ion and redox-active porphyrin. For example, within high-valent Fe–oxido intermediates in heme enzymes, the axial thiolate of the mono-oxygenases cytochrome P450s (P450s) imparts the active intermediate (compound I) with an spin ground state via antiferromagnetic coupling between the FeIV center () and porphyrin cation radical,33 whereas the tyrosinate axial ligand of heme catalases enforces ferromagnetic coupling and therefore an spin ground state.34
To explore whether exchange coupling can similarly be controlled within synthetic non-heme complexes, we have investigated the magnetic properties of CuII complexes with the redox-active ligand N,N’-(azanediyly-bis(2,1-phenylene))bis(2,4,6-triisopropyl-benzene-sulfonamido) ([ibaps]3−, Figure 1E) having a series of ancillary ligands (Figures 1F & 1G). We previously demonstrated that Fe, Ga, and Ni complexes of [ibaps]3− have rich electrochemical properties and that the sulfonamido groups can be H-bond acceptors to promote intramolecular H-bond formation.35,36 Examination of the redox and structural properties of the Cu complexes reported here allowed us to monitor differences in the electronic structure with four different ancillary ligands that have 0, 1, or 2 H-bond donors over three oxidation levels. Owing to the similar energies of the orbital of the CuII center and the redox-active orbital of [ibaps]3−, a nascent bond forms between a tilted orbital and the pz orbital of the central N atom of [ibaps]3−, which we discovered promotes molecular distortions away from the customary square planarity of four-coordinate CuII complexes. These distortions and their effect on the electronic properties of the complexes are shown to correlate with the spin state of the 1e-oxidized complexes. Our findings further illustrate the influence of intramolecular H-bonds on molecular structures and their applicability in regulating the magnetic properties of metal complexes.
The molecular distortions observed in the series of Cu complexes are also shown to correlate with the experimental hyperfine splittings of CuII. The hyperfine interactions in CuII complexes are commonly probed with electron paramagnetic resonance (EPR) spectroscopy, and the spectra typically show resolved hyperfine splittings from only one component of the hyperfine tensor (Az). In our study, EPR data were collected at multiple microwave frequencies, which allowed us to determine the full A-tensor and extract the spin-dipolar contributions to the A-tensors. We introduce a new methodology for connecting experimental measurements to density functional theory (DFT)-calculated structures of copper complexes. With this methodology, we identified the metal orbitals containing the unpaired electrons and determined the relative spin density at the metal ion. This analysis established a link between the molecular distortions and the resultant interactions of the unpaired electron in the orbital of the CuII center with the orbital of [ibaps]3−, and this link was experimentally validated through the magnetic hyperfine tensors and the exchange interactions within the complexes.
Results
Preparative Methods of Bu4N[CuII(ibaps)(L)] (1-L, where L = OH2, DBU, TMG, TBD).
The ligand precursor, H3ibaps, was deprotonated with 3 equiv of KH in tetrahydrofuran (THF) and then treated with CuII(OAc)2 and water to generate K[1-OH2] and 2 equiv of KOAc, which was removed via filtration (Scheme 1). Without isolation, this potassium salt was metathesized with tetrabutylammonium bromide to afford Bu4N[1-OH2] in 85 % crystalline yield after recrystallization. To prepare the CuII–imine complexes, a similar synthetic procedure was followed in which the appropriate imine (DBU, TMG, TBD) was used in place of water – all the Cu-imine complexes were isolated in yields greater than 80 %. We also found that the 1-imine complexes could be prepared by treating 1-OH2 with the appropriate imine ligand in yields that are comparable to those obtained from the more direct route.
Scheme 1.
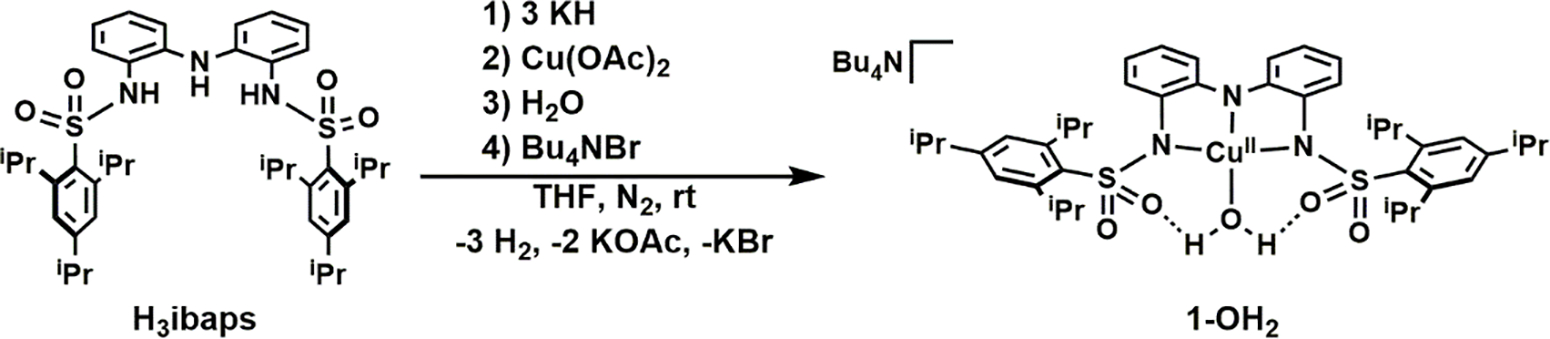
Synthetic route for the preparation of 1-OH2.
Molecular Structures of 1-OH2 and 1-DBU.
The molecular structure of 1-OH2 was determined using X-ray diffraction (XRD) methods to reveal a 4-coordinate CuII center in a geometry slightly distorted from ideal square planar ( = 0.22, where = 0 for square planar and 1 for tetrahedral, Figure 2, Tables 1 and S1).37 The nitrogen donor atoms of [ibaps]3− are coordinated in a near-meridional fashion to the CuII center, with an aqua ligand occupying the final coordination site. The Cu1–N1 bond distance of 1.874(1) Å is significantly shorter than the Cu–N2/3 bond lengths, which average 1.997(1) Å. The Cu–O1 bond length is 1.960(1) Å, which is similar to the distance observed in [Cu(TD1•)(H2O)] (Cu–O bond length of 1.958 – 1.971 Å).7 In addition, the phenyl groups within the backbone of [ibaps]3− are slightly skewed, as indicated by an angle of 158° between the planes of the rings.
Figure 2.
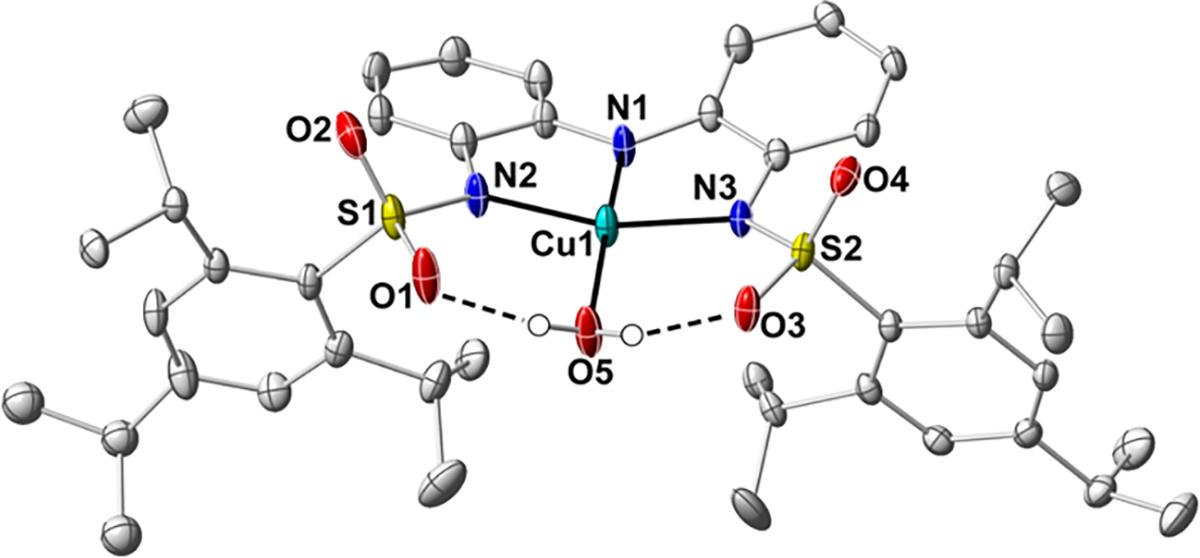
Thermal ellipsoid diagram depicting the molecular structure of 1-OH2 (CCDC Code: 2291042). The thermal ellipsoids are drawn at the 50 % probability level, and implicit hydrogen atoms and the TBA cation are removed for clarity.
Table 1.
Selected Metrical Parameters for 1-OH2
| Bond Distances (Å) and Angles (°) | |
|---|---|
| Cu1-N1 | 1.874(1) |
| Cu1-N2 | 1.993(1) |
| Cu1-N3 | 2.000(1) |
| Cu1-O1 | 1.960(1) |
| O1…O5 | 2.624(1) |
| O3…O5 | 2.610(1) |
| N1-Cu1-O5 | 166.01(6) |
| N2-Cu1-N3 | 163.48(6) |
The structure of 1-OH2 also revealed the existence of two intramolecular H-bonds, which form between the aqua ligand and the sulfonamido groups of [ibaps]3−, as indicated by the O1…O5 and O3…O5 distances of 2.624(2) and 2.610(2) Å, respectively. The presence of these H-bonds influences the overall structure of 1-OH2. For instance, the O atom of the aqua ligand is displaced out of the square plane (N1-Cu1-O5, 166.01(6)°), and an S=O unit on each sulfonamido group is rotated inward to accommodate the formation of the H-bonds. This structural constraint positions the 2,4,6-triisopropylphenyl groups in a syn conformation, and within the crystal lattice, they are nearly coplanar (the angle between the planes of the rings is 172.7°). We note that the molecular structure of a related Ni complex, [NiII(ibaps)(OH2)]−, also has analogous intramolecular H-bonds, but the sulfonamido groups are in an anti-conformation.36 It thus appears that either conformation can support H-bonds to the aqua ligand.
We also obtained the molecular structure of 1-DBU and the overall resolution was low because of solvent disorder within the lattice. Although caution should be taken in comparing metrical parameters, the primary coordination sphere around the Cu was well-ordered, allowing the overall coordination geometry and selected metrical parameters to be qualitatively compared to those of 1-OH2 (Figures 3A and S1A, Tables S1 and S2). The structure of 1-DBU revealed a four-coordinate species in which three coordination sites are again occupied by [ibaps]3−, and the final site is occupied by the external DBU ligand, which is bound trans to N1 (Figures 3A and S1A). As the DBU ligand lacks any H-bond donors, no intramolecular H-bonds are observed in the molecular structure, allowing for more rotation around the Cu ion than is possible in 1-OH2. The bond lengths around the Cu center are similar to those in 1-OH2; however, the overall coordination geometry is distorted further away from square planar. For instance, the N1-Cu1-N4 and N2-Cu1-N3 bond angles contract to 159.3(3) and 154.6(2)°, respectively, and the τ4 value increases to 0.33. Moreover, the appended sulfonamido groups of 1-DBU are in an anti-conformation, again showing their flexibility around the exogenous ligand.
Figure 3.
