

Abstract
Vincristine and bortezomib are effective chemotherapeutics widely used to treat hematological cancers. Vincristine blocks tubulin polymerization, whereas bortezomib is a proteasome inhibitor. Despite different mechanisms of action, the main non-hematological side effect of both is peripheral neuropathy that can last long after treatment has ended and cause permanent disability. Many different cellular and animal models of various aspects of vincristine and bortezomib-induced neuropathies have been generated to investigate underlying molecular mechanisms and serve as platforms to develop new therapeutics. These models revealed that bortezomib induces several transcriptional programs in dorsal root ganglia that result in the activation of different neuroinflammatory pathways and secondary central sensitization. In contrast, vincristine has direct toxic effects on the axon, which are accompanied by changes similar to those observed after nerve cut. Axon degeneration following both vincristine and bortezomib is mediated by a phylogenetically ancient, genetically encoded axon destruction program that leads to the activation of the Toll-like receptor adaptor SARM1 (sterile alpha and TIR motif containing protein 1) and local decrease of nicotinamide dinucleotide (NAD+). Here, I describe current in vitro and in vivo models of vincristine- and bortezomib induced neuropathies, present discoveries resulting from these models in the context of clinical findings and discuss how increased understanding of molecular mechanisms underlying different aspects of neuropathies can be translated to effective treatments to prevent, attenuate or reverse vincristine- and bortezomib-induced neuropathies. Such treatments could improve the quality of life of patients both during and after cancer therapy and, accordingly, have enormous societal impact.
Keywords: chemotherapy-induced neuropathy, neuroinflammation, pain, axon degeneration, SARM1, therapeutics
Introduction
Vincristine (VCR) and bortezomib (BTZ) are powerful chemotherapeutic drugs widely used to treat several hematological cancers. The discovery of VCR dates back to 1958, when two research teams, one from the University of Western Ontario and the other from the pharmaceutical company Eli Lilly, independently tested the biological effects of an extract from the Madagascar rosy periwinkle (Catharanthus roseus, previously named Vinca rosea), a plant with beautiful deep pink flowers, in the hope of identifying new drugs to treat diabetes (Duffin, 2002; Johnson et al., 1963; Noble, 1990). This plant has enjoyed a popular reputation as a medicinal among indigenous peoples in various parts of the world, e.g., as a hypoglycemic by natives of Madagascar, the Philippines and South Africa. Although it turned out that the periwinkle extract was not very effective at lowering the blood sugar levels of mice, it dramatically decreased the number of leukocytes and prolonged survival of mice with leukemia. Subsequent fractionation of the vinca plant extract led to the isolation of an indole alkaloid that was named vincristine (Johnson et al., 1963). Further research showed that VCR inhibits the polymerization of tubulin and its incorporation into microtubules, thereby preventing mitotic spindle assembly and leading to an attenuation of mitosis and consequent apoptosis (Himes et al., 1976; Owellen et al., 1976; Smith et al., 2016). In 1963, VCR was approved by the FDA as Oncovin, and it is still widely used for treating leukemias, lymphomas, brain tumors, and solid tumors in adults (Gidding et al., 1999). It is also administered as part of treatment protocols for children with acute lymphoblastic leukemia, neuroblastoma, Wilms tumor, rhabdomyosarcoma, Ewing sarcoma, and retinoblastoma (Diouf et al., 2015; Gidding et al., 1999; Ness et al., 2013). After VCR came to the market, the survival rates of acute lymphoblastic leukemia in children increased from 10% to 90% (Gatta et al., 2005; Pui et al., 2009; Vora et al., 2013). VCR is now among the most commonly used anticancer drugs worldwide.
Disrupting tubulin polymerization also induces apoptosis in other rapidly dividing cell types and, in addition, blocks anterograde and retrograde axonal transport, resulting in multiple side effects, including myelosuppression, hair loss and neurotoxicity. Neurotoxicity is the most common dose limiting side effect of VCR, manifesting mainly as axonal sensorimotor and autonomic neuropathies, and, less commonly, cranial neuropathies (Deangelis et al., 1991; Lavoie Smith et al., 2015; Madsen et al., 2019). The severity of VCR-induced peripheral neuropathy (VIPN) in patients depends on dose intensity and total cumulative dose wherein higher individual and cumulative doses are associated with more severe neuropathy (Casey et al., 1973a; Sandler et al., 1969; Verstappen et al., 2005). Most adult patients treated with VCR develop sensory symptoms beginning at a cumulative dose of 4–5 mg and motor symptoms at doses around 30–50 mg (Casey et al., 1973). Loss of ankle jerks is usually the first manifestation produced by VCR, followed by paraesthesias (numbness and tingling) in the fingers and a little later in the toes (Casey et al., 1973; Verstappen et al., 2005). In a study of patients receiving high intensity chemotherapy with VCR (1.33 mg VCR weekly) 70% of patients experienced numbness, 62% pain and 60% tingling sensations, whereas low intensity VCR (0.67 mg VCR per week) resulted in numbness, pain and tingling in 43%, 14%, and 34% of patients, respectively. If weakness develops, it affects initially finger and wrist extensor muscles, and later toe and foot dorsiflexion. Patients frequently complain of difficulties writing, buttoning, walking and walking stairs. On neurological examination, patients have an abnormal Romberg sign, tandem walking, decreased sensation to pin prick and vibration and reduced strength of wrist and finger extension and toe- and ankle dorsiflexion (Casey et al., 1973b; Verstappen et al., 2005). Nerve conduction studies demonstrate decreased amplitude of distal muscle and sensory nerve potentials with only slight variations in conduction velocities, consistent with an axonal neuropathy (Casey, 1973; Guiheneuc et al., 1980) In addition, VCR can cause autonomic dysfunction, which leads to constipation, paralytic ileus, urinary retention and orthostatic hypotension (Hirvonen et al., 1989; Roca et al., 1985; Sandler et al., 1969). Cranial neuropathies, described mainly in children, are associated with vocal cord paralysis, ocular muscle paresis and tongue weakness (Dixit et al., 2012; Latiff et al., 2010; Naithani et al., 2009; Ryan et al., 1999). VIPN typically improves in patients after VCR treatment has ended, but further deterioration of neurologic signs continues for about 4 weeks after the last dose of VCR (“coasting” phenomenon) in about 30% of patients (Casey et al., 1973; Verstappen et al., 2005).
