

Abstract
Aim: Castration-resistant prostate cancer (CRPC) eventually becomes resistant to androgen receptor pathway inhibitors like enzalutamide. Immunotherapy also fails in CRPC. We propose a new approach to simultaneously revert enzalutamide resistance and rewire anti-tumor immunity. Methods: We investigated in vitro and in subcutaneous and spontaneous mouse models the effects of combining enzalutamide and GSK-126, a drug inhibiting the epigenetic modulator EZH2. Results: Enzalutamide and GSK-126 synergized to reduce CRPC growth, also restraining tumor neuroendocrine differentiation. The anti-tumor activity was lost in immunodeficient mice. Indeed, the combination treatment awoke cytotoxic activity and IFN-γ production of tumor-specific CD8+ T lymphocytes. Conclusion: These results promote the combination of enzalutamide and GSK-126 in CRPC, also offering new avenues for immunotherapy in prostate cancer.
Keywords: : castration resistance, epigenetic regulation, mouse models, prostate cancer, tumor immunology
Plain language summary
Prostate cancer depends on hormones called androgens for its growth. Therefore, hormonal therapies are commonly used. However, the tumor often does not respond to these treatments and new therapeutic approaches are needed. Here, using cell and mouse models, we have tested a new combination between hormone therapy and a drug that restrains an enzyme regulating gene expression. Our results have shown that this combination therapy not only reduces the growth of the tumor but also stops it from becoming more aggressive. This is really important because aggressive prostate cancer is much harder to treat. We have also found that this approach helps the immune system recognizing and attacking cancer cells. More research is needed to identify the mechanism of action of this treatment. However, our findings suggest that this approach could pave the way for new therapeutic strategies, including using immunotherapy, typically unsuccessful in treating prostate cancer.
Tweetable abstract
Castration-resistant prostate cancer is a fatal disease in which immunotherapy failed. Combined therapy inhibiting the androgen receptor and the epigenetic regulator EZH2 can revert castration resistance and promote anti-tumor immunity.
Graphical abstract
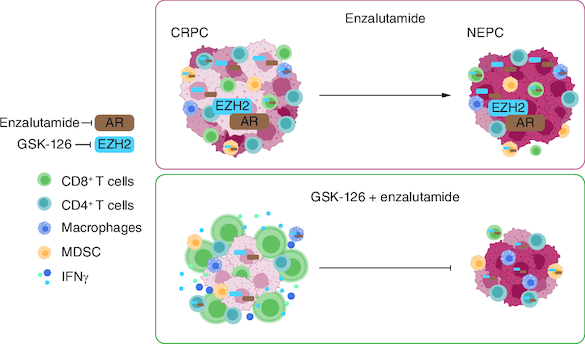
Plain language summary
Summary points.
Castration-resistant prostate cancer (CRPC) is initially responsive to androgen receptor pathway inhibitors, like enzalutamide, but resistance eventually occurs, often associated to the emergence of aggressive neuroendocrine prostate cancer.
Immunotherapy showed no efficacy in prostate cancer so far due to the immunosuppressive microenvironment.
We tested a new therapeutic approach combining enzalutamide and GSK-126, an inhibitor of the epigenetic regulator EZH2 in both in vitro and in vivo models of prostate cancer.
GSK-126 treatment confers enzalutamide sensitivity to otherwise resistant prostate cancer cells.
GSK-126 and enzalutamide combination blocks CRPC growth and neuroendocrine prostate cancer differentiation in mouse models.
Antitumor efficacy of GSK-126 and enzalutamide combination is lost in immunodeficient mice.
GSK-126 and enzalutamide combination restore the activity of tumor-specific CD8+ T cells in the TRAMP spontaneous model.
These results promote future clinical investigation of the combined use of enzalutamide and GSK-126 against CRPC and neuroendocrine prostate cancer, also opening new possibilities for immunotherapy in prostate cancer.
Prostate cancer ranks among the top causes of cancer-related mortality globally [1]. Advanced/metastatic tumors subjected to androgen-deprivation therapy (ADT) ultimately progress to become castration resistant (castration-resistant prostate cancer [CRPC]). Despite availability of novel androgen receptor pathway inhibitors (ARPIs), such as enzalutamide, prognosis of CRPC remains poor. Resistance to ADT/ARPI is linked to cellular plasticity [2] and the development of aggressive, fatal, neuroendocrine prostate cancer (NEPC), which counts for about 20–30% of patients who bypass hormone therapies [3,4]. De novo NEPC can also occur, albeit rarely (<1% of patients at first diagnosis). Thus, effective therapies for CRPC and NEPC are needed [5].
Immune checkpoint blockade is now the mainstream option for several solid tumors, but unsuccessful in prostate cancer [6]. Indeed, despite its potential immunogenicity [7], prostate cancer is a ‘cold’ tumor, with few infiltrating effector T cells and an immunosuppressive microenvironment governed by myeloid-derived suppressor cells (MDSCs) [8,9] and regulatory T cells (Treg) [10], also further recruited following ADT [10,11]. Notably, immune cells have the androgen receptor (AR) [12], and AR signaling can contribute to immunosuppression [13] directly transcribing Foxp3 in Treg cells [14], and blunting effector functions in T cells [15]. AR signaling can also promote M2 polarization of macrophages [16], production of IL-10 and TGF by neutrophils [17], and either foster or inhibit MDSC recruitment [18,19]. Therefore, treatment with ADT/ARPI could potentially reshape the tumor microenvironment.
A second approach to mold the immune system could rely on epigenetic reprogramming. Enhancer of zeste homolog 2 (EZH2) is the catalytic subunit of the polycomb repressive complex 2, a transcriptional repressor [20]. In several preclinical tumor models EZH2 genetic or pharmacologic inhibition induced recruitment and cytokine production of effector T cells, and skewed Treg toward pro-inflammatory functions [21–23]. Moreover, a few studies show either tumor MDSC recruitment [24] or inhibition [25] upon EZH2 targeting. Similarly, it was shown that EZH2 inhibition holds the potential to skew macrophage polarization [26,27].
EZH2 also regulates cellular differentiation and malignant proliferation [28], and phase I/II trials are currently assessing the activity of EZH2 inhibitors in patients with solid tumors [29]. In prostate cancer, EZH2 is a marker of aggressive and metastatic disease [30], known to contribute to cell invasiveness through repression of E-cadherin expression [31]. Furthermore, the expression of a polycomb related signature has been associated with poor outcome in patients [32]. EZH2 can also directly promote the expression of AR, in a polycomb independent manner [33,34]. Finally, it has been found that EZH2 is upregulated in CRPC and involved in NEPC differentiation [35,36]. These data highlight the therapeutic potential of EZH2 inhibitors in prostate cancer. Indeed the concomitant treatment with enzalutamide and EZH2 inhibitors restrained CRPC/NEPC growth in different preclinical models [37,38].
Therefore, in prostate cancer the combination of ADT/ARPI and EZH2 inhibitors could exert a dual effect: restoring hormone sensitivity of tumor cells and awakening the antitumor immune response. This new hypothesis is herein investigated by exploiting in preclinical models, in vitro and in vivo.
Materials & methods
Cell lines
T1525, T23 and ST4787 murine prostate cancer cells were characterized previously [39,40]. All murine cells were cultured in DMEM (Gibco) supplemented with 10% of fetal bovine serum (FBS; Gibco), 200 U/ml penicillin (Cambrex), 150 U/ml streptomycin, 10 mmol/l Hepes, 10 mmol/l sodium pyruvate (Gibco), and 2 mmol/l L-glutamine. LNCaP, 22RV1 and PC3 human prostate cancer cells were purchased from ATCC (cat. no: CRL-1740, CRL-2505D, CRL-1435). Cells were tested for Mycoplasma as in [40]. Human cells were cultured in RPMI-1640 (Gibco) supplemented with 10% of FBS, 200 U/m penicillin, 150 U/ml streptomycin, 10 mM sodium pyruvate, and 2 mM L-glutamine; for LNCaP and 22Rv1 cells, a 2.5 g/l of D-(+)-glucose (Sigma Aldrich) were also added to the culture media. For proliferation experiments, 3.5 × 104 cells were seeded in 6-well plates, and the following day treated with enzalutamide (Selleckchem S1250; 5, 10, 12.5 or 25 μM, as indicated in each experiment), GSK-126 (Selleckchem S7061; 1, 2.5, or 5 μM, as indicated in each experiment) or their combination (hereafter named COMBO). Cell count was evaluated after 96 h with the XTT assay kit (Applichem) or by trypan blue evaluation. For the evaluation of H3K27me3 expression, cells were cultured in 6-well plates, either treated or not with GSK-126 (5 μM) for 96 h, and then lysed for western blot analysis. For evaluation of cytokine production, murine cell lines were seeded in 12-well plates and the following day were either left untreated or treated with enzalutamide (10 μM), GSK-126 (5 μM), or COMBO. Cytokine production was evaluated after 24, 48 and 96 h by flow cytometry.