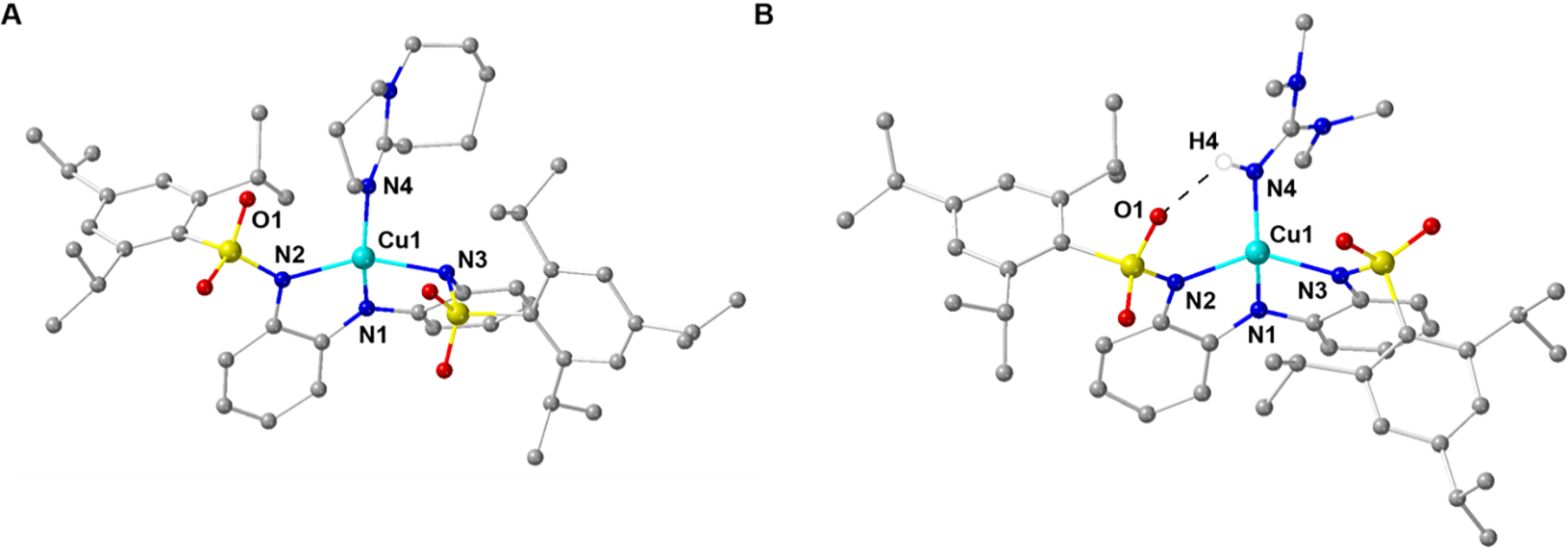
Ball and stick depictions of the molecular structures for 1-DBU and 2-TMG (CDCC Codes: 2291043 and 2291041). The counterion and hydrogen atoms are removed for clarity with the exception of the hydrogen atom in 2-TMG that is involved in an H-bond. Selected bond angles (°) for 1-DBU (2-TMG): N1-Cu1-N4, 159.3(3) (148.5(2)); N2-Cu1-N3, 154.6(2) (142.8(2)).
Electrochemical Properties.
Cyclic voltammetry (CV) was used to evaluate the redox properties of 1-OH2 in solution, which are referenced to the [FeCp2]+/0 couple (Figures 4 and S2, Tables 2 and S3). Two reversible one-electron processes were observed in DCM, with E1/2 values of −0.48 V (ia/ic = 0.81, ΔE = 93 mV) and +0.44 V (ia/ic = 1.1, ΔE = 98 mV); similar electrochemical events were found in THF (Table S3). For comparison, [Cu(TD1•)(H2O)] also has two redox events at E1/2 = −0.6 and −0.052 V that are assigned to the (TD1•) ligand.8 The remaining 1-TBD, 1-TMG, and 1-DBU complexes also exhibited a reversible redox process that is negative of the [FeCp]+/0couple (Figures S3–S5, Tables 2 and S3), with each process occurring at potentials more negative than those in 1-OH2 (Table S3). A second oxidative event was observed for the 1-imine complexes but was only 1-TMG approached reversibility (see SI). We provide spectroscopic evidence that indicates that these redox processes are ligand-localized (see below), which is supported by DFT calculations that reproduce these redox potentials (see SI and Table S4).
Figure 4.
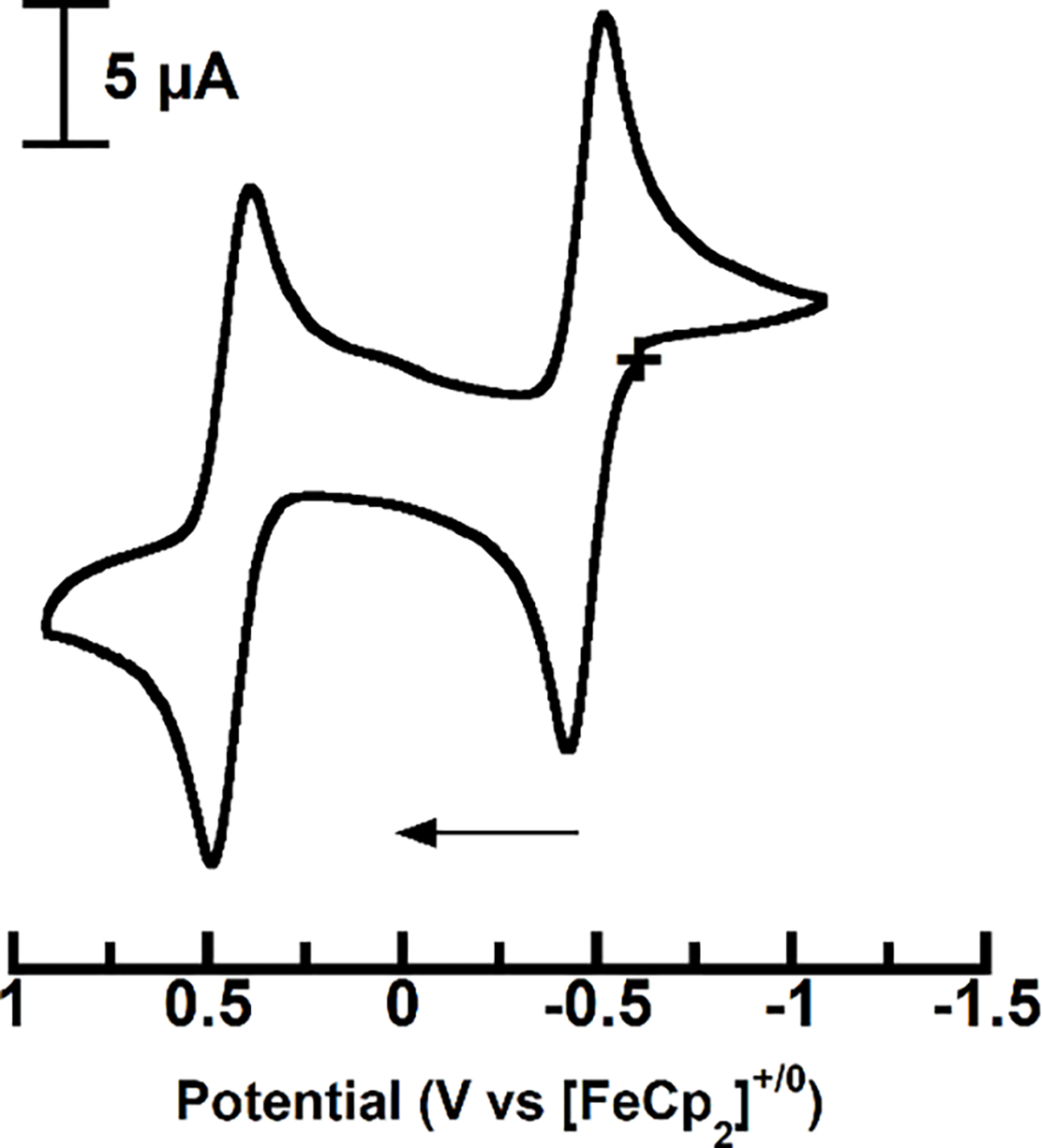
Cyclic voltammogram of 1-OH2 at room temperature in DCM.
Table 2.
Redox Properties of 1-L in DCM and relevant literature values.a
Preparation and Properties of the One-Electron (2-L) and Two-Electron (3-L) Oxidized Complexes.
Synthetic routes to the one-electron-oxidized complexes (2-L) were successfully pursued for all species using ferrocenium as the oxidant in DCM, and the desired complexes were isolated in yields that ranged from 33 – 87 % (Scheme 2A, SI). The singly oxidized complexes are stable in the solid state and in a 1:1 DCM:THF mixture for >24 hours under an anaerobic environment (Figure S6). We obtained single crystals suitable for analysis by XRD methods for only 2-TMG; the crystals were grown at −30°C (Figure 3B, Figure S1B). As expected, the Cu center is bound to four N atoms with a single intramolecular H-bond between the NH group of TMG and an O atom in the sulfonamido group of [ibaps]3−. In comparison to the molecular structure of 1-OH2, the geometry around the Cu center is more distorted from square planar, with N1-Cu1-N4 and N2-Cu1-N3 angles of 148.5(2)° and 142.8(2)°, respectively. The t4 value of 0.49 for 2-TMG is larger than that for 1-OH2 and 1-DBU. These distortions support the premise that the two H-bonds within 1-OH2 are enforcing the nearly planar coordination geometry around the Cu center.
Scheme 2.
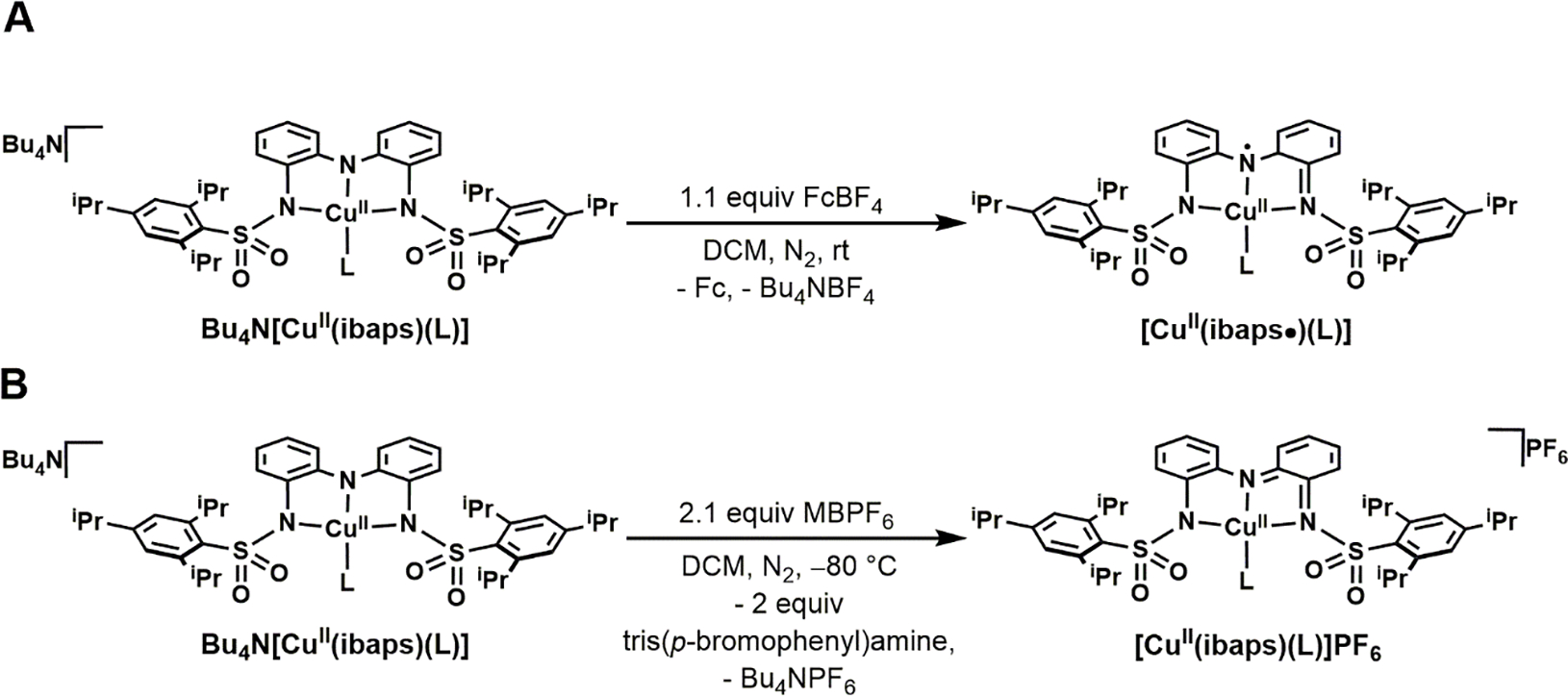
Preparative routes of 2-L (A) and 3-L (B) complexes.
The doubly oxidized complexes were generated using two equivalents of tris(p-bromophenyl)-ammoniumyl hexafluorophosphate (MBPF6) (E1/2 = 0.70 V vs [FeCp2]+/0) in DCM at −80 °C (Scheme 2B). The two systems with reversible redox processes afforded stable complexes, 3-OH2 and 3-TMG; however, 3-TBD and 3-DBU were unstable under these conditions, consistent with the irreversibility of their second oxidation event observed by CV and were not further studied.