While VCR was discovered somewhat serendipitously, BTZ was identified in a targeted screen for inhibition of the proteasome (Adams, 2002). The proteasome plays a critical role in the regulated degradation of more than 80% of cellular proteins and is required for normal function of the cell. Thus, scientists surmised that dysregulating the degradation of such proteins should have profound effects on tumor growth and cause cells to undergo apoptosis. Indeed, compound PS-341 (later called bortezomib) caused cell death in several tumor cell lines maintained by the National Cancer Institute and decreased tumor growth in mice (Adams, 2002; Adams et al., 1999). Importantly, cancerous cells were much more sensitive to blockade of the proteasome than normal cells. BTZ inhibits the 20S core proteasome, resulting in cancer cell death via multiple mechanisms, including suppression of the unfolded protein response, accumulation of ubiquitinated proteins, stabilization of tumor suppressor proteins, such as p21, p27, Bax and p54, and induction of reactive oxygen species (Arkwright et al., 2017; Bladé et al., 2005; Utecht and Kolesar, 2008). A prominent theory regarding the cancer-killing properties of BTZ is the decreased activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB), a transcription factor bound in the cytoplasm to its inhibitory regulatory protein IκB. Cellular stress and stimulation with pro-inflammatory signals induce the ubiquitination and subsequent degradation of IκB, leading to release and translocation of NFκB to the nucleus, where it induces the expression of specific proinflammatory and antiapoptotic genes. BTZ prevents the degradation of IκB and the consequent activation of NFκB, resulting in cell cycle arrest and induction of the apoptotic cascade (Adams, 2002; Arkwright et al., 2017). BTZ was approved by the FDA in 2003 for treatment of multiple myeloma and in 2007 to treat mantle cell lymphoma (Kane et al., 2006). Following FDA approval of BTZ, it has been used in the clinic for a large number of hematologic cancer patients.
Peripheral neuropathy is considered the main non-hematological toxicity of BTZ, and can result in dose modifications depending on its severity (Richardson et al., 2009, 2006). The incidence of bortezomib-induced neuropathy (BIPN) varies depending on the study, assessment method and patient population. A meta-analysis of 25 clinical trials including 3459 patients treated with intravenous BTZ revealed an incidence of all grade peripheral neuropathies of 33.9% and that of high-grade events of 8.1% (Peng et al., 2015). BIPN typically starts in the feet and is characterized by numbness and tingling, moderate to severe burning pain, and imbalance (Bilińska et al., 2013; Chaudhry et al., 2008; Expósito Vizcaíno et al., 2018; Zaroulis et al., 2014). Rarely, BTZ can also cause severe autonomic neuropathy manifesting as orthostatic hypotension (Giannoccaro et al., 2011; Stratogianni et al., 2012). On neurological examination, patients have loss of sensation to pinprick, vibration and proprioception in a stocking-glove distribution and decreased tendon stretch reflexes in their legs (Chaudhry et al., 2008; Richardson et al., 2009, 2006; Zaroulis et al., 2014). Sensory nerve action potential (SNAP) amplitudes of the sural nerve were decreased in 86% of patients with BIPN, indicating degeneration of large myelinated axons in sensory nerves, while a reduction of compound motor action potentials has been observed in up to 50% of patients (Bechakra et al., 2018; Chaudhry et al., 2008; Richardson et al., 2009; Stubblefield et al., 2006). A decrease of unmyelinated intraepidermal nerve fiber density (IENFD) on skin biopsies was noticed in some studies (Giannoccaro et al., 2011), but not others (Bechakra et al., 2020, 2018; Chaudhry et al., 2008; Richardson et al., 2009), which, instead found no change (Chaudhry et al., 2008) or increased IENFD (Bechakra et al., 2020; Richardson et al., 2009) and a decrease of nerve fibers in the subdermal plexus (Bechakra et al., 2020, 2018). In addition to the toxic neuropathy and in contrast to many other chemotherapy-inducing agents, BTZ can cause a severe, immune-mediated, demyelinating neuropathy (Pitarokoili et al., 2017; Ravaglia et al., 2008; Saifee et al., 2010; Schmitt et al., 2011; Xu et al., 2019).