Mice & treatments
C57BL/6 and NSG male mice were purchased from Charles River Laboratories. 2 × 106 T23 or 1 × 106 ST4787 cells were subcutaneously injected in saline solution. When tumor volume reached 40–50 mm3, mice were treated with mock solution (DMSO and cremophor in saline solution), enzalutamide (30 mg/kg), GSK-126 (30 mg/Kg) or COMBO, intraperitoneally (i.p.) 3 days per week, for 2 weeks. Mice were sacrificed the day after the last treatment.
TRAMP mice on C57BL6/J background [C57BL/6-tgN (TRAMP)8247Ng] [41] were maintained as in [42]. For therapy experiments, 20 weeks old TRAMP mice underwent surgical castration as described in [40,42]. Starting from 3 weeks post-castration mice were treated with mock solution, enzalutamide, GSK-126 or COMBO, ip., once per week, and then sacrificed at 35 weeks of age. For cytotoxicity experiments, 16 weeks-old TRAMP mice were treated i.p. twice per week, for 3 weeks.
Animal housing and experimentation were performed following institutional guidelines and the Italian law (D.Lgs. 26/2014). In vivo experiments were approved by the Italian Ministry of Health (authorization number 8/2020-PR).
Antibodies
All the antibodies used in this study are listed in Supplementary Table 1.
Western blot
For AR and EZH2 evaluation, cells were lysed in RIPA buffer containing protease and phosphatase inhibitors. For H3K27me3 evaluation, cells were lysed in a buffer containing 0.5 M TrisHCl (pH 6.8) and 5% SDS plus protease and phosphatase inhibitors. Lysates were boiled for 5 min and then sonicated for 20 s at 25 A. Lysates were quantified (we used the Pierce BCA Protein assay kit catalog no. 23225 Thermo Scientific for RIPA samples and the Euroclone Quantum Micro Protein kit catalog no. EMP015480 for SDS samples), run on NuPAGE Bis-Tris gels (4–12%) and transferred on a nitrocellulose membrane. Membranes were blocked 1 h at room temperature with 5% BSA in PBS- 0.5% tween and then incubated overnight at 4° with primary antibody. HRP conjugated secondary antibody was then incubated 1 h at room temperature. Staining was revealed with ECL solution and images were acquired on an x-ray film or with Azure 600 imager (AZI600-0, Azure Biosystems). Blots were quantified with the ImageJ software.
Classification of tumor lesions in TRAMP mice
Murine urogenital apparata were fixed in formalin and embedded in paraffin (FFPE). Sections (5 μm) were deparaffined and rehydrated, stained with H&E (BioOptica) and evaluated by a pathologist. In serial sections, immunofluorescence for luminal (CK8) and neuroendocrine (SYP) markers was performed (see section 2.7). Murine prostate lesions were scored according to histopathological and immunophenotypical analyses as described in [40] and in the Results section.
Human prostate cancer samples
FFPE prostatectomies from prostate cancer patients (n = 38) were obtained from ASST Valle Olona, Busto Arsizio (VA), Italy (Protocol number 0046679/18), upon informed consent and in accordance with the Helsinki Declaration. Tissue collection, fixation and processing followed standardized protocols as part of routine clinical activity. The Gleason Score of analyzed tumors is reported in Table 1.
Table 1.
Patients' characteristics and EZH2 staining score.
| Patient ID | Gleason score | EZH2 staining score |
|---|---|---|
| 3 | 3+2 | - |
| 4 | 3+2 | - |
| 6 | 3+3 | +/- |
| 24 | 3+3 | - |
| 25 | 3+3 | - |
| 34 | 3+3 | - |
| 35 | 3+3 | - |
| 37 | 3+3 | - |
| 1 | 4+3 | +/- |
| 2 | 4+3 | +/- |
| 5 | 3+4 | + |
| 7 | 3+4 | - |
| 8 | 4+3 | + |
| 9 | 4+4 | + |
| 10 | 3+4 | - |
| 13 | 3+4 | + |
| 14 | 3+3 | - |
| 15 | 3+3 | - |
| 16 | 3+3 | +/- |
| 20 | 4+3 | - |
| 21 | 3+4 | + |
| 26 | 4+3 | + |
| 28 | 4+3 | + |
| 29 | 3+4 | - |
| 30 | 4+3 | + |
| 36 | 4+3 | +/- |
| 38 | 3+4 | + |
| 11 | 4+5 | +/- |
| 12 | 4+5 | ++ |
| 17 | 5+4 | +/- |
| 18 | 4+5 | +++ |
| 19 | 5+4 focal neuroendocrine prostate cancer | +++ |
| 22 | 4+4 | +++ |
| 23 | 5+4 | +++ |
| 27 | 4+4 | +/- |
| 31 | 5+4 | + |
| 32 | 5+4 | +++ |
| 33 | 4+5 | ++ |
The score index was assigned to distinguish intensity of EZH2 staining. - means no staining; +++ means maximum intensity.
Immunohistochemistry & immunofluorescence
FFPE tumor samples were cut in 5 μm sections, which were deparaffined and rehydrated. Antigen retrieval was performed utilizing the Novocastra Epitope Retrival Solution pH6 (Novocastra, Leica Biosystems), at 95°C for 15 min. Sections were brought to room temperature and washed with PBS. For immunohistochemistry, after neutralization of the endogenous peroxidase with 3% H2O2 and Fc blocking by a specific protein block (BSA 1%), the samples were incubated with the primary EZH2, AR or H3K23me3 antibodies overnight at 4° (antibodies are listed in Supplementary Table 1). Staining was revealed using a polymer detection kit (Novocastra, Leica Biosystems) or DAKO EnVison FLEX (Dako Omnis) as chromogenic substrate, followed by counterstaining with Harris hematoxylin (Novocastra, Leica Biosystems). Slides were acquired with a Leica DM4 B microscope equipped with a Leica DFC450 C digital camera, utilizing the LAS 4.8 software (Leica Biosystems). For double CK8/SYP staining, after antigen retrieval sections were blocked with PBS-Tween (0.1%) containing 5% of BSA. Primary conjugated antibodies were simultaneously incubated overnight at 4°C. Staining with DAPI (ThermoFisher Scientific) was performed for 10 min at room temperature. Slides were mounted with ProLong Diamond Antifade Mountant (ThermoFisher Scientific), and acquired with a Leica DM4 B microscope equipped with a Leica DFC450 C digital camera, utilizing the LAS X software (Leica Biosystems). Images were mounted and staining intensity were quantified using the ImageJ software. The EZH2 staining score on human tumor samples (Table 1) was determined in a blind fashion, comparing the staining intensity of all the slides, assigning (+++) to the maximum intensity observed and (-) to the lowest, and then re-assigning the values to all the slides.
RNA extraction & real-time PCR
T23 and ST4787 tumors were stored in RNA later (Thermofisher) at -80°C, and, when needed, were homogenized with TissueRuptor® (Qiagen) in RNA Lysis Buffer. For the isolation of CD45+ cells, T23 tumors were instead digested with collagenase I (1 mg/ml Gibco™ by Life Technologies, catalog no. 17018-029) for 2 h at 37°C. Cell suspensions were then labeled with CD45+ antibody and then sorted with a BD FACSMelody™ Cell Sorter. Dead cells were excluded by 7AAD staining.