Infrared Studies.
Solid-state Fourier transform infrared (FTIR) spectra of 1-L and 2-L were obtained to investigate the H-bonding interactions of the exogenous ligands (Figure S7). The 1-OH2 complex showed a broad, weak ν(OH) at 3180 cm–1 that shifted to 2424 cm−1 upon deuteration (ν(OH)/ν(OD) = 1.31; calcd. 1.37, Table 3, Figure S7). The feature for 2-OH2 was observed at a slightly higher energy of 3225 cm–1 and was also isotopically sensitive. FTIR spectra of the imine compounds excluding 1-DBU also revealed broad, weak features, which are attributed to the NH moieties; these features appear at 3300 cm–1 and 3335 cm–1 for 1-TBD and 1-TMG, respectively. Oxidation of the imine complexes also shifted the ν(NH) bands to slightly higher energies (Table 3). The broadness and relatively low energies of the and ν(NH) features are attributed to the presence of intramolecular H-bonds to the exogenous ligand that were observed in the molecular structures of 1-OH2 and 2-TMG and that are presumed to be present in the other complexes.38–40 Moreover, the changes observed in these frequencies upon oxidation could be caused by a weakening of the H-bonds. The FTIR spectra of 1-DBU and 2-DBU do not contain any features higher in energy than 3050 cm–1, consistent with the absence of NH bonds in these systems.
Table 3.
Select Spectroscopic Properties of the 1-L, 2-L, and 3-L Complexes.
| Complex | λmax/nm (εM/M−1 cm−1) | g-value(s)b | ACu (MHz)a,b | AN b,d (MHz) | ν(OH or NH) (cm−1c | ν(OD or ND) (cm−1)c |
|---|---|---|---|---|---|---|
| 1-OH2 | 316 (24000), 341 (30000), 500 (360), 540 (330), 990 (260)f | 2.03, 2.06, 2.20 | −50, −50, −570 (−220)a | 39, 44 | 3180 | 2424 |
| 1-TBD | 316 (20000), 340 (25000), 430 (sh), 545 (900), 730 (680), 995 (390)f | 2.04, 2.05, 2.21 | −175, 40, −375 (−170) | 41, 35 | 3300 | 2480 |
| 1-TMG | 347 (29000), 340 (25000), 425 (2100), 525 (2100), 1000 (700)f | 2.04, 2.06, 2.20 | −10, −20, −460 (−160) | 40, 29 | 3330 | 2480 |
| 1-DBU | 317 (23000), 340 (29000), 488 (500), 550 (600), 697 (320), 990 (300)f | 2.03, 2.06, 2.21 | −110, 55, −420 (−160) | 41, 35 | NA | NA |
| 2-OH2 | 346 (20000), 380 (16000), 480 (2800), 532 (3300), 685 (3300), 965 (8400)f | 4.1e | 195e | NA | 3225 | 2424 |
| 2-TBD | 343 (21000), 472 (5200), 731(10000), 910sh (2700)f | NA | NA | NA | 3330 | 2480 |
| 2-TMG | 342 (26000), 450 (6900), 665 (7100), 795 (8100), 920 (5600)f | NA | NA | NA | 3360 | 2490 |
| 2-DBU | 341 (24000), 472 (6100), 718 (10600), 865 (5300)f | NA | NA | NA | NA | NA |
| 3-OH2 | 387 (12000), 485 (28000), 530sh (17000), 610 (8900), 726 (9100), 840sh (14000), 938 (20000), 1075 (22000)g | 2.04, 2.08, 2.26 | −45, −30, −480 (−185) | 37, 36 | NA | NA |
| 3-TMG | 479 (20000), 525sh (13000), 582sh (7100), 735 (7700), 920 (12000), 1010 (13000), >1100 (~15000)g | 2.02, 2.06, 2.22 | 0, 0, −410 (−140) | - | NA | NA |
(Aiso).
In DCM:THF.
In solid-state.
Isotropic values of the 2 (1-OH2) or 3 equivalent 14N atoms, the second value is for the lone inequivalent 14N atom.
Observed z-component values from S = 1.
In DCM:THF at 22 °C
In neat DCM at −80
Electronic Absorption Properties.
The electronic absorption spectra of the 1-L complexes in 1:1 DCM:THF at room temperature exhibited relatively weak features centered around 340 nm, ~530 nm, and 1000 nm (Figure 5A, Table 3). Time-dependent (TD)-DFT analysis (see below) assigned these bands to primarily ligand to CuII charge transfer transitions. The absorption spectra for the 2-L complexes contain several new, more intense bands, notably a low-energy feature with values that range from 865 to 965 nm and molar extension coefficients between 2700 and 8400 M–1cm–1 (Figure 5B, Table 3, Figures S8–S11). Complexes 2-TBD, 2-TMG, and 2-DBU could also be generated through the addition of the imine reagent to 2-OH2 (Figures S12–14). Note that the one-electron oxidation processes to generate 2-L are reversible: reversion to the spectra of the 1-L complexes was accomplished using cobaltocene (Figures S15–S18). Bands with similar values and extinction coefficients as those of the 2-L complexes have been reported for other complexes with [NNN]3− redox-active ligands and have been assigned to ligand-centered transitions. For instance, comparable features have been observed in complexes from the Heyduk group with (4500 M–1cm–1),21 the [Cu(TD1•)(H2O)] complex from the Tomat group with ,8 and tBuCuOH from the Garcia-Bosch group at (4000 M–1cm–1).9 Moreover, we have previously characterized the one-electron-oxidized complex [Fe(ibaps)(bpy)], which also exhibited an NIR feature at (4080 M–1cm–1) that was attributed to a ligand-centered radical.35 Finally, the spectral features of the two-electron-oxidized species 3-OH2 and 3-TMG (Figure 5C, Table 3) are similar to those of the [CuII(RL)]+ complexes from Storr and Thomas, where R = H or Ph (Figure 1D).10,41 Both of these [CuII(RL)]+ species from Storr and Thomas were determined to have ligands that were oxidized by two electrons, and their electronic absorption characteristics were assigned to ligand-based transitions.
Figure 5.
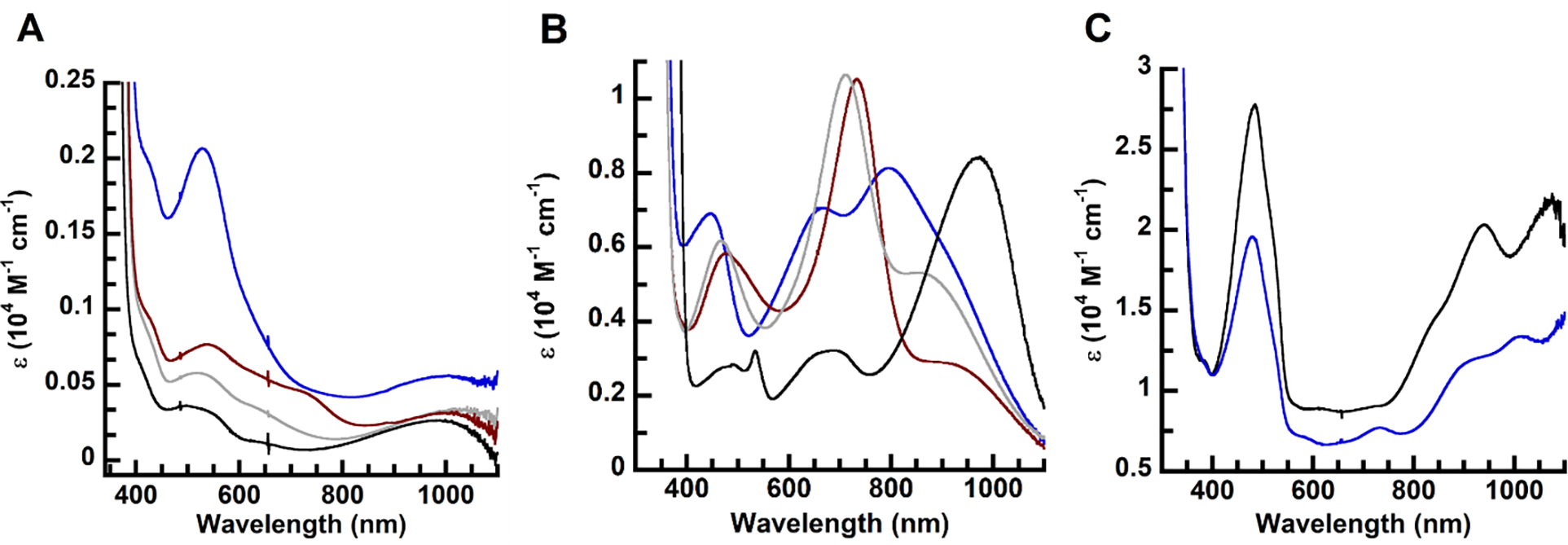
Electronic absorbance spectra of the (A) 1-L species and (B) 2-L species in 1:1 DCM:THF at 22 °C and (C) the 3-L species in neat DCM at −80 °C. L= aqua (black), DBU (gray), TMG (blue), and TBD (red).
Magnetic Properties.
X- and S-band EPR spectra (Figures 6, 7) of the 1-L complexes showed signals indicative of spin ground states. From the spectra collected at two microwave frequencies, the full g and hyperfine A-tensors of the complexes could be determined (Table 3). As expected, the lower microwave frequency (S-band) enhanced the resolution of the 63/65Cu and 14N hyperfine splittings. For each complex, the second derivative of the S-band spectrum further enhanced the resolution of the 14N hyperfine splitting (Figure 7, green spectra). Simulations were generated for the spectra via simultaneous least-squares fitting of the X-band, S-band, and S-band 2nd derivative spectra and are overlaid on the data (thin black lines). The spectral fits included second-order perturbation of the Cu hyperfine term in the spin Hamiltonian because of the large 63/65Cu hyperfine constants. The simulations were sensitive to the number of 14N nuclei within each complex, allowing us to determine the total number of N atom donors coordinated to the Cu center in each complex. For 1-OH2, the best fits occurred when three 14N nuclei were included and two of them were assumed to be equivalent. DFT calculations supported this assumption, as the N atoms of the sulfonamido groups had approximately equal spin densities, which were smaller than that found on the central amine nitrogen. For the 1-imine complexes, the simulations required four 14N nuclei, which was the initial indication that the added imines had substituted for the water ligand rather than deprotonating it. DFT calculations found that the coordinated N atoms of the imine ligands (TBD, TMG, and DBU) gave a significantly lower spin density than the N atoms of [ibaps]3−. Based on this finding, the 1-imine simulations assumed that three of the four coordinated N atoms were equivalent. The isotropic values of their 14N A-tensors (Table 3) are typical of Cu complexes with nitrogen-donating ligands.42
Figure 6.