VCR- and BTZ-induced neuropathies can last long after chemotherapy has ended and cause permanent disability and reduced quality of life. This is an especially significant problem in children with a long expected lifespan after treatment with VCR (Goldsby et al., 2010; Kandula et al., 2018; Kirchhoff et al., 2011; Lieber et al., 2018; Wright et al., 2017). Indeed, the clinical significance of chemotherapy-induced neuropathy (CIPN) is increasing proportionately with the increasing number of cancer survivors. While the mechanisms of action of both VCR and BTZ on cancer cells are well established, the molecular mechanisms that lead to peripheral neuropathy represent an area of intense research. Cellular (in vitro) and animal (in vivo) models have been generated to reveal mechanisms and screen for therapeutics (Bruna et al., 2020; Höke and Ray, 2014; Lehmann et al., 2020). More recently, these models have been complemented by genetic studies in patients (Campo et al., 2018; Diouf et al., 2015; Mahmoudpour et al., 2018), which generate new hypotheses that can then be taken back to the lab and tested in appropriate in vitro and in vivo models.
Here, I describe key discoveries that emerged from consideration of different in vitro and in vivo models of VCR- and BTZ induced neuropathies in the context of clinical findings. I then summarize recent findings pertinent to a common axon degeneration program that underlies VCR- and BTZ induced neuropathies and discuss how our understanding of molecular mechanisms of neuropathies can be translated to effective treatments for VIPN and BIPN.
Modeling BTZ-induced neurotoxicity
In vitro models of BTZ-induced neurotoxicity
Cellular models provide simple, well controlled and cost-effective systems that can be used as screening platforms to investigate cell-autonomous processes (Lehmann et al., 2020). Several in vitro model systems have been employed to evaluate BTZ-induced neurotoxicity, including primary neurons, murine neural stem cells, neurons derived from human induced pluripotent stem cells and neurons differentiated from rat pheochromocytoma- and human neuroblastoma cell lines (Meregalli et al., 2014; Poruchynsky et al., 2008; Staff et al., 2013; Wing et al., 2017). BIPN is sensory predominant and affects sensory dorsal root ganglion neurons (DRGs) and their processes early in the disease. Neurons derived from DRGs of embryonic, postnatal and adult animals can be easily grown in culture. Embryonic rodent DRG neuron cultures have the advantage of higher cell yields and produce greater proportions of neurons compared to DRGs cultures obtained from adults, but the cells are more immature and require neurotrophin supplementation during the first week of culture and, thus, may not fully represent the changes seen in adult neurons. In embryonic neuron cultures of mouse and rat DRGs, 50–100 nM BTZ, but not 10 nM, induces a dose-dependent, long-lasting inhibition of the proteasome (Meregalli et al., 2014), which leads to differential expression of 92 proteins related to cellular component organization (protein processing in the endoplasmic reticulum), macromolecular complex subunit organization, protein complex subunit organization, and cellular component assembly (Karademir et al., 2018). Proteasome inhibition in DRGs is associated with axon injury, as shown either by fragmented neurites (Geisler et al., 2019a) or decreased neurite outgrowth (Staff et al., 2013), indicating that BTZ affects neurites, which is similar to the clinical phenotype of axonal sensory neuropathy seen in patients treated with BTZ. The dose of BTZ in these in vitro models is clinically relevant, as the standard dose of BTZ (1.3 mg/m2) given to patients yields a median peak blood concentration that corresponds to a free concentration of 110 nM BTZ at peak (Staff et al., 2013).
BIPN symptoms in patients start in the feet, which may indicate perturbed axonal transport possibly due to interference with tubulin dynamics. Indeed, increased tubulin polymerization has been observed within 24 hours after adding 100 nM BTZ to embryonic DRGs (Meregalli et al., 2014; Staff et al., 2013). Similarly, neurons derived from human SH-SY5Y and KCNR neuroblastoma cells treated with 100nM BTZ show an increase of the polymerized tubulin fraction (Poruchynsky et al., 2008). The magnitude of tubulin polymerization is comparable to the effect of the microtubule stabilizer paclitaxel, suggesting that this might contribute to BTZ neurotoxicity (Poruchynsky et al., 2008). In contrast to paclitaxel, however, BTZ does not lead to significant microtubule bundling or polymerization of purified microtubule protein in solution, which suggests that the effect of BTZ is indirect and not a result of direct binding to microtubulin as occurs with other microtubulin stabilizing agents, such as paclitaxel (Poruchynsky et al., 2008). In a human cortical cell line (HCN2), but not embryonic DRGs, increased acetylation of alpha-tubulin was observed (Poruchynsky et al., 2008; Staff et al., 2013). Acetylation of α-tubulin is a post-translational modification indicative of stabilized microtubules. The tubulin polymerization is a class effect of proteasome inhibitors, because six different proteasome inhibitors induce tubulin polymerization in three different cell lines and embryonic rat DRGs (Meregalli et al., 2014; Poruchynsky et al., 2008; Staff et al., 2013). Increased tubulin polymerization and acetylated tubulin may cause decreased axonal transport. Indeed, decreased mitochondrial transport and increased histone deacetylase 6 (HDAC6) aggresomes in the soma were observed within the first 24 hours after BTZ was added to embryonic DRGs and HCN2 neurons (Poruchynsky et al., 2008; Staff et al., 2013).