Total RNA was extracted with Quick-RNA MicroPrep or MiniPrep kits (Zymo Research), quantified by Nanodrop 2000c spectrophotometer (ThermoFisher Scientific), and reverse transcribed using the High-Capacity cDNA Reverse Transcription Kit (TermoFisher Scientific, catalog no. 4368814). Real time-PCR was performed in a total volume of 20 μL using the Taqman® Fast Universal PCR Master Mix no Amperase UNG (ThermoFisher Scientific, catalog no. 4352042), 20 ng of cDNA, and Taqman probes for Ar (Mm00442688_m1), Ezh2 (Mm00468464_m1) and Gadph (Mm99999915_g1). Values were normalized to internal control (Gapdh) and analyzed using the ΔCT method.
Flow cytometry
Cell suspensions were obtained from mechanical disgregation of murine spleens or from digestion of prostates and tumors with collagenase I (1 mg/ml) incubated 2 h at 37°C. For surface staining, cells were labeled 15 min at 4°C with the desired antibody mix. Fixable Viability Stain (BD Bioscience; cat. 565388) was added to exclude dead cells from the analysis. For intracellular cytokine detection, tumor or spleen derived cells were stimulated for 4 h with PMA/Ionomycin or with the Tag-IV404-411 peptide (1 μg/ml), respectively, adding brefeldin A in the last 3 h. Cells were stained with surface markers and fixed with Foxp3/Transcription Factor Staining Buffer kit (Tonbo Biosciences, TNB-1022-L160 and TNB-1020-L050), respectively. Then, they were permeabilized (Perm buffer; TNB-1213-L150; Tonbo Biosciences) and stained with the desired intracellular antibody. Samples were acquired with BD LSRII Fortessa™ and analyzed with the Flow Jo software.
Immunization protocol & in vivo cytotoxicity assay
Dendritic cells (DCs) were prepared culturing bone marrow precursor isolated from the flushing of the tibias and femurs of a C57BL/6 mouse for 7 days in complete IMDM containing 10% of FBS, IL-4 (5 ng/ml; Peprotech) and GM-CSF (25 ng/ml; Peprotech). At day 7, DCs were stimulated for 7 h with LPS (1 μg/ml; Sigma), pulsed 1 hour with Tag-IV404-411 peptide (2 μg/ml) and injected (2 × 105 DC/mouse) intradermal into the right flank of the mice. 6 days later mice were injected intravenously with 107 cells containing equal numbers of splenocytes labeled with either 1.25 μg/ml (CFSEhi) or 0.125 μg/ml of CFSE (CFSElow). CFSEhi cells were previously pulsed 1 h with Tag-IV404-411 peptide. Mice were sacrificed the following day and spleens and prostates were collected and analyzed by flow cytometry for the evaluation of the presence of CFSEhi and CFSElow cells. Tag-specific cytolytic activity was calculated as: (percentage CFSEhi cells) × 100/(percentage CFSElow cells), as in [9]. Spleen and prostates of mice were also analyzed by flow cytometry for immune infiltrate and cytokine production, as described above.
Magnetic beads purification & in vitro experiments
Splenocytes from C57BL/6 mice were stained with CD19 and CD11b APC-conjugated antibodies, labeled with anti-APC Microbeads (Miltenyi Biotec; 130-090-855) and loaded onto magnetic columns for T cell enrichment through negative selection. CFSE-labeled purified T cells were activated in vitro with anti-CD3 (2 μg/ml) and anti-CD28 (1 μg/ml), alone or with enzalutamide (10 μM), GSK-126 (5 μM) or COMBO. After 3 days, we tested proliferation (CFSE dilution) and IFN-γ production by flow cytometry.
Regulatory T cells were isolated from the spleen of naive TRAMP mice through the CD4+CD25+kit (Miltenyi; 130-091-041) and cultured in vitro overnight, alone or with enzalutamide, GSK-126 or COMBO. Cytokine production was evaluated through flow cytometry.
MDSC were isolated from the spleen of TRAMP mice treated in vivo with mock solution, enzalutamide, GSK-126 or COMBO. MDSC purification and suppressive assay were performed as described in [9].
Statistical analyses
We utilized the GraphPad Prism9 software. All experiments were performed at least two times. Pools of biological replicates obtained in all the independent experiments were performed and statistical analysis was run considering all the samples. Histograms report means ± SD of all biological replicates, which are represented by dots. Data were analyzed using two-way ANOVA for tumor growth comparison in subcutaneous models, the Fisher test for comparison of tumor frequency in castrated TRAMP mice and one-way ANOVA with post hoc Tukey's test for multiple comparisons for other experiments. *p < 0.05; **p < 0.01; ***p < 0.001, and ****p < 0.0001. Where p-value is not indicated, the comparison between groups is not statistically significant.
Results
A drug inhibiting EZH2 restores enzalutamide sensitivity in CRPC cells
For in vitro experiments, we utilized a panel of human prostate cancer cell lines, known to be either sensitive (LNCaP) or resistant (22Rv1, and PC3) to enzalutamide [44–46], and a panel of in-house generated murine prostate adenocarcinoma (T1525 and T23), and NEPC (ST4787) cells [39,40]. All the cells express AR and EZH2, except human PC3 cells that, as known [44], are AR negative (Figure 1A & B). Dose–response experiments showed that T1525 are sensitive to enzalutamide (IC50 10.86 μM), whereas both T23 and ST4787 are not (Figure 1C). We also performed dose–response experiments with the EZH2-inhibitor GSK-126 that, according to what previously shown in a different prostate cancer model [37], did not affect the proliferation of neither human nor murine prostate cancer cells (Figure 1D & E), despite effectively reducing histone H3 lysine 27 tri-methylation (H3K23me3), measured as readout of EZH2 methyltransferase activity (Figure 1F & G).
Figure 1.
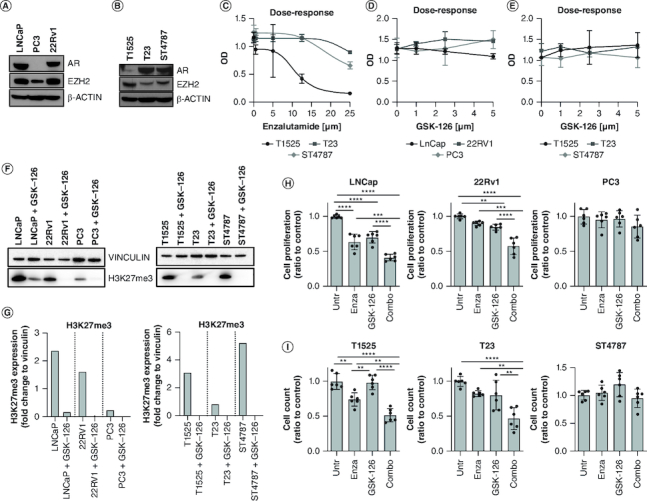
EZH2 inhibition restores enzalutamide sensitivity in castration-resistant prostate cancer cells.
(A & B) Human (LNCaP, PC3, 22Rv1) and murine (T1525, T23, ST4787) prostate cancer cell lines were assessed for AR and EZH2 expression through western blot analysis. β-actin was used as housekeeping. (C) Murine cells were cultured with different doses of enzalutamide (0, 5, 12.5, and 25 μM) for 96 h and tested for proliferation by XTT assay. (D & E) Human and murine cells were cultured with different doses of GSK-126 (0, 1, 2.5, and 5 μM) for 96 h and tested for proliferation by XTT assay. (F) Human and murine cells were either left untreated or treated with GSK-126 (5 μM). After 96 h, H3K27me3 levels were tested by western blot. Vinculin was used as housekeeping. The western blot was repeated twice (G). Quantification of F. (H & I) Human and murine cells were cultured in the presence of enzalutamide (10 μM), GSK-126 (5 μM), or their combination (Combo) as indicated, for 96 h and tested for proliferation through trypan blue cell counting. Histograms report mean ± SD of biological replicates (represented by dots), pooled from two independent experiments. One-way Anova followed by Tukey's multiple comparison test is used, **p < 0.01; ***p < 0.001, and ****p < 0.0001. Where p-value is not indicated, the comparison between groups is not statistically significant.
We then evaluated the effects of the combination of enzalutamide (10 μM) and GSK-126 (5 μM) against cell proliferation. In LNCaP and T1525 cells, only enzalutamide as single agent decreased cell proliferation but this effect was significantly increased by the combination of enzalutamide and GSK-126 (hereafter named COMBO; Figure 1H & I). Notably, the COMBO significantly reduced the proliferation of both T23 and 22Rv1 CRPC cells, which were insensitive to any of the two drugs given alone (Figure 1H & I). Neither PC3 nor ST4787 cells were affected by the treatments (Figure 1H & I). Therefore, inhibition of EZH2 can restore sensitivity to enzalutamide in both human and murine CRPC cells lines, but not in established NEPC cells.