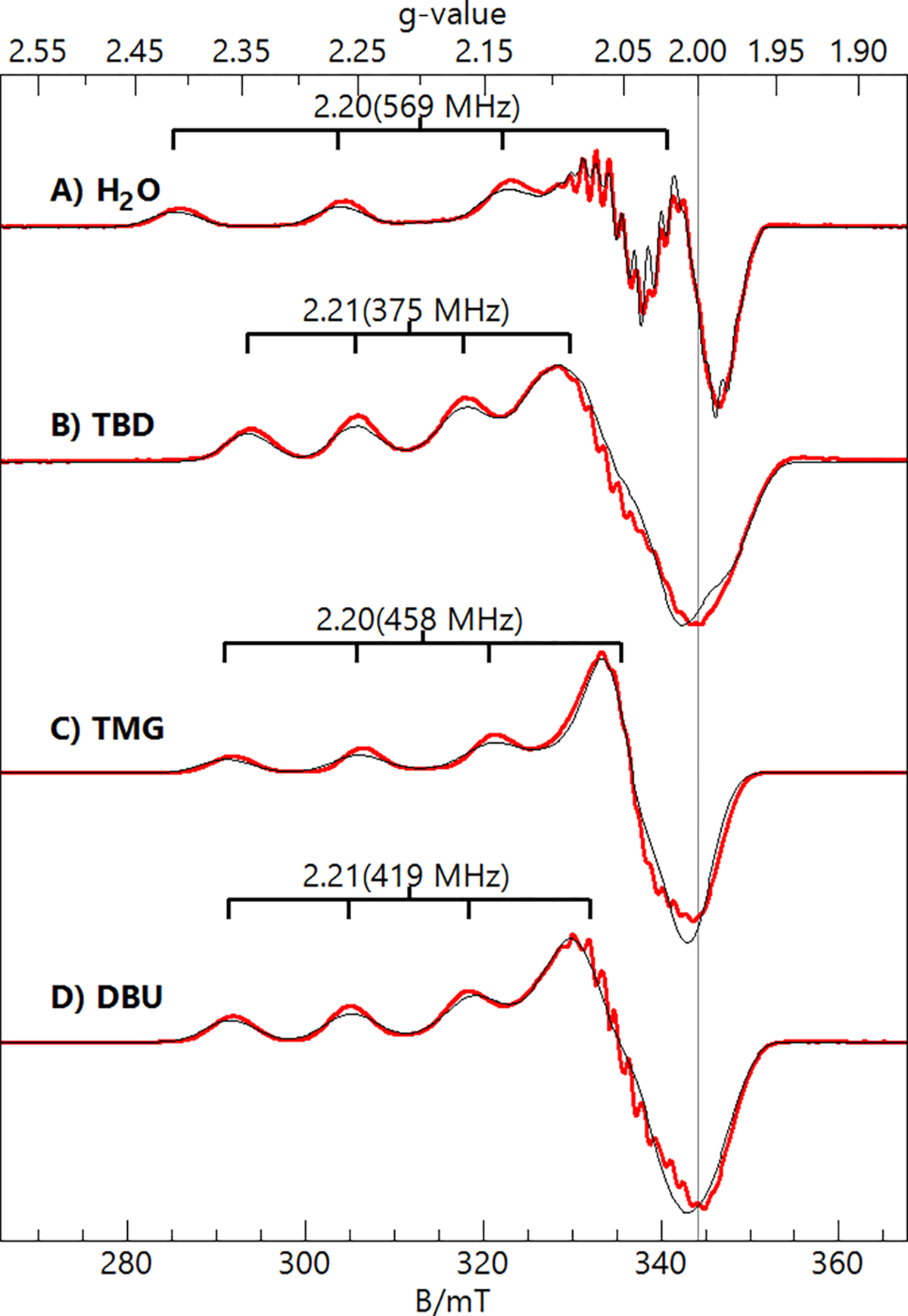
X-band (9.64 GHz, 0.2 mW) EPR spectra (red traces) and simulations (black traces) of the 1-L complexes in DCM:THF. The simulation parameters are listed in Table 3. Sample temperature, 12 K.
Figure 7.
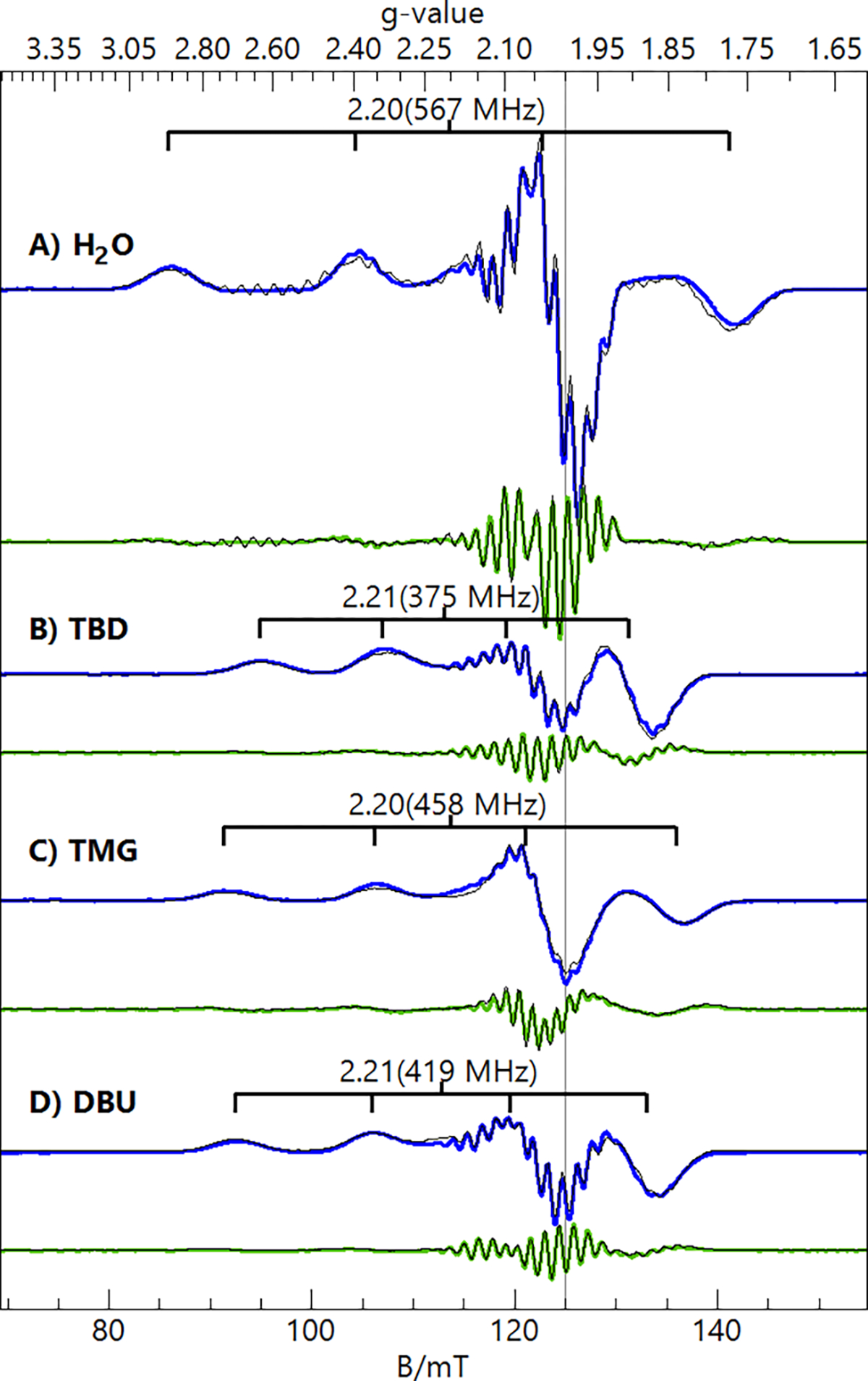
S-band (3.50 GHz, 0.03 mW) EPR spectra (1st derivative, blue traces; 2nd derivative, green traces) and simulations (black traces) of the 1-L complexes in DCM:THF. The simulation parameters are listed in Table 3. Sample temperature, 30 K.
In general, we obtained excellent fits of the 14N hyperfine structure, which were important in our determination of accurate values for the x and y components of the Cu A-tensor (Table 3). For the 1-imine complexes, the Az values and isotropic value of the A-tensor (Aiso) for the Cu centers were significantly lower than those of 1-OH2. We attribute this difference to the donation of more electron density from the [ibaps]3− ligand into the CuII orbital (see Discussion). While the magnitude of the A-tensor components can be determined from the spectra, their signs cannot be obtained directly. However, CuII Aiso values are negative,43 and we used this fact in our analyses. The values of Ax and Ay for 1-TMG are near zero, and therefore, the value of Az must be negative, and Aiso = −160 MHz. The values of Ay for 1-TBD and 1-DBU required a positive sign (Table 3) to give values of Aiso close to that of 1-TMG.
Magnetic Properties of 2-L.
Upon oxidation of the 1-L complexes, the EPR spectra associated with the CuII species vanished. Only 2-OH2 produced a new EPR signal that was observed in parallel mode at g = 4.1 with a 4-line hyperfine splitting of 6.5 mT (Figure 8). The signal position is indicative of a transition between the and levels of an spin system, where are the zero-field spin states. The state is produced from ferromagnetic exchange coupling between the CuII site and an ligand radical ([ibaps·]2−) with the 4-line hyperfine pattern arising from the 63/65Cu nucleus, which has a nuclear spin (I) of 3/2. The temperature dependence of the signal (see inset) was analyzed with an spin Hamiltonian to give a zero-field splitting constant of +3.4(4) cm−1. The positive sign of is indicative of a spin system in which the ms = 0 level is the ground state and the levels are excited states within the manifold. The simulation overlaid on the spectrum (Figure 8) is for with gz = 2.06 and Az = 195 MHz and is quantitative for the sample concentration. The conversion from the system spin to the local CuII site gives and (AS=1/2 = 2AS=1).51 The large Cu hyperfine coupling is consistent with the oxidation of the [ibaps]3− ligand. The large zero-field splitting is due to spin-orbit coupling with an excited orbital configuration (see below). The significant change in the and values of 2-H2O from those in 1-H2O is caused by a rotation of the -tensor relative to the axis of the doubly occupied orbital owing to mixing of the and orbitals arising from the distortion of the ligand plane. For a rotation of 45° relative to the CuII electronic coordinate system, the g and A values observed for the local CuII site of 2-H2O are reduced from 2.20 and 570 MHz in 1-OH2 to 2.12 and 390 MHz. The addition of cobaltocene to the 2-L complexes regenerated the EPR spectra of the respective 1-L complexes (Figures S19–S22).
Figure 8.
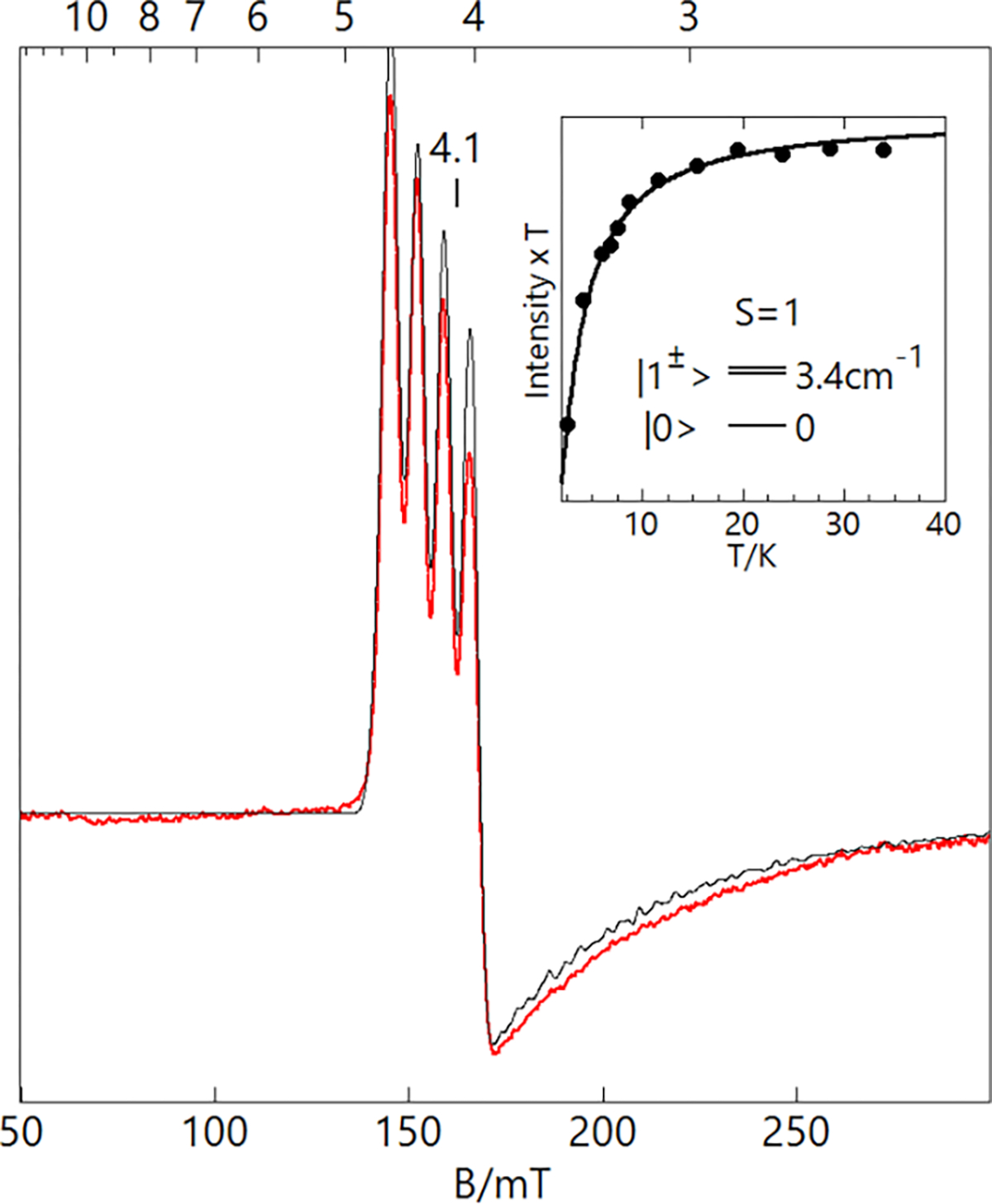
Parallel-mode X-band (9.337 GHz) EPR spectrum (red trace) of 2-OH2, 12 mM in DCM:THF. The simulation (black trace) uses the parameters listed in Table 3 and is quantitative for the sample concentration. Sample temperature: 6 K, microwave power: 2 mW. The inset shows the temperature dependence of the signal, plotted as signal intensity × temperature (•) versus temperature. The curve is calculated from the spin Hamiltonian for and D = +3.4 cm−1.