Embryonic DRG sensory neurons are particularly suitable to be cultured in compartmentalized chambers, which allows for the isolation of axon and soma compartments to determine the subcellular site of action of the drug. Surprisingly, although BTZ induces an axonal neuropathy with symptoms starting in the feet, axon fragmentation was not induced by adding BTZ to the axon compartment (Geisler et al., 2019a). However, BTZ applied to the soma produced axon degeneration within 24 hours. These results indicate that the cell body is necessary for BTZ-induced axon degeneration, perhaps by transcribing a factor that is transported from the soma into the axon to activate a degeneration program, which has been shown for degeneration induced by local deprivation of nerve growth factors (NGF; Simon et al., 2016). In this regard it may be interesting that serum NGF is significantly decreased in patients who developed BIPN (Yan et al., 2020; Youk et al., 2017), while NGF levels were unchanged in patients who did not develop BIPN (Youk et al., 2017), suggesting that low NGF contributes to BIPN (Yan et al., 2020). Intramuscular injection of rat NGF in combination with oral vitamin B supplements for two months led to alleviation of symptoms and improvement of electromyography in patients with BIPN (Yan et al., 2020). However, whether NGF can be used therapeutically to decrease BIPN will require further investigation as a large phase III clinical trial of 1019 patients with diabetic neuropathy randomized to receive either recombinant human NGF or placebo for 48 weeks failed to show efficacy and was associated with high toxicity manifesting as hyperalgesia and allodynia at the injection site (Apfel, 2002a, 2002b).
DRG neuron cultures derived from adult animals (eight weeks) also exhibit proteasome inhibition, decreased neurite outgrowth and tubulin polymerization, but with greater vulnerability than embryonic neurons, because as little as 10 nM BTZ is sufficient to induce these changes in adult DRG culture (Meregalli et al., 2014).
BTZ also induces dose-dependent reduction of axon outgrowth and cell viability in human iPSC derived neurons, which may be especially useful to screen for therapeutics (Wing et al., 2017).
In vivo models of BTZ-neurotoxicity
While mechanistic studies of BTZ neurotoxicity performed in vitro are valuable, their inability to capture the inherent complexity of organ systems is a major drawback. Accordingly, results obtained from in vitro studies frequently benefit from in vivo verification. While animal models address many of the shortcomings of in vitro studies, they are time and resource-intensive, require advanced personnel training and must adhere to strict ethical guidelines (Bruna et al., 2020). To increase translatability to clinical practice, outcome measures of preclinical studies should focus on parameters and biomarkers with proven relevance in clinical settings (Cavaletti, 2014). Most animal models use intraperitoneal (i.p.) or intravenous (i.v.) injections of the chemotherapeutic drug. However, i.p. administration is not a mode of delivery in patients because of local toxic effects. Intravenous injections in animal models more closely resembles the situation in human patients. While it is difficult to evaluate spontaneous pain and tingling in rodents, the withdrawal of the hind paw in response to touch by a graded series of filaments (von Frey testing and its variations) is frequently used as a proxy to test for mechanical hyper- and hypoalgesia (Chaplan et al., 1994). More recently, a place escape avoidance paradigm and place preference tests were introduced to better reflect the affective/aversive component of neuropathy pain (Hamity et al., 2020, 2017). As an objective measure for axon loss and demyelination, nerve conduction studies are performed in patients and can also be done in mice and rats using the same set up and similar parameters (Argyriou et al., 2019). Similarly, skin biopsies from the ankle and distal thigh are obtained in patients to evaluate for loss of unmyelinated or thinly myelinated fibers to diagnose small fiber neuropathy (Lauria et al., 2005; McArthur et al., 1998; McCarthy et al., 1995). Using the footpad and the same stains and counting rules, these measures are also easily obtained in rodents and can be compared to findings in humans. If the animal models mirror key features observed in human patients, they can be exploited to provide information that is difficult to obtain in patients, including, particularly, insights into disease mechanisms.
An overview of different in vivo models to investigate BIPN is provided in table 1. In rats, commonly employed treatment regimens include: i) BTZ given daily over five days intraperitoneally (i.p.) or intravenously (i.v.) at doses of 0.2 or 0.4 mg/kg/day to a cumulative dose of 1 or 2 mg/kg, or ii) 0.2 mg/kg BTZ administered three times a week for 8 weeks to a cumulative dose of 4.8 mg/kg. The latter more reflects doses given to human patients in whom BTZ is infused twice weekly for two weeks (days 1, 4, 8 and 11), which is repeated after a 10-day rest period to complete one treatment cycle. Rats that receive BTZ for a short duration and with lower cumulative doses (1–2 mg/kg) show mechanical hyperalgesia that starts around day five and continues to at least day 35 (Stockstill et al., 2018). Similarly, these rats develop cold, but not heat, allodynia (Duggett and Flatters, 2017; Yamamoto et al., 2015). Administering BTZ over an eight week period, in addition, results in a decrease of the nerve conduction velocity of the caudal (tail) nerve and degeneration of myelinated and unmyelinated axons of the caudal and sciatic nerves, cytoplasmic vacuolization in satellite glia cells that envelope DRG neurons, detachment of satellite glia cells from the nerve cell body, and occasional morphological alterations in DRG neurons (Meregalli et al., 2010, 2014, 2018b).