Enzalutamide & GSK-126 combination restrains CRPC growth & NEPC differentiation in vivo
We then moved in vivo, subcutaneously injecting T23 or ST4787 cells in syngeneic C57BL/6 immunocompetent mice. Differently from in vitro experiments, in mice enzalutamide alone significantly reduced the growth of CRPC T23-derived tumors (Figure 2A), suggesting that the microenvironment could play a role in this effect. Tumors in the COMBO group were also significantly smaller than controls. Indeed, adding GSK-126 to enzalutamide seemed to further reduce tumor volumes, albeit the difference between the enzalutamide and the COMBO group was not statistically significant (Figure 2A). None of the treatments was effective in mice bearing ST4787 tumors (Figure 2B). Nevertheless, the concomitant treatment with GSK-126 inhibited NEPC differentiation of T23-derived tumors induced in vivo by enzalutamide (Figure 2C & Supplementary Figure 1A & B).
Figure 2.
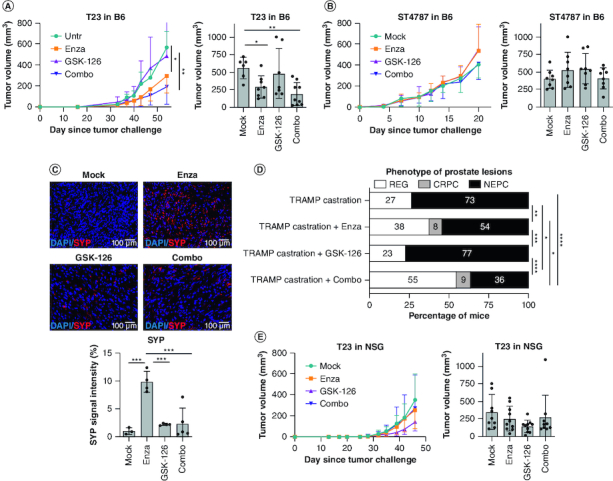
Antitumor effect of concomitant AR and EZH2 inhibition restrain castration-resistant prostate cancer and neuroendocrine prostate cancer in vivo and the effect is immune-dependent.
(A & B) Tumor growth in C57BL/6 mice injected subcutaneously with T23 (A) or ST4787 (B) cells and treated with enzalutamide, GSK-126, or their combination (Combo). N = 8 mice for each group, pool of two independent experiments. Left graphs represent mean tumor volume; right histograms report mean tumor volume ± SD at the day of sacrifice, each dot is a mouse. Two-way Anova followed by Tukey's multiple comparison test: *p < 0.05. (C) SYP expression (red) was assessed in excised subcutaneous tumors of (A) by immunofluorescence. Blue staining is DAPI. Experiment was repeated two-times. SYP signal intensity was evaluated with ImageJ software. Histogram report mean ± SD, each dot is a mouse. One-way Anova followed by Tukey's multiple comparison test: ***p < 0.001. (D) TRAMP mice were surgically castrated at 20 weeks of age, and after 3 weeks either left untreated (n = 11) or treated with enzalutamide (n = 13), GSK-126 (n = 13), or their combination (Combo; [n = 11]) and sacrificed at 35 weeks of age. Histograms report the frequency of tumor lesions scored as neuroendocrine prostate cancer (NEPC), castration-resistant prostate cancer (CRPC) or regression (REG); numbers written in the bars indicate the relative percentages of each type of lesion. Murine prostate lesions were scored according to histopathological and immunophenotypical analyses (Supplementary Figure 1C & D) as follows: i) CRPC: lesions of high-grade prostatic intraepithelial neoplasia or adenocarcinoma characterized by CK8 positive atypical cells forming distorted/ill-defined glands within the stroma. ii) NEPC: composed of sheets and nests of medium-sized to large cells with high nuclear to cytoplasmic ratio and/or anaplastic morphology. Cells were immunoreactive for SYP and either negative (in case of pure small-cell NEPC) or positive for CK8 (in case of tumors with mixed adenocarcinoma and NEPC features, ref. [43]). When NEPC areas and CRPC or REG lesions were present in different lobes, the tumor was classified as NEPC. iii) Regression (REG): prostates with variable degree of glandular distortion characterized by dilated lumina with flattened or focally hyperplastic epithelia in the absence of overt nuclear atypia. Fisher test: *p < 0.05; **p < 0.01; ***p < 0.001, and ****p < 0.0001. (E) Tumor growth in immunodeficient NSG mice injected subcutaneously with T23 cells and treated with enzalutamide, GSK-126, or their combination (Combo). N = 8 mice for each group, pool of two independent experiments. Left graphs represent mean tumor volume; right histograms report mean tumor volume ± SD at the day of sacrifice, each dot is a mouse. Two-way Anova followed by Tukey's multiple comparison test was performed. In all graphs, where p-value is not indicated, the comparison between groups is not statistically significant.
To evaluate the therapeutic activity of enzalutamide and GSK-126 combination in a setting resembling a clinical situation where enzalutamide is administered after ADT, we then moved to the spontaneous TRAMP prostate cancer model, in which we can mimic ADT by surgical castration (Figure 2D & Supplementary Figure 1C & D). Results showed that castration caused tumor regression in 27% of mice, whereas the remaining 73% of castrated animals developed tumors scored as NEPC. The frequency of NEPC induced by castration was slightly reduced by administration of enzalutamide (54%) and not altered by GSK-126 (77%). Compared with castration only and to castration plus enzalutamide, the COMBO treatment respectively halved and reduced by a third the rate of NEPC (36% in castrated + COMBO vs 73% in castrated vs 54% in castrated + enzalutamide), significantly increasing the frequency of cured mice (55% of regression). Some CRPC were detected in the enzalutamide and COMBO groups (8% and 9% respectively). Thus, the COMBO treatment can efficiently restrain NEPC differentiation in mouse models.
Antitumor effect of the COMBO treatment requires proficient immune response
To test whether immune cells contribute to the antitumor efficacy observed in vivo, we injected T23 cells in NSG mice, lacking lymphoid cells and having defective myeloid cells [47]. In these immunodeficient mice, the therapeutic effect of the enzalutamide alone and of the COMBO regimen was lost (Figure 2E). By immunohistochemistry, we checked that both the GSK-126 and the COMBO treatment reduced the levels of H3K23me3 in T23 tumors, both when grown in C57BL/6 and NSG mice, as well as in ST4787 tumors grown in C57BL/6 mice (Figure 3A). We also confirmed AR and EZH2 expression in tumor and stroma cells of T23 and ST4787-derived lesions by immunohistochemistry (Figure 3B). To best appreciate the expression of the two markers in tumor-infiltrating immune cells, we also performed a real time PCR to measure Ar and Ezh2 transcript levels in RNA obtained from either whole murine tumors or FACS sorted infiltrating CD45+ cells (Figure 3C). By immunohistochemistry, we also identified EZH2 positive tumor and stroma cells in high grade human prostate cancer (Figure 3D). Notably, EZH2 staining intensity was almost negative or dim in human tumors with low Gleason score, but increased in tumors with high Gleason score (4+5, 5+4) and NEPC features (Figure 3D & Table 1).
Figure 3.
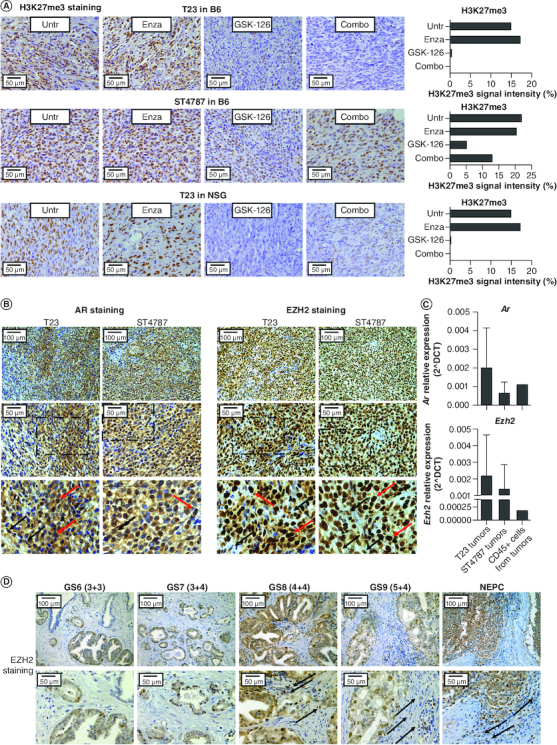
EZH2 is expressed in murine and human prostate cancer lesions.