The 2-imine complexes were investigated using NMR spectroscopy, which revealed diamagnetic spectra for all three species (Figures S23–S26). These results are consistent with the lack of observable EPR signals and suggest that spin ground states are achieved through antiferromagnetic coupling between the CuII center and [ibaps·]2−. Using a combination of 1H, gCOSY, and gNOESY spectroscopies and the addition of D2O, the proton assignments were made for each complex (Figures S27–S32). To corroborate the electronic assignments established by UV-vis, EPR, and NMR experiments, X-ray absorption near-edge spectral (XANES) data were collected for a subset of 1-L and 2-L complexes (see SI). For all the complexes analyzed (1-OH2, 1-TBD, 2-OH2, and 2-TBD), the pre-edge features were centered at ~8979 eV (Figure S33, Table S5). This value falls in the range for CuII ions and is consistent with the valence at the Cu centers being unchanged by oxidation.44,45
Magnetic Properties of 3-L.
The two-electron-oxidized complexes, 3-OH2 and 3-TMG, gave CuII X- and S-band EPR spectra that were different from those of their precursors (Figures S34 and S35). Both complexes showed spectra with large hyperfine constants (Table 3), indicating that the second oxidation event also occurs in the [NNN] ligand, generating [ibaps]−.
Computational Analyses.
DFT calculations were performed to obtain the optimized structures of the 1-L complexes. The DFT-calculated structure of 1-OH2 agrees with that obtained by XRD methods and has a distorted square planar coordination geometry. The N1–Cu–O1 and N2–Cu–N3 bond angles are greater than 160°, and the two aryl groups of [ibaps]3− are slightly skewed in the same manner as observed experimentally (see above). The unpaired electron occupies the Cu orbital, which is antibonding to the sp2/sp3 orbitals of the [ibaps]3−/aqua ligands (Figures 9, S36–S37). The ligand orbitals transfer -electron density into the half-filled Cu orbital, resulting in spin polarization on the N atoms, which contributes to the observed 14N hyperfine splitting in the EPR spectra. The largest spin polarization appears on the central N donor (N1).
Figure 9.
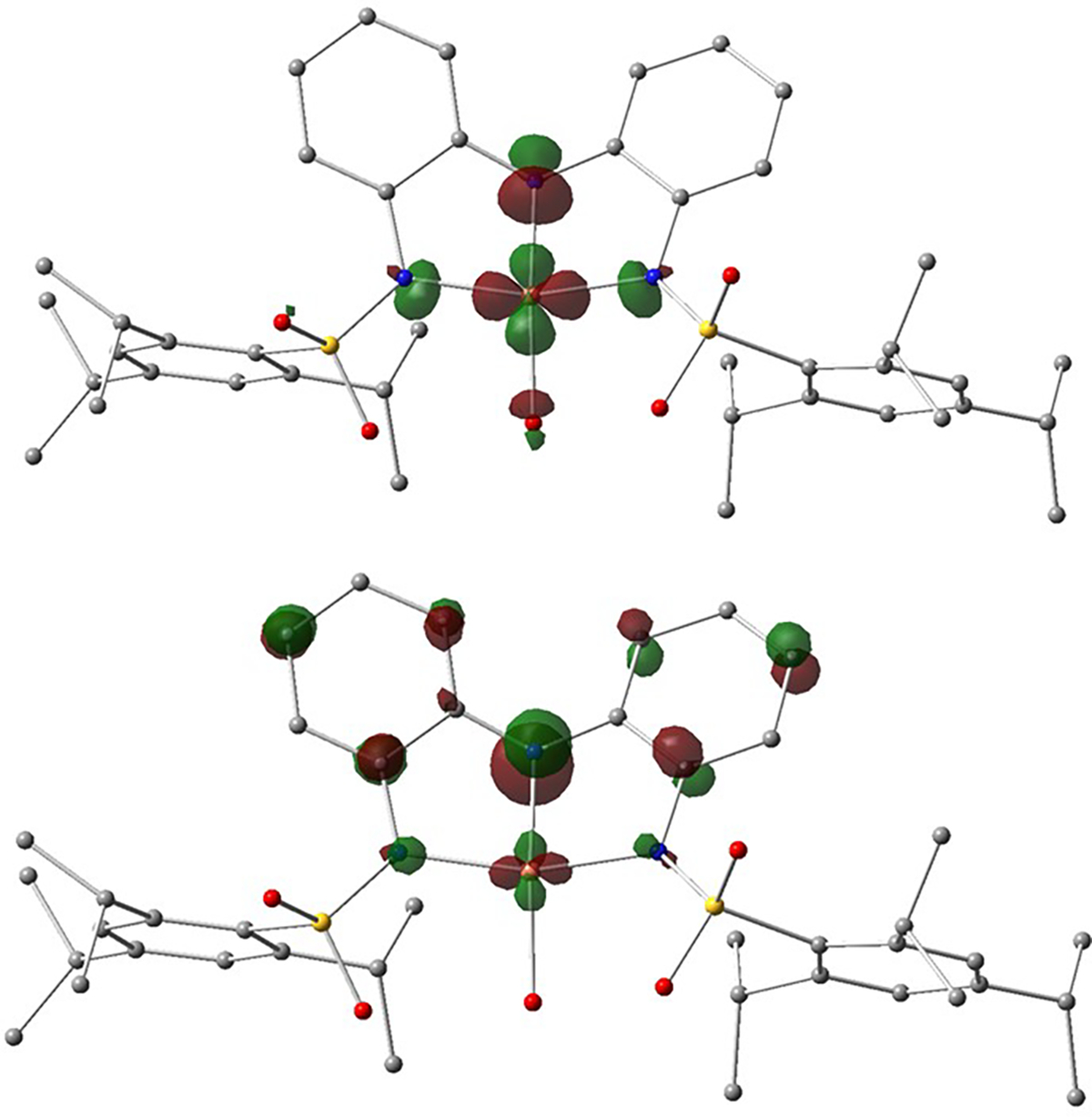
Molecular orbitals of 1-OH2 containing the unpaired electron (top, primarily ) and the redox-active HOMO (bottom, primarily pz of the central N atom).
Similar to the XRD structure of 1-DBU, DFT calculations found that the structures of the 1-imine complexes are more distorted from square planar than that of 1-OH2 (Table 4). The smaller N2-Cu-N3 angle is principally caused by a shift of the Cu atom away from N1 within the plane defined by the three N atoms of the [ibaps]3− ligand. The changes in the molecular structures from 1-OH2 to the 1-imine complexes depend on the steric and H-bonding interactions between the ancillary ligand and [ibaps]3− and the electronic interactions affecting the bonds between the Cu center and the [ibaps]3− ligand. We will show that the H-bonds within 1-OH2 are the main cause of the structural differences, but first, we will explore the specifics of the electronic structure of [ibaps]3− that led to a distortion away from a square planar coordination geometry. The HOMO in all the 1-L complexes is a p orbital principally centered on [ibaps]3−, and the largest amplitude is at the N1 pz orbital (Figure 9, bottom). For 1-OH2, s-bonding dominates, and the N1 pz orbital is barely involved in bonding because the Cu orbital is nearly aligned with the nodal plane of the redox-active p orbital of [ibaps]3−. The spin density on N1 thus more closely aligns with the Cu–N1 vector to form a predominately s-bond between the N1 sp2 and Cu orbitals (Figure 10, top). For the 1-imine complexes, the Cu orbital is tilted away from the nodal plane, which creates a non-orthogonal orbital arrangement (an overlap analysis is given in the SI), resulting in bond formation between the tilted Cu orbital and the N1 pz orbital (Figure 10, bottom). The additional d-pz bonding interaction derived from this distortion results in the transfer of -electron density from the pz orbital into the half-filled Cu orbital. For 1-TBD (Figure 10, bottom-right), the density transfer from both the sp2 and pz orbitals causes the net spin density on N1 to be rotated away from the Cu-N1 vector. The additional spin transfer reduces the spin population of the Cu orbital, which lowers the magnitude of the experimental A values for the CuII center (Table 4). These findings indicate that the rotation of the spin density at N1 is not due to a structural change within [ibaps]3− (e.g., change in the orientation of the C-N1-C angle) but rather the resultant combination of spin densities from both sp2 and pz orbitals.
Table 4.
Selected Bond Lengths (Å) and Angles (°) for 1-L calculated from DFT
| L | Cu-N1 | Cu-O1/N | Cu-N2/N3 | N1-Cu-O5/N | N2-Cu-N3 |
|---|---|---|---|---|---|
| H2O | 1.915 | 2.019 | 2.030/2.045 | 166 | 162 |
| TMG | 1.945 | 1.996 | 2.098/2.108 | 156 | 148 |
| TBD | 1.974 | 2.032 | 2.110/2.076 | 137 | 146 |
| DBU | 1.966 | 2.043 | 2.108/2.114 | 147 | 147 |
Figure 10.
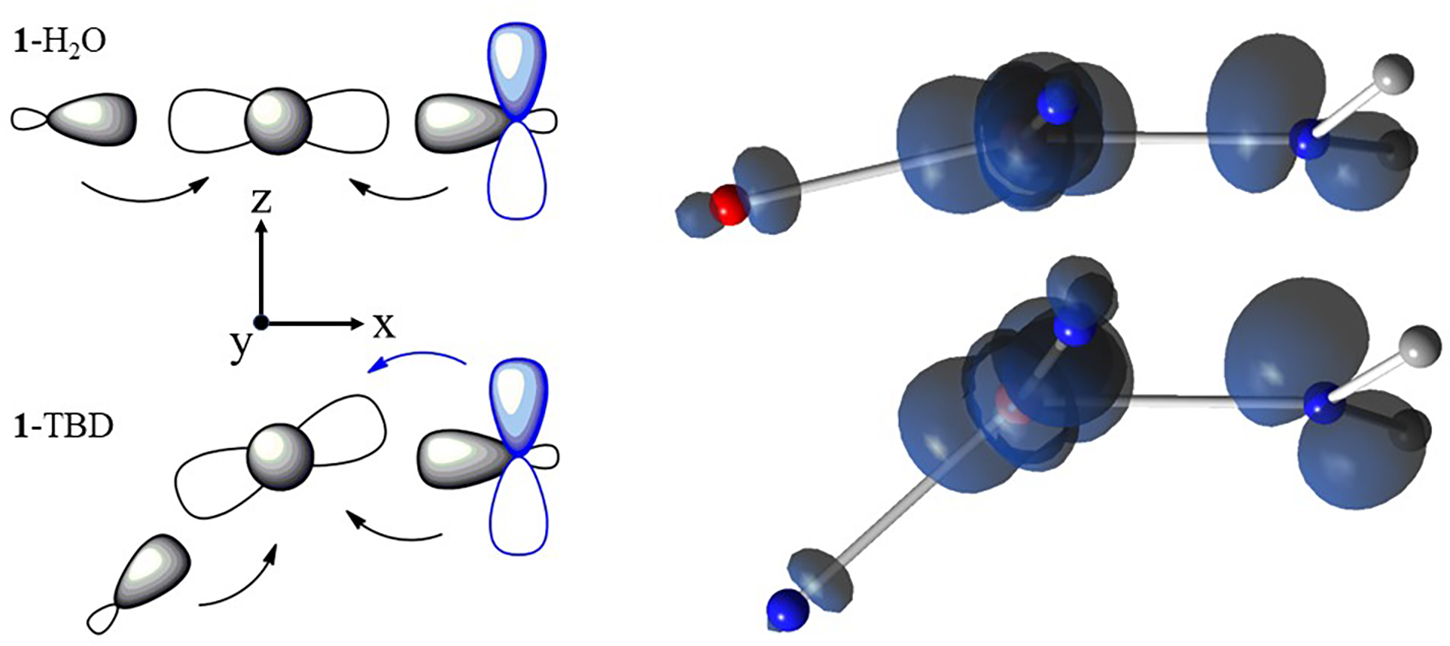
Left: Orbital interactions between and the ligand/N1 atom of [ibaps]3−. The component of the redox-active orbital at N1 is shown in blue. Right: corresponding spin densities from DFT showing spin populations in Cu and sp2 and pz orbitals of N1, the latter arising from the transfer of minority spin density into the orbital. For clarity, only the Cu center, its coordinating atoms, and the C atoms of [ibaps]3− are displayed.