Table 1:
Animal models of bortezomib-induced neuropathy
| Strain | Gender | Age | Regimen | Route | Cumulative dose | Findings | References |
|---|---|---|---|---|---|---|---|
| Mouse | |||||||
| ICR | male | 3–4 weeks | 0.2 mg/kg/d × 5 | i.p. | 1 mg/kg | mechanical hyperalgesia transcription in DRG neurons | (Ludman and Melemedjian, 2019) |
| C57Bl/6 | male | 9 weeks | 0.4 mg/kg 3x/week × 4 | i.p. | 4.8 mg/kg | mechanical and thermal hyperalgesia decreased sensory nerve action potential axon loss in the sciatic nerve neuroinflammation in DRGs, sciatic nerve, and spinal cord satellite glia cell degeneration, mitochondrial swelling in DRG neurons | (Boehmerle et al., 2015; Moschetti et al., 2019) |
| C57Bl/6 | male | 8 weeks | 0.8 mg/kg twice weekly × 4 | i.v. | 6.4 mg/kg | decreased IENF density | (Geisler et al., 2019) |
| OF1 | female | 10–12 weeks | 1 mg/kg twice weekly × 6 | i.p. | 12 mg/kg | mechanical hyperalgesia abnormalities on rotarod testing decreased sensory nerve amplitude potentials and conduction velocity decreased IENF density neuroinflammation | (Alé et al., 2014; Bruna et al., 2011) |
| OF1 | n/a | 10 weeks | 1 mg/kg 2x/week × 8 weeks | i.p. | 16 mg/kg | mechanical and thermal hyperalgesia decreased sensory nerve action potential axon loss in the sciatic nerve neuroinflammation in DRGs | (Alé et al., 2016) |
| Rat | |||||||
| SD | male | 300–350g | 0.15 mg/kg days 0,3,5,7 | i.p. | 0.6 mg/kg | mechanical hyperalgesia, no change to cold or heat allodynia increased neuronal firing in the dorsal horn of the spinal cord | (Caleb R. Robinson et al., 2014; C. R. Robinson et al., 2014; Robinson and Dougherty, 2015) (Duggett and Flatters, 2017) |
| SD | both | 180–200 g | 0.2 mg/kg days 0,3,7,10 | i.p. | 0.8 mg/kg | normal weight gain, mechanical and cold hyperalgesia no change to heat stimulation no effect on motor coordination | (Duggett and Flatters, 2017) |
| Wistar | male | 200–250 g | 0.2 mg/kg twice weekly × 2 weeks (days 1,4,8,11) | ip | 0.8 mg/kg | mechanical hyperalgesia and cold allodynia decreased circularity index of the axons of the sciatic nerve | (Yamamoto et al., 2015) |
| SD | male | 200–220 | 0.2 mg/kg/day × 5 days | i.p. | 1 mg/kg | mechanical hyperalgesia astrocyte activation and neuroinflammation dorsal horn spinal cord | (Stockstill et al., 2018; Zhang et al., 2014) |
| SD or Wistar | male only or female only | 220–250 | 0.4 mg/kg/day × 5 | i.p. | 2 mg/kg | mechanical and cold allodynia activation of transcriptional programs in DRG neurons neuroinflammation in DRGs | (Duan et al., 2020; Li et al., 2018; Liu et al., 2016, 2019; Zhang et al., 2014) |
| Wistar | female | Adult | 0.2 mg/kg 3x/wk × 4 weeks | i.v. | 2.4 mg/kg | decreased sensory nerve conduction velocity vacuolization of Schwann cells and myelin sheath degeneration, no change in unmyelinated axons in sciatic nerve damage satellite glia cells | (Cavaletti et al., 2007) |
| Wistar | female | 175–200g | 0.2 mg/kg 3x/wk × 8 weeks | i.v. | 4.8 mg/kg | mechanical hyperalgesia, no change in heat sensitivity decreased sensory nerve conduction velocity loss of myelinated axons and morphological alterations of unmyelinated axons in the sciatic nerve, cytoplasmic vacuolization of satellite glia cells, shrinkage of DRG neurons persistent proteasome inhibition | (Chiorazzi et al., 2013; Meregalli et al., 2014, 2012, 2010) |
Similar to rat, many different models are employed in mice with cumulative doses ranging from 1 mg/kg to 16 mg/kg. The findings in mice are similar to those obtained in rats and include mechanical hyperalgesia in shorter treatment protocols (Ludman and Melemedjian, 2019a), whereas longer protocols, in addition, report decrease of sensory nerve amplitudes and degeneration of myelinated axons in the sciatic nerve and of unmyelinated/thinly myelinated intraepidermal nerve fibers (Alé et al., 2015; Boehmerle et al., 2015; Bruna et al., 2011; Carozzi et al., 2010; Carozzi et al., 2013; Geisler et al., 2019a).