(A) Pictures show immunohistochemistry for H3K27me3 in T23 and ST4787 tumors collected from the experiments reported in Figure 2A, B and E. Histograms report staining intensity quantification, evaluated with ImageJ software. (B) Immunohistochemistry for AR and EZH2 on subcutaneously T23 and ST4787 lesions. Scale bars indicate magnifications. The lower panels are digital magnifications of selected areas indicated by a dotted line in the middle panels; red and black arrows indicate selected tumor and stromal cells, respectively (C). Real time PCR for Ar and Ezh2 on RNA samples collected from whole T23 and ST4787 tumors and from CD45+ cells FACS sorted from T23 tumors (n = 2). (D) Immunohistochemistry for EZH2 on human prostate cancer lesions with different Gleason Score. Scale bars indicate magnifications. Black arrows indicate stromal cells.
Results suggest that in vivo enzalutamide might rewire the immune microenvironment toward CRPC inhibition, an effect that could potentially be enhanced by concomitant administration of GSK-126. We therefore investigated by flow cytometry the immune milieu of T23 and ST4787 derived tumors (Figure 4 & Supplementary Figure 2). Total CD45+ immune cells were higher in T23 samples. Among CD45+ cells, in both tumor types CD11b+ myeloid cells were predominant than CD3+ lymphoid cells. CD8+ T cells and CD4+ Treg were prevalent in T23 and ST4787 tumors, respectively. Macrophages were the most representative cells in both tumor types, but skewed to M2 in ST4787 tumors. PMN- and M-MDSC equally infiltrated T23 and ST4787 samples. These results suggest that ST4787 NEPC tumors have a ‘colder’ microenvironment than T23 tumors. In both models none of the treatments altered the frequency of the immune cell population analyzed, neither in tumor (Figure 4) nor in spleen (Supplementary Figure 3), except for a slight increase of tumoral PMN-MDSC (Figure 4) and splenic M2 macrophages (Supplementary Figure 4), after both GSK-126 and COMBO treatment.
Figure 4.
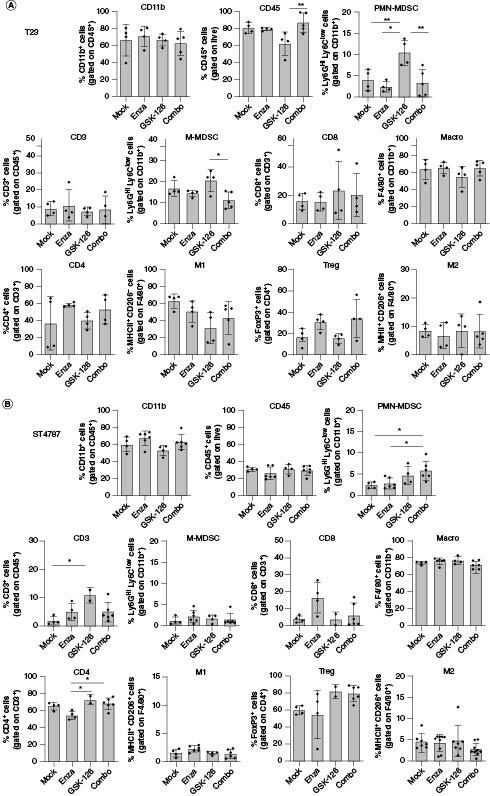
The COMBO treatment does not affect the frequency of immune subpopulations in subcutaneous tumors.
Analysis of immune infiltrate of subcutaneous T23 (A) and ST4787 (B) tumors by flow cytometry. Histograms report mean ± SD, each dot is a mouse. One-way Anova followed by Tukey's multiple comparison test: *p < 0.05; **p < 0.01. Where p-value is not indicated, the comparison between groups is not statistically significant.
We therefore hypothesized that the antitumor effect seen in immunocompetent mice was related to differences in the function rather than in the frequency of immune cells.
Enzalutamide & GSK-126 COMBO enhances tumor-specific CD8+ T cell activity in vivo
To dissect the activity of tumor-specific T cells we exploited TRAMP mice, where the SV40-Large T antigen (Tag) oncogene acts as a tumor-associated antigen, leading to persistent Tag-specific tolerance of CD8+ T cells [9,48]. To test whether we could restore T cell functionality, we treated 16 weeks-old TRAMP mice with control mock solution, enzalutamide, GSK-126 or the COMBO. After 2 weeks, all mice were immunized with DCs pulsed with the CD8-immunodominant TagIV peptide [49] and killed one week later to test in vivo specific cytotoxic activity against Tag-expressing target cells, injected the day before (Figure 5A & Supplementary Figure 4). As known [9], in vivo Tag-specific CD8+ T cell cytotoxic activity was very low in untreated TRAMP mice, but it significantly increased in TRAMP mice treated with the COMBO (Figure 5B). We also observed an increase of T cell cytotoxicity in the enzalutamide group, albeit not statistically significant. Furthermore, the effect was tumor specific, as the COMBO did not further increase T cell cytotoxicity in control wild type C57BL/6 mice, not tolerant against Tag (Figure 5B).
Figure 5.
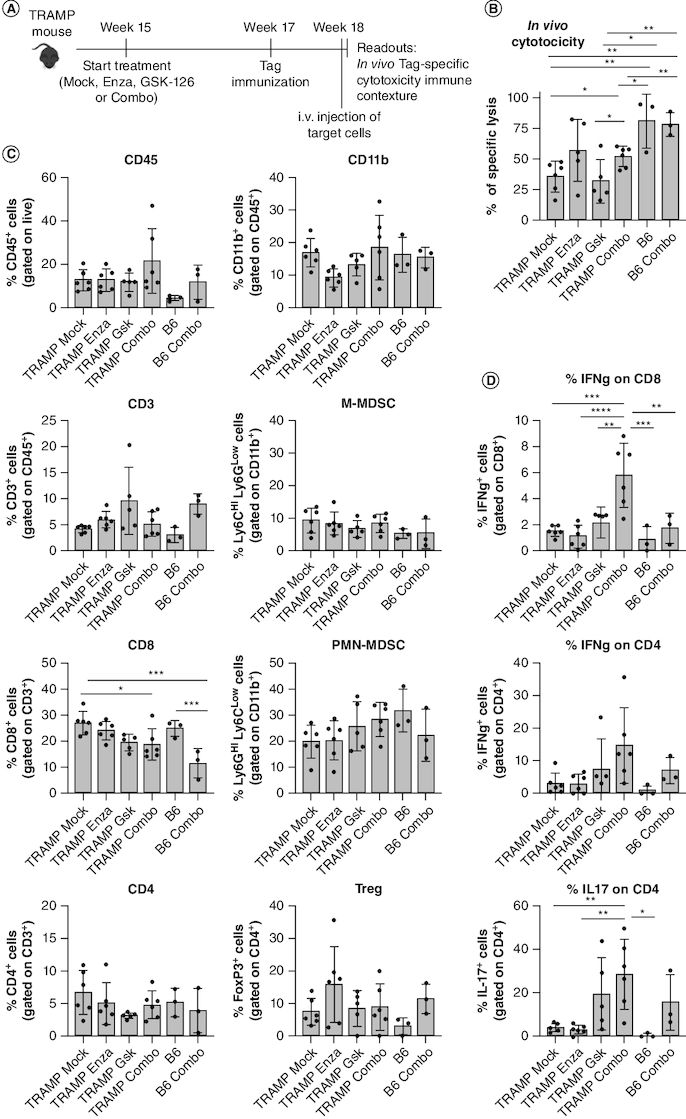
Concomitant inhibition of AR and EZH2 enhances cytotoxic activity and cytokine production of tumor-specific CD8+ T cells.