We also considered whether the observed structural and electronic properties of the Cu complexes could be sterically induced rather than rooted in the redox-active nature of [ibaps]3−. To distinguish between these two possibilities, we performed DFT calculations for a truncated model in which the sulfonamido O atoms and aryl groups have been replaced by hydrogen atoms (denoted [ibapsT]3−, Figures S38–S40). These calculations were performed over the three experimentally investigated oxidation levels with water, ammonia, imine H2C=NH, and the TBD and DBU molecules as ligands ([Cu(ibapsT)L]n, Table 5). For the Cu complexes of both [ibaps]3− and [ibapsT]3−, the one- and two-electron oxidations remove electrons from the ligand p HOMO with the largest amplitude at the central N atom of the tridentate ligands. A key outcome of the computational studies is the similarity in the properties, including coordination geometries, of the [Cu(ibapsT)L]n complexes to those found for [Cu(ibaps)L]n, with the exception of L = H2O. (Figures S38–S40, Tables 5). For instance, the computed structures of both the truncated 1T-imine and 1-imine complexes gave similar N2–Cu–N3 angles and L-Cu-N1 angles of less than 160°. These calculations indicate that intrinsic electronic factors play a role in the control of the L-Cu-N1 angle. Compared to the 1T-OH2 complex, which has a small H2O-Cu-N1 angle (131°) like those found in the 1T-imine complexes, the optimized 1-OH2 complex has a markedly larger angle (166°, Table 5). We attribute the large L-Cu-N1 angle in 1-OH2 to geometric constraints imposed by the intramolecular H-bonds of the aqua ligand to the O atoms of the sulfonamido groups. We note that in the DFT calculations of 1T-OH2, one of the lone pairs of the aqua ligand was constrained to be directed toward the Cu center throughout the optimization (see SI). Without this constraint, the Cu–O bond breaks, and the water molecule reorients to form an H-bond with N2 or N3, thus indicating that the water is weakly coordinated to Cu.
Table 5.
DFT-calculated X-Cu-N1 bond angles and relative energies for the [Cu(ibaps)L]n and [Cu(ibapsT)X]n complexes.a
| Complex | 1-L | 2-L (broken symmetry) | 2-L (ferromagnetic) | ΔEc | 3-L |
|---|---|---|---|---|---|
| [Cu(ibaps)OH2]n | 166 | 171 | 172 | −450 | 172 |
| [Cu(ibapsT)OH2]n | 131d | 137 | 180 | 370 | 176 |
| [Cu(ibaps)TMG]n | 156 | 153 | 162 | 830 | 158 |
| [Cu(ibaps)DBU]n | 147 | 143 | 165 | 560 | 149 |
| [Cu(ibapsT)DBU]n | 159 | 152 | 180 | 532 | 179 |
| [Cu(ibaps)TBD]n | 137 | 144 | 152 | 1070 | 152 |
| [Cu(ibapsT)TBD]n,b | 123 | 140 | 175 | 581 | 179 |
| [Cu(ibapsT)N(H)CH2]n,b | 156 | 141 | 180 | 800 | 178 |
| [Cu(ibapsT)NH3]n | 156 | 145 | 178 | 491 | 175 |
T is truncated ligand with the sulfonamido aryl groups replaced by hydrogen atoms. Angles in degrees. Broken symmetry refers to antiparallel spins, and ferromagnetic refers to complexes with the S = 1 spin ground state. For selected computed bond lengths in 1-X and 2-X (X = OH2 and DBU) see Table S10.
During optimization, the imine and TBD plane was kept orthogonal to the ibaps plane by imposing suitable dihedral constraints (see Supporting Information).
Difference in the energies of the ferromagnetic and the broken symmetry states, Ererro - EBroken Symm, in wavenumbers.
In this optimization, one of the lone pairs of the water ligand was directed toward the Cu throughout the optimization by imposing suitable angular and dihedral constraints (see Supporting Information).
Our computational studies considered two possibilities for the one-electron-oxidized species (Tables 5 and S9): 1) the antiparallel spin configuration (broken symmetry) in which the 2-imine complexes retained the L-Cu-N1 angles of < 160°, and 2) the parallel spin state (ferromagnetic coupling) where the angle is consistently near 180° for the truncated species. The relative energies and bond angles of these two calculated states were compared, and our analysis found that the smaller angles in the antiparallel spin configuration are a consequence of the nascent tilted bonding interaction, as discussed above for 1-L and 1T-L. This tilted bond is prevented by the Pauli principle in the parallel spin state, resulting in nearly linear L-Cu-N1 angles. Again, the broken symmetry state of 2-OH2 is the exception owing to the two intramolecular H-bonds, and thus, the parallel spin configuration is favorable with a lower energy than the broken symmetry configuration (ΔE < 0 in Table 5), as observed using EPR spectroscopy.
For the two-electron-oxidized complexes (3-L), DFT results gave different L-Cu-N1 angles for the Cu complexes with [ibaps]1− versus [ibapsT]1− ligands (Table 5). For the 3T-L complexes, the now-empty orbital cannot donate into the Cu orbital, resulting in L-Cu-N1 angles of nearly 180°. The same angle computed for the 3-L complexes deviates from linearity with angles between 149° and 172°; thus, the calculations of these complexes indicate that the imine ligands have either steric or H-bond interactions that also contribute to the smaller L-Cu-N1 angles.
Electronic Absorption Properties.
Computed electronic absorption spectra obtained from TD-DFT calculations for the 1-OH2 and 1-TMG complexes (see SI, Figures S41–S42) revealed that the absorption bands between 500 and 1000 nm are ligand-to-metal charge transfer (LMCT) transitions () involving the orbitals of [ibaps]3−. The lowest-energy band at is predominately the LMCT transition . The overall greater absorption of 1-TMG relative to 1-OH2 is ascribed to a higher degree of non-planarity in the Cu coordination of the Cu-imine complex. The 2-L and 3-L complexes, in which electrons are removed from the redox-active orbital, introduces additional intra-ligand charge transfer transitions in the visible range – these transitions involve the transfer of an electron into the vacated orbital on [ibaps]2−/1− (Figure S43).
We used DFT computations to also account for aspects of our EPR results. In particular, the experimentally determined isotropic and Az values of 1-OH2 are significantly larger in magnitude than those of the 1-imine complexes (Table 3). To understand this trend, we performed a comparative analysis of the values (SD, spin-dipole) evaluated by DFT. The A-tensor has three contributions: , from isotropic contact, spin-dipolar, and spin-orbit coupling, respectively, and thus, . The values of and are experimentally determined (Table 3), and the spin-orbit contribution was obtained from eq 1
| (1) |
where (Tables 3 and S6).46 The value of , which is a function of the radial distribution of d electrons in a free metal ion, has been treated as an adjustable parameter for incorporating the influence of the covalency of the Cu-ligand bonds on the magnetic hyperfine interactions. For 1-L and 3-L, the values of that gave the best agreement with the DFT results were 1000 MHz and 1300 MHz, respectively, which are in the expected range for CuII complexes.46 As stated above, the signs of the A values are not determined from experiment, but CuII values are known to be negative. We considered all possible sign combinations of the components of the A-tensor (see SI, Tables S7 & S8) to obtain the experimental values for each complex (Table 6), which agreed with those found by DFT calculations (Table 6). The large negative value reflects the predominant Cu character of the unpaired electron. Complex 1-OH2 has an L-Cu-N1 bond angle > 160° and a large coupling of MHz. In contrast, the analogous 1-imine complexes have L-Cu-N bond angles < 160° and have smaller couplings, with values of significantly below 450 MHz.
Table 6.
Comparison of experimental and DFT-calculated AzSD values (in MHz) for select [Cu(ibaps)L]n complexes.
| Complexes | AzSD, experimental | AzSD, DFT |
|---|---|---|
| 1-OH2 | −470 | −460 |
| 1-TMG | −420 | −390 |
| 1-TBD | −330 | −350 |
| 1-DBU | −350 | −350 |
| 3-OH2 | −500 | −500 |
| 3-TMG | −450 | −460 |
Discussion
Cu Complexes that Span Three Oxidation Levels.
The Cu complex of [ibaps]3− can readily coordinate an external ligand to produce a series of four-coordinate, mononuclear Cu complexes ([CuII(ibaps)L]−) in which the fourth ligand (L) is either water or an imine (DBU, TBD, or TMG). We also found that the 1-imine complexes could be prepared directly from 1-OH2 via ligand substitution – we recognize that imines are often used as Brønsted bases, but in the case of 1-OH2, substitution chemistry is more favorable than deprotonation of the aqua ligand. The relatively short Cu-N1 bond (1.874(1) Å) found in the structure of 1-OH2 would lead to a considerable trans effect that resulted in the facile displacement of the aqua ligand. Moreover, our computational studies suggested that the Cu–O1 interaction is weak, and the isolation of 1-OH2 was aided by intramolecular H-bonds involving the aqua ligand.
All the 1-L complexes possess two redox processes, most of which were accessed using chemical oxidants to afford one-electron (2-L) and two-electron-oxidized (3-L) species. Our spectroscopic and computational studies agree that both oxidations are centered on the [ibaps]3− ligand, with the Cu center remaining in the 2+ valence state. For instance, the absorption spectra of the 2-L and 3-L complexes resemble those reported for the oxidized forms of complexes with [NNN] redox-active ligands.9,10,21,35,36,41 The isolation of 1-L, 2-L, and 3-L provided an opportunity to examine the effects of the ancillary ligands on both the molecular and electronic structures and the impact of intramolecular H-bonds in regulating magnetic coupling between a metal center and a multidentate, redox-active ligand.
Interplay Between the Cu Center and [ibaps]3−.
Distortion away from square planarity in four-coordinate CuII complexes is often achieved through secondary coordination sphere effects, such as steric constraints imposed by ligands. Our studies on [CuII(ibaps)L]− discovered another contributor to molecular non-planarity which involves the redox-active electron(s) of [ibaps]3− that occupy a orbital. The interplay of this ligand orbital with the Cu orbital has a profound effect on the structure and electronic properties of the complexes. We computationally explored these effects for the 1-L complexes. For planar structures, the Cu orbital is centered at the nodal plane of the orbital of [ibaps]3−, leaving the two orbitals orthogonal because they have different symmetries under reflection with the xy-plane. For non-planar structures, the mirror symmetry is broken, and a d-pz bond occurs between the tilted Cu orbital and the orbital of [ibaps]3− (Figure 10, bottom). CuII complexes are generally square planar because of strong bonding. The energies of the ligand orbitals are generally significantly lower than that of the Cu orbital containing the unpaired electron, which precludes additional bonding interactions. This is not the case for the 1-L complexes because the redox-active orbital on [ibaps]3−, of which the largest amplitude is at the central nitrogen atom, N1, is close in energy to the Cu orbital. As the angle L-Cu-N1 deviates appreciably from 180°, the strength of the d-pz bond increases and stabilizes a non-planar Cu coordination geometry. However, the angular bending also weakens the s bonds, and thus, the stability of the complexes depends on the L-Cu-N1 angle that best balances the and d-pz bonding interactions to give the lowest overall energy. The outlier in this series is 1-OH2, which has a relatively large L-Cu-N1 angle of 166° that is caused by the structural constraints imposed by the presence of two intramolecular H-bonds. Removal of these interactions in the truncated complex ([Cu(ibapsT)OH2]n− resulted in a substantially smaller angle of 131° (Table 6). The presence of a H-bond in 1-TMG, illustrated in 2-TMG (Figure 3), prompted us to examine why this interaction does not lead to a more linear N1-Cu-L angle like in 1-OH2. Our studies on 1-OH2 (Figure 2) show that it has two intramolecular H-bonds which appear to be stronger (DFT: H · O 1.71 Å) than the single H-bond in 1-TMG (DFT: H · O 2.07 Å). In addition, 1-TMG has, among the three 1-imine complexes, the largest N1-Cu-L angle which may influence the H-bond. The H · O H-bond in 1-TMG would have been stronger with a more linear N1-Cu-L angle, but the d-p2 bonding interaction (Figure 10) prevents this from occurring; thus, the observed angle is a compromise between these two competing bonding interactions. For 1-TBD, the N1-Cu-L angle is smallest among the Cu-imine complexes and may be influenced by a weak H-bond between the NH group of the TBD ligand and the lone pair at N1 (DFT: H · N1 2.59 Å) that could contribute to the shortening the N1-Cu-L angle. There are no intramolecular H-bonds in 1-DBU and the N1-Cu-L angle is between those in 1-TMG and 1-TBD because it is solely regulated by the orbital mechanism outlined above.