Another important consideration is that cancer patients given BTZ, particularly multiple myeloma patients, frequently have neuropathy at baseline, likely due to the hematological malignancy (Stubblefield et al., 2006). While most protocols evaluate BTZ in non-cancerous mice, Meregalli et al. established a protocol in which immunocompromised (SCID) mice were injected with human multiple myeloma cells and treated with 1 mg/kg BTZ i.v. weekly for 5 weeks (Meregalli et al., 2015), a dose that effectively decreased tumor growth in these mice. Multiple myeloma bearing mice had signs of mild axon degeneration in the sciatic nerve and a decreased soma size of DRG neurons at baseline. Despite these changes, the neurotoxic effects induced by BTZ were similar between tumor-bearing and non-tumor bearing mice and included decreased nerve conduction velocity, mechanical hyperalgesia, mild axon degeneration in the sciatic nerve and vacuolization of satellite glia cells (Meregalli et al., 2015).
Thus, different treatment regimens are being utilized to evaluate mechanisms for BTZ-induced neurotoxicity, some of which are discussed below.
BTZ-induced neuroinflammation and pain
Bortezomib-induced neurotoxicity is peculiar among different types of chemotherapy-induced neuropathy as it can be extremely painful (Bilińska et al., 2013). Many patients with BIPN suffer from intense, persistent burning pain that significantly reduces quality of life long after chemotherapy has ended. Because currently available drugs treat only symptoms with only partial success (Dorsey et al., 2019; Majithia et al., 2016), many studies on BIPN aim to reveal molecular mechanisms that lead to BTZ-induced pain in order to develop more effective therapies. Much progress has been made in recent years elucidating mechanisms underlying neuropathic pain after peripheral nerve injury, and it has become clear that BTZ activates some of the same pathways. Pain results from the detection of noxious stimuli by sensory neurons in the DRG, relay of action potentials from DRGs to the spinal cord, and transmission of the warning signal to the brain (Jain et al., 2020; Scholz and Woolf, 2007; von Hehn et al., 2012). As discussed in more detail below, the development of neuropathic pain after BTZ involves dysregulation of transcriptional programs in DRGs, activation of the peripheral immune system, altered synaptic transmission in the dorsal horn of the spinal cord, and transformation of spinal astrocytes (Fig. 1).
Figure 1: Molecular mechanisms of BIPN and sites of therapeutic intervention based on results obtained from rodent models.
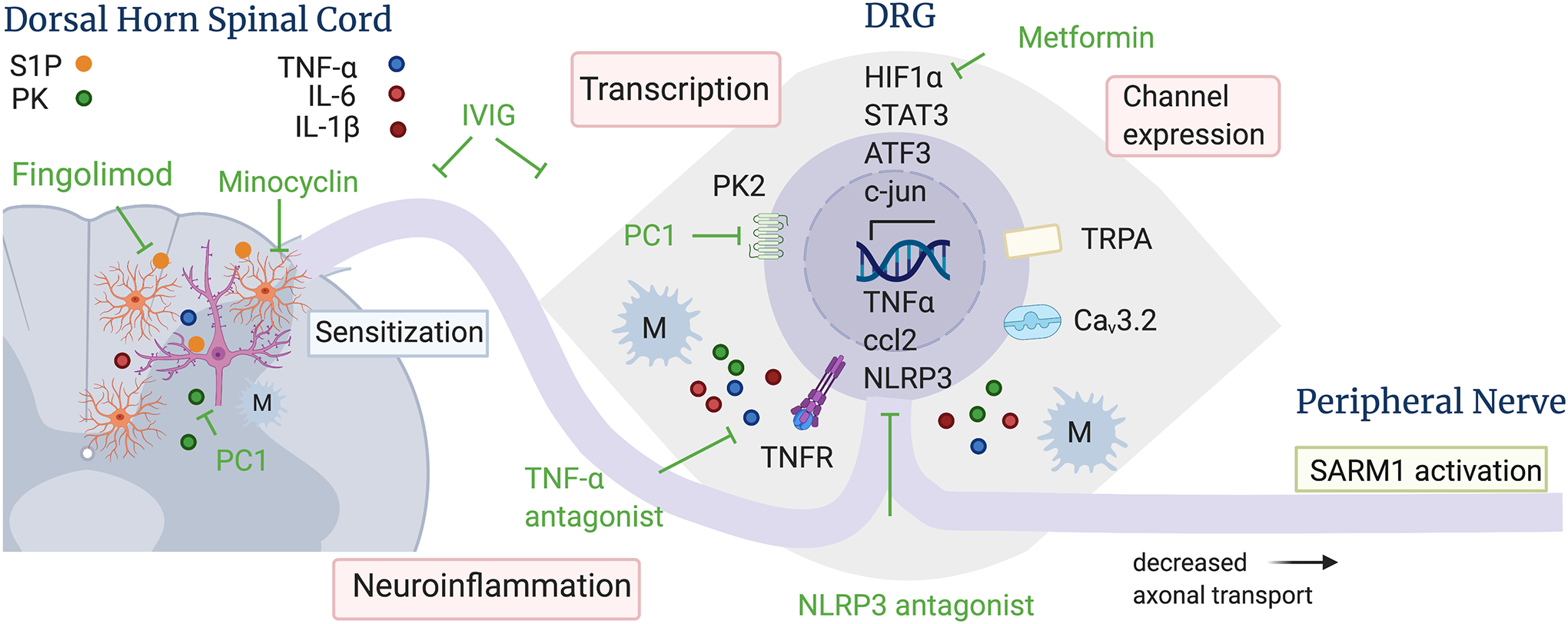
BTZ leads to activation of transcriptional programs and channel overexpression in DRG neurons, which results in neuroinflammation and increased neuronal firing. In the dorsal horn of the spinal cord, BTZ induces astrocyte, but not microglia, activation, dysregulation of sphingolipid metabolism and neuroinflammation, which is associated with increased firing of wide dynamic range neurons and pain sensitization. Several pharmacological interventions indicated in green have been shown to reduce BTZ-induced hyperalgesia in rodent models, including TNFalpha and NLRP3 antagonists, Fingolimod (blocks sphingosine-1-phosphate receptor signaling in astrocytes), PC1, (inhibits prokineticin receptors), metformin (blocks the binding of HIF1alpha to the HIF1-alpha responsive binding element), and IVIg (blocks neuroinflammation). Mechanisms that lead to BTZ-induced axon degeneration and treatment strategies