(A) Scheme of the in vivo cytotoxicity assay in TRAMP mice. 15-week-old TRAMP mice were treated with enzalutamide, GSK-126, or their combination (Combo). 2 weeks later treated TRAMP and control non-tumor-bearing naive C57BL/6 (B6) mice were immunized with DCs pulsed with TagIV peptide. 1 week later, in vivo cytotoxicity was tested as described in methods (B). From these mice we also evaluated frequency (C) and cytokine production (D) of prostate-infiltrating immune cells, after 4 h stimulation with PMA-ionomycin and brefeldin A. Histograms report mean ± SD, each dot is a mouse. One-way ANOVA followed by Tukey's multiple comparison test: *p < 0.05; **p < 0.01; ***p < 0.001 and ****p < 0.0001. Where p-value is not indicated, the comparison between groups is not statistically significant.
In the same cohorts, we tested frequency (Figure 5C) and cytokine production (Figure 5D) of prostate-infiltrating T cells. As experiments in the subcutaneous models (Figure 4) showed no modulation in the myeloid compartment, we spared macrophages from this analysis, but we still tested MDSC frequency, knowing from our previous studies that PMN-MDSC can restrain T cell activity in TRAMP mice [9]. None of the treatments altered immune cell recruitment (Figure 5C). However, the COMBO significantly increased IFN-γ production by CD8+ T cells (Figure 5D), and IL-17 production by CD4+ T cells (Figure 5D) in TRAMP mice. These changes were confined to the tumor microenvironment, as they were not detected in the spleen of TRAMP mice (Supplementary Figure 5) nor in the prostates of wild type, tumor free, C57BL/6 mice (Figure 5D).
These data indicate that the COMBO can increase the activity of tumor-specific T cells in prostate cancer.
The effect of the COMBO on T cell function depends on microenvironment
In vivo experiments indicate that the COMBO treatment can increase cytotoxicity and IFN-γ production of tumor-specific CD8+ T cells (Figure 5). This could be either a direct effect or an indirect effect involving their crosstalk with other cells in the microenvironment. In vitro experiments excluded the first hypothesis, as no differences on proliferation and IFN-γ production were detected in purified CD8+ or CD4+ T cells treated in vitro with the COMBO (Figure 6A). As CD4+ T cells in TRAMP prostates treated with COMBO produce more IL-17, we also tested such induction in either conventional T (CD4+ FoxP3neg) or Treg cells (CD4+ FoxP3+) treated in vitro, observing no significant changes (Figure 6B).
Figure 6.
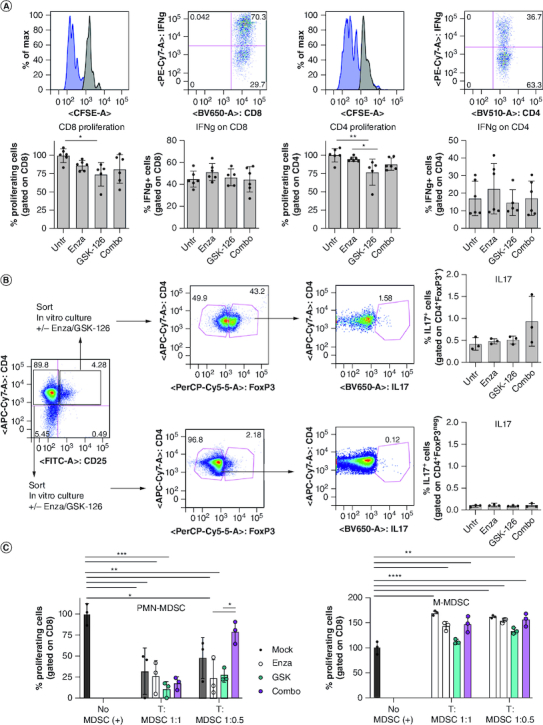
The COMBO treatment does not affect T cell and MDSC activity in vitro.
(A) Naive T lymphocytes purified from the spleen of C57BL/6 mice were labeled with CFSE, activated in vitro with CD3/CD28 and treated with enzalutamide, GSK-126 or the Combo. 4 days later, proliferation (CFSE dilution) and IFN-γ production were evaluated by flow cytometry. (B) Flow cytometry evaluation of IL-17 production by T regulatory (CD4+CD25+FoxP3+) or effector (CD4+CD25negFoxP3neg) T cells purified from tumor bearing mice and treated in vitro with enzalutamide, GSK-126 or combo. (C) PMN-MDSCs (CD11b+Ly6GhiLy6Cint) and M-MDSC-like cells (CD11b+Ly6GlowLy6Chi) isolated from TRAMP mice previously treated with enzalutamide, GSK-126 or the Combo were tested in vitro for suppressive activity against responder T cells. MDSC: responder ratio 1:1, 1:0.5 as indicated. All histograms report mean ± SD of biological replicates, represented by dots. One-way ANOVA followed by Tukey's multiple comparison test *p < 0.05; **p < 0.01; ***p < 0.001, and ****p < 0.0001. Where P-value is not indicated, the comparison between groups is not statistically significant.
Having described that PMN-MDSC contribute to CD8+ T cell tolerance in TRAMP mice [9], we also investigated the suppressive activity of PMN-MDSC isolated from TRAMP mice previously treated with enzalutamide, GSK-126 or the COMBO, finding no significant modulation (Figure 6C). As for M-MDSC, not suppressive in TRAMP mice [9], none of the treatments altered their functions (Figure 6C).
We also measured the production of CD8+ T-cell stimulating cytokines, IFN-γ and IL-2 [50,51], by T1525, T23 and ST4787 tumor cells after in vitro treatment. Interestingly, the COMBO significantly increased the production of both IFN-γ and IL-2 in all tumor cells, albeit the former reached only very low levels (Supplementary Figure 6). However, the COMBO also seemed to enhance the release of IL-17, which can instead blunt Th1 activity [52]. This result suggests that, in tumor bearing mice treated with the COMBO, tumor cell could play a role in shaping T cell activation. This possible crosstalk would be the object of future investigation.
In conclusion, our results show that the simultaneous administration of enzalutamide and GSK-126 can restore tumor cell sensitivity to enzalutamide, reduce NEPC differentiation and simultaneously awake the anti-tumor specific T-cell response, in vivo, such to restrain the growth of prostate cancer. They also suggest that the increased T cell activity that we see in vivo as a consequence of the combination therapy could be related to the crosstalk with other cells in the microenvironment, including tumor cell themselves, however excluding MDSC as a relevant player in this setting.
Discussion
Effective treatment of CRPC and NEPC remains an unmet clinical need. Immune-checkpoint blockade revolutionized the therapy of many solid tumors, but proved scarce success in ‘cold’ prostate cancer. To identify new approaches for reverting castration resistance and simultaneously blunting immune suppression, we here investigated in preclinical models the combination of enzalutamide with the EZH2 inhibitor GSK-126.
In vitro, the COMBO treatment restored enzalutamide sensitivity in otherwise resistant cells, as similarly shown in other preclinical models [37,38]. Importantly, our results moved the finding further ahead proving the effectiveness of this combination in vivo against NEPC differentiation, and establishing the importance of the immune system to gain therapeutic efficacy of both enzalutamide alone and the COMBO. It is worth to say that in our in vivo experiments we used a relatively low dose of GSK-126 (30 mg/Kg) compared with that administered in other studies. Yet, we think that our results are not in contrast with the existing literature for several reasons. Indeed, we did not demonstrate any in vivo antitumor activity by GSK-126 when administered alone; rather, the effect was only observed when we combined GSK-126 and enzalutamide. Thus, our data do not contradict the findings from the paper of Huang et al. [25], which tested GSK-126 as a single agent (showing that only doses higher than 50 mg/Kg have antitumor activity). A different paper evaluated a single dose of GSK-126 in a completely different model (iv. injection of human tumor cells in nude mice, to model experimental metastases) [53]; therefore, it is difficult to make a comparison with our subcutaneous prostate cancer models. Lastly, as far as combination with either castration or enzalutamide was tested in immunodeficient mice [27,37], these were performed on prostate cancer models different from those used in our study. It is conceivable that different cells may have different sensitivity to GSK-126 doses. Alternatively, we could speculate that the relatively low dose of GSK-126 we administered might have unveiled the possibility to appreciate its direct effects on the microenvironment, which might have been otherwise masked by a strong direct activity on tumor cells exerted by higher doses of GSK-126. These results would require further investigation to determine the possible benefit of adding immunotherapy after enzalutamide and GSK-126 conditioning of the microenvironment. However, it is worth noting that both the manuscripts from Bolis and Ku [27,37] lack a comparison with a syngeneic, immunocompetent model.