Evidence for the importance of the L-Cu-N1 angle in the determination of the electronic properties comes from correlations between the computed structural parameters and the experimentally determined spectroscopic data. In particular, the interplay between the redox-active ligand orbital and the tilted Cu orbital affects the observed hyperfine constants of the Cu centers. For L-Cu-N1 angles with large deviations from 180° (that is, for the 1-imine complexes), the additional d-pz bonding interaction increases the transfer of electron density from the ligand p orbital into the Cu orbital. This transfer thus results in a reduction of the spin population at the Cu center, which lowers the magnitude of the Cu Aiso and AzSD values (spin densities are listed in SI). In addition, the DFT-calculated AzSD values agree with those values determined experimentally, supporting the correlation between the experimental L-Cu-N1 angle and the observed A values (Tables 3 and 6). For 1-OH2, the magnitudes of the A values are larger because the two intramolecular H-bonds enforce an O5-Cu-N1 angle closer to linear. Furthermore, our analysis also applies to the CuII–hydroxide complex of Garcia-Bosch (tBuCuOH, Figure 1) that has a similar design as 1-OH2 with two intramolecular H-bonds between urea groups and a hydroxide ligand. The molecular structure of tBuCuOH obtained from DFT calculations showed a nearly square planar coordination geometry, including an O-Cu-N1 angle of 160° with an observed Az value (540 MHz) close to that of 1-OH2 (570 MHz).
The important role of d-pz bonding found for the 1-L complexes is absent in nearly all other Cu complexes reported in the literature. For instance, the Cu-aqua complex with the tridentate ligand N,N’-bis(2,6-diisopropylphenyl)-2,6-pyridinedicarboxamide (4-OH2, Figure S44) has an O-Cu-Npy angle of ~180° despite the lack of any enforcing H-bonds as in 1-OH2.47 The tridentate ligand in 4-OH2 utilizes a central pyridine that interacts differently with the Cu orbital because the occupied orbitals of the pyridine are delocalized and significantly lower in energy relative to the redox-active orbital in [ibaps]3−. This difference can be observed in the energies of the LMCT bands for the two complexes. The absorption spectrum of 1-OH2 shows a band with that is predominately a N1 pz to Cu charge transfer transition. In contrast, the lowest pyridine to Cu charge transfer transition in 4-OH2 has a peak with (see SI for details, Figure S45), indicating that the energy gap between the p orbitals and the CuII orbital in this complex is much larger than that in 1-OH2. Consequently, distortions of the complex that would allow Npy /Cu overlap do not effectively compete with the bonds that prefer an O-Cu-Npy angle of ~180°. To provide further evidence for the role of the [NNN] ligands in the bending mechanism, we have also investigated the L-Cu-Npy angle when the water ligand in 4-OH2 is replaced by the TBD ligand. DFT optimization showed that the structure of this hypothetical complex has a linear L-Cu-Npy unit and a large value typical for square planar CuII complexes, unlike the DFT structure of 1-TBD in which the L-Cu-N1 unit is bent and is small. As the two species have the same ancillary TBD ligand, any difference between them must be rooted in the properties of the [NNN] ligands supporting these systems.
Oxidized Complexes.
Further evidence for the interplay between the redox-active ligand orbital and Cu orbital is observed from the oxidized complexes. The 2-L complexes have an unpaired electron in the SOMO orbital of [ibaps·]2− that couples with the unpaired electron in the Cu orbital to afford either an or state. For 2-OH2, the near-linear O1-Cu-N1 angle enforced by the intramolecular H-bonds causes the Cu and p orbitals to be nearly orthogonal. According to the Kanamori-Goodenough rules,48–50 orthogonal magnetic orbitals give ferromagnetic coupling of electron spins (, ground state). Indeed, the predicted ground state for 2-OH2 is observed with parallel-mode EPR methods.
Complex 2-OH2 has a large zero-field splitting () that is a consequence of anisotropic exchange owing to spin-orbit coupling at the Cu center. Experimental evidence for such large values of for CuII-radical complexes is rare; for example, values from CuII-nitroxide complexes are less than 0.2 cm−1.51 The 2-OH2 complex has a set of d-d transitions similar to those in 1-OH2, with , and . To understand the magnitude and sign of this , we derived eq 2 (see SI) for determining values in terms of the g value for the coupled spin state (g = 2.06) and the exchange coupling () for the excited electronic configuration, which is admixed into the ground state by spin-orbit coupling, thereby yielding unquenched orbital momentum along .
| (2) |
The orbital has non-orthogonal overlap with the central pz orbital, which contains most of the unpaired electron in [ibaps·]2−, thus giving a strong antiferromagnetic interaction (). Meanwhile, the , and orbitals, which are perpendicular to the pz orbital, give weak ferromagnetic couplings. Equation 2 predicts in agreement with the experimental data and using the experimental value for gives cm−1. The exchange coupling is expected to have a magnitude typical of a coordination bond (~1 eV) given the large overlap between the Cu and redox-active pz orbitals.
The state, which has the two-electron spin function , cannot be rigorously treated by DFT, and instead, we use the broken symmetry state with spin function . Like the true state, the broken symmetry state allows N1 pz – Cu charge transfer and nascent d-pz bond formation, resulting in the small L-Cu-N1 angles (Table 5). In contrast, the N1 pz – Cu charge transfer interaction is forbidden by the Pauli principle in the ferromagnetic state (S = 1). Thus, the L-Cu-N1 angle is strongly correlated with the magneto-chemical properties of the complexes. A nearly linear angle leads to the orthogonality of the magnetic orbitals and results in a paramagnetic complex, whereas smaller angles cause a switch to antiferromagnetic coupling to give diamagnetic complexes. The DFT-calculated energy differences between the two spin states (, Table 5) show that only 2-OH2 is ferromagnetic (). The literature reports several four-coordinate Cu-ligand radical complexes that exhibit ferromagnetic exchange coupling ().7,8,13,52 The metal-ligand bonds in these complexes are dominated by interactions that produce planar coordination geometries and orthogonal magnetic orbitals (Figure 10, top left)
The two-electron oxidation of 1-L removes two electrons from the HOMO orbital of [ibaps]3−, leaving this orbital unoccupied in the 3-L complexes. The d-pz bonding described in Figure 10 is then absent, and the complex cannot lower its energy by decreasing the L-Cu-N1 angle. Thus, the DFT calculations for the truncated 3T-L complexes gave L-Cu-N1 angles that are nearly linear. However, our DFT calculations for the 3-imine complexes gave nonlinear L-Cu-N1 angles (Table S11). Furthermore, the experimental and values for the Cu center in 3-TMG were lower than the values for 3-OH2, and the values agree with the DFT-calculated values (Tables 3 and 6). The L-Cu-N1 angle in the 3-L complexes may have become more susceptible to steric influences than in the 1-L complexes, as the two-electron oxidation of the ligand has lessened the σ donations that stabilize the square planar coordination geometry. The lower σ electron donation and the accompanying increase in the spin population of the CuII site is in keeping with the increase in the P value for the two-electron-oxidized complexes that was noted above.
Summary and Conclusions
Copper(II) complexes have long been known to form mononuclear, square planar complexes in which bonding dominates because the orbitals of the ligands are usually at a significantly lower energy than the Cu d orbitals. The present studies have shown that this bonding scenario is not the case in Cu complexes with the [NNN] redox-active ligand [ibaps]3−: the significant degree of localization of the redox-active orbital at the central nitrogen atom of the [ibaps]3− ligand brings the orbital energetically and spatially close to the half-filled Cu d orbital. The similarity of these orbital energies allows π interactions to compete with bonding in the [Cu(ibaps)L)]n complexes, causing deviations from planarity. These deviations enable the redox-active ligand orbital in 1-L and 2-L to interact with the half-occupied Cu orbital, thereby lowering the energy of the system. In the absence of steric or H-bonding interactions of the ancillary ligand with [ibaps]3−, we have pinpointed that changes in the L-Cu-N1 angle are caused by a nascent d-pz bond, producing the observed non-planar coordination geometries. The effects of this additional bonding interaction are experimentally evident from changes in both the copper-ligand radical exchange interactions and the copper hyperfine values. The S = 1 state of 2-OH2 is attributed to the aqua ligand forming intramolecular H-bonds with the [ibaps·]2− ligand that enforce an O-Cu-N1 angle of greater than 160°. In contrast, the 2-imine complexes are all diamagnetic at room temperature because distortions in the equatorial plane around the Cu center (e.g., Nimine-Cu-N1 angles of less than 160°) lead to strong antiferromagnetic coupling between the spins at the CuII center and [ibaps·]2−. The smaller Nimine-Cu-N1 angles in the 2-imine complexes are also shown to closely correlate with the copper hyperfine values. The discovery of this correlation required the introduction of a new methodology to establish a quantitative connection between the experimental hyperfine measurements and DFT structural calculations for copper complexes.
Prior to this investigation there were no reports of metal-[NNN] complexes that support both ferromagnetic and antiferromagnetic couplings depending on the choice of the exogenous ligand. In heme enzymes, it is suspected that the coordination of exogenous ligands controls the exchange coupling between Fe and ligand-based spins (see above),34 although the mechanism for regulating the exchange-coupling constant has yet to be established. Our proposed exchange mechanism is sensitive to modulations of a single metal-ligand bond angle (by <20 ° between resulting spin states changes) which for the [Cu(ibaps)L] complexes, is influenced by intramolecular H-bonds to cause different magnetic characteristics. Therefore, the balance between a nascent d-pz bond and H-bonds illustrates a new type of control over the structure and properties of metal complexes and adds a conceptual tool in the design of molecular devices and reactive reagents.
Supplementary Material
ACKNOWLEDGMENT
The authors acknowledge the NIH (GM050781 to A.S.B. and GM077387 to M.P.H.) for funding and the UCI Department of Chemistry for fellowships to D.L.R.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publication website at DOI: The following file is available free of charge.
Experimental and theoretical descriptions and characterization data for all new compounds, Tables S1–S11, Figures S1–S46, and details for the molecular structures from XRD methods in cif format.
Accession Codes
CCDC 2291041–2291043 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Notes
The authors declare no competing financial interests.