are shown in more detail in figure 2. Abbreviations: ATF3 – activating transcription factor 3; Ccl2 – chemokine ligand 2; Cav3.2 – voltage gated calcium channel 3.2; HIF1alpha – Hypoxia inducible factor 1 subunit alpha; IL – interleukin; M – macrophage; NLRP3 – nod like receptor protein 3; PC1 – prokineticin receptor antagonist; PK – prokineticin; S1P – sphingosine 1 phosphate, STAT3 – signal transducer and activator of transcription 3; TNF-alpha – tumor necrosis factor alpha; TNFR – TNF receptor; TRPA - transient receptor potential ankyrin 1 ion channel.
BTZ does not penetrate into the central nervous system, but crosses the blood-nerve barrier to accumulate in DRGs and peripheral nerves, such that these are the main anatomical sites of direct neurotoxicity (Adams et al., 1999; Meregalli et al., 2014). In line with in vitro findings indicating that BTZ-induced neurotoxicity is at least in part transcriptionally regulated (Geisler et al., 2019a), thus requiring the cell body involvement to induce axon degeneration, in vivo studies have revealed induction of several transcription programs underlying BTZ-induced pain in DRG neurons (Fig. 1).
One of the earliest activated transcription factors following BTZ administration is the cytosolic Hypoxia-Inducible Factor 1 Subunit alpha (HIF1α; Ludman and Melemedjian, 2019b). Upon hypoxia and metabolic stressors, HIF1α translocates to the nucleus where it binds to the hypoxia response element (HRE) to regulate the expression of mitochondrial and metabolic factors. In the absence of these stressors, HIF1α remains in the cytosol where it undergoes continual proteasomal degradation. Five days after initiation of BTZ in mice, HIF1α protein levels are increased in lumbar (L4–6) DRG neurons. The increase of HIF1α is associated with mechanical hyperalgesia, whereas suppression of HIF1α with siRNAs or blocking the binding of HIF1α to HRE with the antibiotic echinomycin prevents BTZ-induced hyperalgesia (Ludman and Melemedjian, 2019b). These results suggest that transcription of genes that are under HIF1α control is crucial for the development of BTZ-induced neuropathic pain. Importantly, the commonly used anti-diabetic drug metformin suppresses the translation of HIF1α in DRGs and thereby prevents the development of BTZ-induced pain in this mouse model (Ludman and Melemedjian, 2019b). Blockade of HIF1α has no effect once pain is established, indicating that the binding of HIF1α to HRE plays a role in the development of BTZ-induced hyperalgesia, but not in its maintenance.
A hallmark of neuropathic pain after nerve cut is a robust immune response that includes infiltration by activated immune cells, activation of glial cells and increased production of proinflammatory mediators at the injury site, which, collectively, have been shown to sensitize nociceptors and induce pain (Ji et al., 2016; Scholz and Woolf, 2007; Shamash et al., 2002). The pro-inflammatory cytokine tumor necrosis factor alpha (TNFα), in particular, has a lead role in activating a cascade of other cytokines, notably interleukin (IL) 1β, IL-6, IL-8 and transforming growth factor (TGF) β. Similar to nerve injury, BTZ leads to increased expression of mRNAs encoding several cytokines. However, in contrast to nerve injury that is associated with a pronounced immune response in the peripheral nerve (Shamash et al., 2002), the immune response after BTZ occurs first and most potently in DRGs and only subsequently and to a lesser extent in the peripheral nerve and dorsal horn of the spinal cord (Alé et al., 2014).