Furthermore, the 30 mg/Kg dosage was also used in the paper by Peng et al. [21], which showed that the combination of GSK-126 and a second epigenetic modulator (the DNMT inhibitor 5-AZA-dC) synergized with adoptive T cell transfer, causing increased tumor T cell infiltration, production of Th1-type chemokines, and reduction of tumor growth. Indeed, this and other papers showed that either EZH2 or AR inhibitors, alone, can trigger the production of inflammatory cytokines by CD8+ and CD4+ T cells in other tumor models [15,21,23]. Remarkably, while we observed that an intact immune system is needed to gain in vivo effectiveness of enzalutamide in a model that proved to be resistant in vitro, we found that only the enzalutamide and GSK-126 COMBO can awake tumor infiltrating T cells. Also, our observation that the two drugs did not systemically modulate immune cells, except for splenic macrophages, and did not directly alter T cell activity, suggests that an interplay with tumor cells and/or with other elements of the microenvironment is necessary to restore anti-tumor T cell function, in vivo. Nevertheless, our effort to identify the nature of such supporting cells has been so far elusive and will be object of future studies.
Differently from what shown in another NEPC model [37], in the ST4787 NEPC cell line GSK-126 administration did not restore sensitivity to enzalutamide. As NEPC emerges from CRPC in a lineage-plasticity process occurring through several intermediate states [2,4], it is conceivable that the DKO and TKO models used by Ku [37] and our ST4787 cell line represent different intermediates in this transition, with ST4787 closer to fully committed NEPC and no longer sensitive to epigenetic modulation. Nevertheless, as already shown in vitro [54], EZH2 inhibition can efficiently restrain NEPC differentiation induced in vivo by ADT/ARPI, underscoring that the COMBO should be initiated before completion of the CRPC-to-NEPC transition to be effective.
Furthermore, in vivo, the microenvironment of ST4787 NEPC tumors was infiltrated by low numbers of immune cells, and, among the few infiltrating, by a marked reduction of CD8+ T cells and a strong increase in Treg cells, in comparison to T23-derived CRPC tumors. In such a ‘cold’ NEPC microenvironment, the potential benefic effect of the COMBO treatment on T cell activation would not be sufficient to overcome immunosuppression. These differences in the CRPC and NEPC immune milieu would require further investigation in additional prostate cancer models and in patient-derived specimens, such to establish the possible benefit of adding immunotherapy (i.e., immune-checkpoint blockade therapy) after enzalutamide and GSK-126 conditioning of the antitumor immune response. Further investigation is also required to dissect the alterations induced in the tumor microenvironment by either ADT or ARPI alone, as well as their combination. These analyses will be the focus of future research, building upon the findings presented in this paper.
Conclusion
We here provide evidence that the concomitant inhibition of AR and EZH2 can restrain CRPC growth and NEPC differentiation, restoring tumor cell sensitivity to enzalutamide, and also awakening anti-tumor T-cell response, opening new roads for immunotherapy in prostate cancer.
Supplementary Material
Acknowledgments
We thank Fondazione IRCCS Istituto Nazionale dei Tumori: Ester Grande for administrative assistance and Lorena Ventura for technical assistance with immunohistochemistry.
Funding Statement
This work was supported by Grants from Fondazione IRCCS Istituto Nazionale dei Tumori, Milano (Bando per la Valorizzazione della Ricerca Istituzionale, BRI, 2018 to E.J.), Italian Ministry of Health (GR-2016-02362484 to E.J. and “Ricerca Corrente Funds”) and Associazione Italiana per la Ricerca sul Cancro (AIRC; Investigator Grant 24363 to MPC). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Author contributions
I Fischetti: investigation, methodology, writing-original draft, visualization. L Botti: investigation. R Sulsenti: investigation. V Cancila: investigation. C Enriquez: investigation. R Ferri: investigation, methodology. M Bregni: resources. F Crivelli: resources. C Tripodo: investigation, validation. MP Colombo: supervision, funding acquisition, writing reviewing and editing. E Jachetti: conceptualization, investigation, supervision, funding acquisition, writing-original draft, writing review and editing, visualization.
Financial disclosure
This work was supported by Grants from Fondazione IRCCS Istituto Nazionale dei Tumori, Milano (Bando per la Valorizzazione della Ricerca Istituzionale, BRI, 2018 to E.J.), Italian Ministry of Health (GR-2016-02362484 to E.J. and “Ricerca Corrente Funds”) and Associazione Italiana per la Ricerca sul Cancro (AIRC; Investigator Grant 24363 to MPC). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Competing interests disclosure
The authors have no competing interests or relevant affiliations with any organization or entity with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Writing disclosure
No writing assistance was utilized in the production of this manuscript.
Ethical conduct of research
Prostatectomies from prostate cancer patients were obtained from ASST Valle Olona, Busto Arsizio (VA), Italy (Protocol number 0046679/18), upon informed consent and in accordance with the Helsinki Declaration. Tissue collection, fixation and processing followed standardized protocols as part of routine clinical activity. Animal housing and experimentation were performed following institutional guidelines and the Italian law (D.Lgs. 26/2014). In vivo experiments were approved by the Italian Ministry of Health (authorization number 8/2020-PR).
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Sung H, Ferlay J, Siegel RLet al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 71(3), 209–249 (2021). [DOI] [PubMed] [Google Scholar]
- 2.Davies AH, Beltran H, Zoubeidi A. Cellular plasticity and the neuroendocrine phenotype in prostate cancer. Nat. Rev. Urol. 15(5), 271–286 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Aggarwal R, Huang J, Alumkal JJet al. Clinical and genomic characterization of treatment-emergent small-cell neuroendocrine prostate cancer: a multi-institutional prospective study. J. Clin. Oncol. 36(24), 2492–2503 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beltran H, Hruszkewycz A, Scher HIet al. The role of lineage plasticity in prostate cancer therapy resistance. Clin. Cancer Res. 25(23), 6916–6924 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drake CG, Sharma P, Gerritsen W. Metastatic castration-resistant prostate cancer: new therapies, novel combination strategies and implications for immunotherapy. Oncogene 33(43), 5053–5064 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bilusic M, Madan RA, Gulley JL. Immunotherapy of prostate cancer: facts and hopes. Clin. Cancer Res. 23(22), 6764–6770 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drake CG. Prostate cancer as a model for tumour immunotherapy. Nat. Rev. Immunol. 10(8), 580–593 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Idorn M, Kollgaard T, Kongsted P, Sengelov L, Thor Straten P. Correlation between frequencies of blood monocytic myeloid-derived suppressor cells, regulatory T cells and negative prognostic markers in patients with castration-resistant metastatic prostate cancer. Cancer Immunol. Immunother. 63(11), 1177–1187 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jachetti E, Cancila V, Rigoni Aet al. Cross-Talk between myeloid-derived suppressor cells and mast cells mediates tumor-specific immunosuppression in prostate cancer. Cancer Immunol. Res. 6(5), 552–565 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Sorrentino C, Musiani P, Pompa P, Cipollone G, Di Carlo E. Androgen deprivation boosts prostatic infiltration of cytotoxic and regulatory T lymphocytes and has no effect on disease-free survival in prostate cancer patients. Clin. Cancer Res. 17(6), 1571–1581 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Calcinotto A, Spataro C, Zagato Eet al. IL-23 secreted by myeloid cells drives castration-resistant prostate cancer. Nature 559(7714), 363–369 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cioni B, Zwart W, Bergman AM. Androgen receptor moonlighting in the prostate cancer microenvironment. Endocr. Relat. Cancer 25(6), R331–R349 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Gubbels Bupp MR, Jorgensen TN. Androgen-induced immunosuppression. Front. Immunol. 9, 794 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walecki M, Eisel F, Klug Jet al. Androgen receptor modulates Foxp3 expression in CD4+CD25+Foxp3+ regulatory T-cells. Mol. Biol. Cell 26(15), 2845–2857 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Preclinical study showing that androgen receptor (AR) can control FoxP3 expression in Tregs.