References
- 1.McEvoy JP; Brudvig GW Water-splitting chemistry of photosystem II. Chem Rev 2006, 106, 4455–4483. [DOI] [PubMed] [Google Scholar]
- 2.Einsle O; Rees DC Structural Enzymology of Nitrogenase Enzymes. Chem Rev 2020, 120, 4969–5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Labinger JA; Bercaw Arnold JE; Beckman M Understanding and exploiting C-H bond activation. Nature 2002, 417, 507–514. [DOI] [PubMed] [Google Scholar]
- 4.Chirik PJ; Wieghardt K Radical Ligands Confer Nobility on Base-Metal Catalysts. Science (1979) 2010, 327, 794–795. [DOI] [PubMed] [Google Scholar]
- 5.Pegis ML; Wise CF; Martin DJ; Mayer JM Oxygen Reduction by Homogeneous Molecular Catalysts and Electrocatalysts. Chem Rev 2018, 118, 2340–2391. [DOI] [PubMed] [Google Scholar]
- 6.Nguyen AI; Blackmore KJ; Carter SM; Zarkesh RA; Heyduk AF One- and Two-Electron Reactivity of a Tantalum(V) Complex with a Redox-Active Tris(amido) Ligand. J Am Chem Soc 2009, 131, 3307–3316. [DOI] [PubMed] [Google Scholar]
- 7.Bahnmüller S; Plotzitzka J; Baabe D; Cordes B; Menzel D; Schartz K; Schweyen P; Wicht R; Bröring M Hexaethyltripyrrindione (H3Et6tpd): A Non-Innocent Ligand Forming Stable Radical Complexes with Divalent Transition-Metal Ions. Eur J Inorg Chem 2016, 4761–4768. [Google Scholar]
- 8.Gautam R; Astashkin AV; Chang TM; Shearer J; Tomat E Interactions of Metal-Based and Ligand-Based Electronic Spins in Neutral Tripyrrindione π Dimers. Inorg Chem 2017, 56, 6755–6762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu T; Macmillan SN; Rajabimoghadam K; Siegler MA; Lancaster KM; Garcia-Bosch I Structure, Spectroscopy, and Reactivity of a Mononuclear Copper Hydroxide Complex in Three Molecular Oxidation States. J Am Chem Soc 2020, 142, 12265–12276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lecarme L; Kochem A; Chiang L; Moutet J; Berthiol F; Philouze C; Leconte N; Storr T; Thomas F Electronic Structure and Reactivity of One-Electron-Oxidized Copper(II) Bis(phenolate)-Dipyrrin Complexes. Inorg Chem 2018, 57, 9708–9719. [DOI] [PubMed] [Google Scholar]
- 11.Kaim W Electron Transfer Reactivity of Organometallic Compounds Involving Radical-Forming Noninnocent Ligands. Proceedings of the National Academy of Sciences India Section A - Physical Sciences 2016, 86, 445–457. [Google Scholar]
- 12.Kaim W The Shrinking World of Innocent Ligands: Conventional and Non-Conventional Redox-Active Ligands. Eur J Inorg Chem 2012, 343–348. [Google Scholar]
- 13.Chaudhuri P; Nazari Verani C; Bill E; Bothe E; Weyhermüller T; Wieghardt K Electronic Structure of Bis(o-iminobenzosemiquinonato)metal Complexes (Cu, Ni, Pd). The Art of Establishing Physical Oxidation States in Transition-Metal Complexes Containing Radical Ligands. J Am Chem Soc 2001, 123, 2213–2223. [DOI] [PubMed] [Google Scholar]
- 14.Luca OR; Crabtree RH Redox-active ligands in catalysis. Chem Soc Rev 2013, 42, 1440–1459. [DOI] [PubMed] [Google Scholar]
- 15.Speier G; Csihony J; Whalen AM; Pierpont CG Studies on Aerobic Reactions of Ammonia/3,5-Di-tert-butylcatechol Schiff-Base Condensation Products with Copper, Copper(I), and Copper(II). Strong Copper(II)-Radical Ferromagnetic Exchange and Observations on a Unique N-N Coupling Reaction. Inorg Chem 1996, 35, 3519–3524. [Google Scholar]
- 16.Bill E; Müller J; Weyhermüller T; Wieghardt K Intramolecular Spin Interactions in Bis(phenoxyl)metal Complexes of Zinc(II) and Copper(II). Inorg Chem 1999, 38, 5795–5802. [Google Scholar]
- 17.Van Leest NP; De Zwart FJ; Zhou M; De Bruin B Controlling Radical-Type Single-Electron Elementary Steps in Catalysis with Redox-Active Ligands and Substrates. JACS Au 2021, 1, 1101–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Broere DLJ; Plessius R; van der Vlugt JI New avenues for ligand-mediated processes – expanding metal reactivity by the use of redox-active catechol, o-aminophenol and o-phenylenediamine ligands. Chem Soc Rev 2015, 44, 6886–6915. [DOI] [PubMed] [Google Scholar]
- 19.Kessler BJO; Mansoor IF; Wozniak DI; Emge TJ; Lipke MC Controlling Intramolecular and Intermolecular Electronic Coupling of Radical Ligands in a Series of Cobaltoviologen Complexes. J Am Chem Soc 2023, 145, 15924–15935. [DOI] [PubMed] [Google Scholar]
- 20.van der Vlugt JI Radical-Type Reactivity and Catalysis by Single-Electron Transfer to or from Redox-Active Ligands. Chemistry - A European Journal 2019, 25, 2651–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Munhá RF; Zarkesh RA; Heyduk AF Tuning the Electronic and Steric Parameters of a Redox-Active Tris(amido) Ligand. Inorg Chem 2013, 52, 11244–11255. [DOI] [PubMed] [Google Scholar]
- 22.Barman SK; Jones JR; Sun C; Hill EA; Ziller JW; Borovik AS Regulating the Basicity of Metal-Oxido Complexes with a Single Hydrogen Bond and Its Effect on C-H Bond Cleavage. J Am Chem Soc 2019, 141, 11142–11150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Drummond MJ; Ford CL; Gray DL; Popescu CV; Fout AR Radical Rebound Hydroxylation Versus H-Atom Transfer in Non-Heme Iron(III)-Hydroxo Complexes: Reactivity and Structural Differentiation. J Am Chem Soc 2019, 141, 6639–6650. [DOI] [PubMed] [Google Scholar]
- 24.Shook RL; Borovik AS Role of the secondary coordination sphere in metal-mediated dioxygen activation. Inorg Chem 2010, 49, 3646–3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee JL; Ross DL; Barman SK; Ziller JW; Borovik AS C-H Bond Cleavage by Bioinspired Nonheme Metal Complexes. Inorg Chem 2021, 60, 13759–13783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Borovik AS Role of metal–oxo complexes in the cleavage of C–H bonds. Chem Soc Rev 2011, 40, 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bhadra M; Lee JYC; Cowley RE; Kim S; Siegler MA; Solomon EI; Karlin KD Intramolecular Hydrogen Bonding Enhances Stability and Reactivity of Mononuclear Cupric Superoxide Complexes. J Am Chem Soc 2018, 140, 9042–9045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sinha S; Das S; Sikari R; Parua S; Brandaõ P; Demeshko S; Meyer F; Paul ND Redox Noninnocent Azo-Aromatic Pincers and Their Iron Complexes. Isolation, Characterization, and Catalytic Alcohol Oxidation. Inorg Chem 2017, 56, 14084–14100. [DOI] [PubMed] [Google Scholar]
- 29.Balaghi SE; Safaei E; Chiang L; Wong EWY; Savard D; Clarke RM; Storr T Synthesis, characterization and catalytic activity of copper(II) complexes containing a redox-active benzoxazole iminosemiquinone ligand. Dalton Transactions 2013, 42, 6829–6839. [DOI] [PubMed] [Google Scholar]
- 30.Leipzig BK; Rees JA; Kowalska JK; Theisen RM; Kavčič M; Poon PCY; Kaminsky W; DeBeer S; Bill E; Kovacs JA How Do Ring Size and π-Donating Thiolate Ligands Affect Redox-Active, α-Imino-N-heterocycle Ligand Activation? Inorg Chem 2018, 57, 1935–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Costentin C; Savéant JM; Tard C Ligand “noninnocence” in coordination complexes vs. Kinetic, mechanistic, and selectivity issues in electrochemical catalysis. Proc Natl Acad Sci U S A 2018, 115, 9104–9109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burns KT; Marks WR; Cheung PM; Seda T; Zakharov LN; Gilbertson JD Uncoupled Redox-Inactive Lewis Acids in the Secondary Coordination Sphere Entice Ligand-Based Nitrite Reduction. Inorg Chem 2018, 57, 9601–9610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Onderko EL; Silakov A; Yosca TH; Green MT Characterization of a selenocysteine-ligated P450 compound I reveals direct link between electron donation and reactivity. Nat Chem 2017, 9, 623–628. [DOI] [PubMed] [Google Scholar]
- 34.Benecky MJ; Frew JE; Scowen N; Jones P; Hoffman BM EPR and ENDOR Detection of Compound I from Micrococcus lysodeikticus Catalase. Biochemistry 1993, 32, 11929–11933. [DOI] [PubMed] [Google Scholar]
- 35.Cook SA; Bogart JA; Levi N; Weitz AC; Moore C; Rheingold AL; Ziller JW; Hendrich MP; Borovik AS Mononuclear complexes of a tridentate redox-active ligand with sulfonamido groups: Structure, properties, and reactivity. Chem Sci 2018, 9, 6540–6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brazzolotto D; Bogart JA; Ross DL; Ziller JW; Borovik AS Stabilizing a NiII-aqua complex via intramolecular hydrogen bonds: Synthesis, structure, and redox properties. Inorganica Chim Acta 2019, 495, 118960–118965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang L; Powell DR; Houser RP Structural variation in copper(I) complexes with pyridylmethylamide ligands: Structural analysis with a new four-coordinate geometry index, τ4. Journal of the Chemical Society. Dalton Transactions 2007, 955–964. [DOI] [PubMed] [Google Scholar]
- 38.Gardner EJ; Cobb CR; Bertke JA; Warren TH Tris(pyrazolyl)borate Copper Hydroxide Complexes Featuring Tunable Intramolecular H-Bonding. Inorg Chem 2019, 58, 11248–11255. [DOI] [PubMed] [Google Scholar]
- 39.Jones JR; Ziller JW; Borovik AS Modulating the Primary and Secondary Coordination Spheres within a Series of CoII-OH Complexes. Inorg Chem 2017, 56, 1112–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wojtulewski S; Grabowski SJ Different donors and acceptors for intramolecular hydrogen bonds. Chem Phys Lett 2003, 378, 388–394. [Google Scholar]
- 41.Kunert R; Philouze C; Berthiol F; Jarjayes O; Storr T; Thomas F Distorted copper(II) radicals with sterically hindered salens: electronic structure and aerobic oxidation of alcohols. Dalton Transactions 2020, 49, 12990–13002. [DOI] [PubMed] [Google Scholar]
- 42.Hoffman BM; DeRose VJ; Doan PE; Gurbiel RJ; Houseman ALP; Telser J Metalloenzyme Active-Site Structure and Function through Multifrequency CW and Pulsed ENDOR. in EMR of Paramagnetic Molecules. Biological Magnetic Resonance (eds. Berliner LJ & Reuben J) (Springer, 1993). vol. 13 151–218. [Google Scholar]
- 43.Abragam A; Bleaney B Electron Paramagnetic Resonance of Transition Ions. (Clarendon P., 1970). [Google Scholar]
- 44.DuBois JL; Mukherjee P; Stack TDP; Hedman B; Solomon EI; Hodgson KO A systematic K-edge X-ray absorption spectroscopic study of Cu(III) sites. J Am Chem Soc 2000, 122, 5775–5787. [Google Scholar]
- 45.Tomson NC; Williams KD; Dai X; Sproules S; Debeer S; Warren TH; Wieghardt K Re-evaluating the Cu K pre-edge XAS transition in complexes with covalent metal-ligand interactions. Chem Sci 2015, 6, 2474–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hitchman MA Interpretation of Copper(II) Hyperfine Parameters. Inorg Chem 1985, 24, 4762–4765. [Google Scholar]
- 47.Donoghue PJ; Tehranchi J; Cramer CJ; Sarangi R; Solomon EI; Tolman WB Rapid C-H Bond Activation by a Monocopper(III)-Hydroxide Complex. J Am Chem Soc 2011, 133, 17602–17605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kanamori J Superexchange Interaction and Symmetry Properties of Electron Orbitals. J. Phys. Chem. Solids 1959, 10, 87–98. [Google Scholar]
- 49.Goodenough JB Theory of the Role of Covalence in the Perovskite-Type Manganites [La, M(II)]MnO3. Physical Review 1955, 100, 564–573. [Google Scholar]
- 50.Goodenough JB Spin-Orbit-Coupling Effects in Transition-Metal Compounds. Physical Review 1968, 171, 466–479. [Google Scholar]
- 51.Bencini A; Gatteschi D Electron Paramagnetic Resonance of Exchange Coupled Systems. (Springer Berlin Heidelberg, 1990). [Google Scholar]
- 52.Okazawa A; Hashizume D; Ishida T Ferro-and antiferromagnetic coupling switch accompanied by twist deformation around the copper(II) and nitroxide coordination bond. J Am Chem Soc 2010, 132, 11516–11524. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.