- 15.Guan X, Polesso F, Wang Cet al. Androgen receptor activity in T cells limits checkpoint blockade efficacy. Nature 606(7915), 791–796 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Seminal paper showing that AR can directly prevent the expression of IFN-γ and granzyme B in CD8 T cells, thus blunting T cell activation.
- 16.Becerra-Diaz M, Strickland AB, Keselman A, Heller NM. Androgen and androgen receptor as enhancers of M2 macrophage polarization in allergic lung inflammation. J. Immunol. 201(10), 2923–2933 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scalerandi MV, Peinetti N, Leimgruber Cet al. Inefficient N2-like neutrophils are promoted by androgens during infection. Front. Immunol. 9, 1980 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trigunaite A, Khan A, Der E, Song A, Varikuti S, Jorgensen TN. Gr-1(high) CD11b+ cells suppress B cell differentiation and lupus-like disease in lupus-prone male mice. Arthritis Rheum. 65(9), 2392–2402 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Consiglio CR, Udartseva O, Ramsey KD, Bush C, Gollnick SO. Enzalutamide, an androgen receptor antagonist, enhances myeloid cell-mediated immune suppression and tumor progression. Cancer Immunol. Res. 8(9), 1215–1227 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; • This work in mouse models shows that enzalutamide enhances myeloid-derived suppressor cells recruitment and favors the growth of AR-negative tumors.
- 20.Vire E, Brenner C, Deplus Ret al. The polycomb group protein EZH2 directly controls DNA methylation. Nature 439(7078), 871–874 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Peng D, Kryczek I, Nagarsheth Net al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 527(7577), 249–253 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• First paper demonstrating that the inhibition of EZH2 enhances T cell activation and response to anti-PD-1 immunotherapy.
- 22.Wang D, Quiros J, Mahuron Ket al. Targeting EZH2 reprograms intratumoral regulatory T cells to enhance cancer immunity. Cell Rep. 23(11), 3262–3274 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goswami S, Apostolou I, Zhang Jet al. Modulation of EZH2 expression in T cells improves efficacy of anti-CTLA-4 therapy. J. Clin. Invest. 128(9), 3813–3818 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Paper confirming that EZH2 inhibition stimulates T cell function and improves the effectiveness of anti-CTLA-4 immunotherapy in mouse models.
- 24.Zhou J, Liu M, Sun Het al. Hepatoma-intrinsic CCRK inhibition diminishes myeloid-derived suppressor cell immunosuppression and enhances immune-checkpoint blockade efficacy. Gut 67(5), 931–944 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang S, Wang Z, Zhou Jet al. EZH2 Inhibitor GSK126 suppresses antitumor immunity by driving production of myeloid-derived suppressor cells. Cancer Res. 79(8), 2009–2020 (2019). [DOI] [PubMed] [Google Scholar]
- 26.Li C, Song J, Guo Zet al. EZH2 inhibitors suppress colorectal cancer by regulating macrophage polarization in the tumor microenvironment. Front. Immunol. 13, 857808 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bolis M, Bossi D, Vallerga Aet al. Dynamic prostate cancer transcriptome analysis delineates the trajectory to disease progression. Nat. Commun. 12(1), 7033 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamagishi M, Uchimaru K. Targeting EZH2 in cancer therapy. Curr. Opin. Oncol. 29(5), 375–381 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat. Med. 22(2), 128–134 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Varambally S, Dhanasekaran SM, Zhou Met al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 419(6907), 624–629 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Cao Q, Yu J, Dhanasekaran SMet al. Repression of E-cadherin by the polycomb group protein EZH2 in cancer. Oncogene 27(58), 7274–7284 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu J, Yu J, Rhodes DRet al. A polycomb repression signature in metastatic prostate cancer predicts cancer outcome. Cancer Res. 67(22), 10657–10663 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Liu Q, Wang G, Li Qet al. Polycomb group proteins EZH2 and EED directly regulate androgen receptor in advanced prostate cancer. Int. J. Cancer 145(2), 415–426 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim J, Lee Y, Lu Xet al. Polycomb- and methylation-independent roles of EZH2 as a transcription activator. Cell Rep. 25(10), 2808–2820; e2804 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu K, Wu ZJ, Groner ACet al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is polycomb-independent. Science 338(6113), 1465–1469 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dardenne E, Beltran H, Benelli Met al. N-Myc induces an EZH2-mediated transcriptional program driving neuroendocrine prostate cancer. Cancer Cell 30(4), 563–577 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Elegant demonstration in mouse models that EZH2 mediates neuroendocrine prostate cancer development.
- 37.Ku SY, Rosario S, Wang Yet al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 355(6320), 78–83 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• First demonstration in the PBCre4:Ptenflfl:Rb1flfl:Trp53flfl mouse model that EZH2 inhibition restores sensitivity to enzalutamide.
- 38.Xiao L, Tien JC, Vo Jet al. Epigenetic reprogramming with antisense oligonucleotides enhances the effectiveness of androgen receptor inhibition in castration-resistant prostate cancer. Cancer Res. 78(20), 5731–5740 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pittoni P, Tripodo C, Piconese Set al. Mast cell targeting hampers prostate adenocarcinoma development but promotes the occurrence of highly malignant neuroendocrine cancers. Cancer Res. 71(18), 5987–5997 (2011). [DOI] [PubMed] [Google Scholar]
- 40.Sulsenti RFB, Bongiovanni L, Cancila Vet al. Repurposing of the Antiepileptic Drug Levetiracetam to Restrain Neuroendocrine Prostate Cancer and Inhibit Mast Cell Support to Adenocarcinoma. Front. Immunol. 12, 622001 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Greenberg NM, Demayo F, Finegold MJet al. Prostate cancer in a transgenic mouse. Proc. Natl Acad Sci. USA 92(8), 3439–3443 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Enriquez C, Cancila V, Ferri Ret al. Castration-induced down-regulation of SPARC in stromal cells drives neuroendocrine differentiation of prostate cancer. Cancer Res. 81(16), 4257–4274 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beltran H, Tomlins S, Aparicio Aet al. Aggressive variants of castration-resistant prostate cancer. Clin. Cancer Res. 20(11), 2846–2850 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, Dehm SM. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 73(2), 483–489 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith R, Liu M, Liby Tet al. Enzalutamide response in a panel of prostate cancer cell lines reveals a role for glucocorticoid receptor in enzalutamide resistant disease. Sci. Rep. 10(1), 21750 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Masoodi KZ, Eisermann K, Yang Zet al. Inhibition of androgen receptor function and level in castration-resistant prostate cancer cells by 2-[(isoxazol-4-ylmethyl)thio]-1-(4-phenylpiperazin-1-yl)ethanone. Endocrinology 158(10), 3152–3161 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ishikawa F, Yasukawa M, Lyons Bet al. Development of functional human blood and immune systems in NOD/SCID/IL2 receptor gamma chain(null) mice. Blood 106(5), 1565–1573 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Degl'innocenti E, Grioni M, Boni Aet al. Peripheral T cell tolerance occurs early during spontaneous prostate cancer development and can be rescued by dendritic cell immunization. Eur. J. Immunol. 35(1), 66–75 (2005). [DOI] [PubMed] [Google Scholar]
- 49.Mylin LM, Bonneau RH, Lippolis JD, Tevethia SS. Hierarchy among multiple H-2b-restricted cytotoxic T-lymphocyte epitopes within simian virus 40 T antigen. J. Virol. 69(11), 6665–6677 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bhat P, Leggatt G, Waterhouse N, Frazer IH. Interferon-gamma derived from cytotoxic lymphocytes directly enhances their motility and cytotoxicity. Cell Death Dis. 8(6), e2836 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ross SH, Cantrell DA. Signaling and function of interleukin-2 in T lymphocytes. Annu. Rev. Immunol. 36, 411–433 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mills KHG. IL-17 and IL-17-producing cells in protection versus pathology. Nat. Rev. Immunol. 23(1), 38–54 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen YT, Zhu F, Lin WR, Ying RB, Yang YP, Zeng LH. The novel EZH2 inhibitor, GSK126, suppresses cell migration and angiogenesis via down-regulating VEGF-A. Cancer Chemother. Pharmacol. 77(4), 757–765 (2016). [DOI] [PubMed] [Google Scholar]
- 54.Puca L, Bareja R, Prandi Det al. Patient derived organoids to model rare prostate cancer phenotypes. Nat. Commun. 9(1), 2404 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.