During a course of administration of 1mg/kg BTZ i.p. twice weekly for 5 weeks, expression of TNFα mRNA increased in DRGs after only two BTZ injections (Alé et al., 2014). This was followed by a peak in IL-6 mRNA and an increase of the TNF receptor 1 in the second week, suggesting auto-induction of TNFα at later time points (Alé et al., 2014). In contrast to nerve injury, in which TNFα is released at the injury site by Schwann cells and macrophages (Shamash et al., 2002), high TNFα immune reactivity was especially abundant in small DRG neurons, whereas DRG satellite glia cells did not exhibit increased TNFα expression following BTZ administration (Alé et al., 2014). At later time points, the levels of TNFα and IL-6 decreased in DRGs, but to levels still higher than observed in control animals. In the peripheral nerve and dorsal horn of the spinal cord, TNFα and IL-6 mRNA expression peaked later and to a lesser extent than in DRGs. Co-administration of antibodies against TNFα prevented BTZ-induced hyperalgesia, sensory-motor dysfunction and even reduction in sensory nerve amplitude (SNAP; Alé et al., 2014; Chiorazzi et al., 2013; Li et al., 2018; Zhang et al., 2014). While the increase in TNFα after nerve cut is a consequence of the axon injury, the preservation of the SNAP amplitude following block of TNFα after BTZ suggests that axon loss after BTZ treatment is, at least in part, secondary to TNFα activation and consequent neuroinflammation.
The binding of TNFα to its cell surface receptors engages multiple signal transduction pathways, including activation of mitogen-activated protein (MAP) kinases, such as cJun NH2-terminal kinases (JNKs). Accordingly, phosphorylated JNK1 and 2 are found in TNFα positive DRG neurons on days 5 and 10 (but not day 1), during daily i.p. administration of BTZ for 5 days at a dose of 0.2 mg/kg (Zhang et al., 2014). Knockout of TNF receptor 1 and 2 or injection of thalidomide, an inhibitor of TNFα synthesis, significantly blocks BTZ-induced JNK activation and the development of allodynia (Zhang et al., 2014). JNK is a critical upstream messenger of the transcription factor c-jun, which can bind to the promoter of the small chemokine ligand 2 (ccl2), a potent monocyte-attracting chemokine that greatly contributes to the recruitment of blood monocytes into sites of inflammatory responses. Following BTZ, c-jun and ccl2 mRNA are upregulated in DRG neurons, but not local glia cells, and remain elevated for at least 5 days after discontinuation of BTZ treatment (Liu et al., 2016). The binding of c-jun to the ccl2 promoter is enhanced by the Activating Transcription Factor (ATF) 3, which is also increased in DRG neurons at that time (Liu et al., 2016). Blocking c-jun prevents BTZ-induced ccl2 upregulation and attenuates macrophage infiltration into DRGs and mechanical hyperalgesia. However, blocking ccl2 does not completely rescue BTZ-induced hyperalgesia, indicating that other mechanisms are also involved in BTZ-induced pain (Liu et al., 2016).
TNFα also activates p38 MAP kinase, which leads to increased expression of the non-selective cation channel transient receptor potential ankyrin 1 (TRPA1). Expressed by nociceptors, TRPA1 is activated by various irritants, including mustard oil and capsaicin, and integrates a variety of noxious stimuli. Blocking p38 MAP kinase blocks BTZ-induced increased expression of TRPA1 and hyperalgesia (Guo et al., 2020; Liu et al., 2019). Similarly, the IL-6 inhibitor SC144 reduces BTZ-induced upregulation of pJNK, p38 MAP kinase and TRPA1 and mechanical hyperalgesia and block or genetic deletion of TRPA1 prevents BTZ-induced mechanical and cold hyperalgesia, highlighting the importance of this pathway in the development of BTZ-induced pain (Liu et al., 2019; Trevisan et al., 2013; Wang et al., 2017). The expression of TNFα appears to be partially regulated by micro-RNA155 (miR155), because inhibition of miR155 decreases BTZ-induced expression of TNFα, p38MAPK and TRPA1 and decreases mechanical hyperalgesia (Duan et al., 2020).
Thus, several animal models revealed early and robust BTZ-induced enhancement of TNFα and its effect on downstream signaling cascades that lead to mechanical hyperalgesia, whereas inhibiting TNFα signaling at different points in the cascade attenuates BTZ-induced pain. Interestingly, patients who develop grade 2 or higher peripheral neuropathy during BTZ therapy show a distinct increase of serum TNFα level, which is observed before development of peripheral neuropathy and increases further after peripheral neuropathy is detected (W. Zhao et al., 2019). Because TNFα levels correlate so closely to neuropathy symptoms, the early increase in TNFα level can be used as an effective biomarker to predict the development of peripheral neuropathy, which would allow intervention in the disease process early, possibly by using one of several FDA-approved TNFα inhibitors. As BTZ decreases TNFα in cancer cells (Hideshima et al., 2001), but increases it in nervous tissue (Alé et al., 2014; Li et al., 2018; Zhang et al., 2014), treating BIPN with TNFα antagonist would produce synergistic effect on BIPN and cancer.
Similar to its effect on cancer cells, BTZ leads to activation and upregulation of Signal Transducer and Activator of Transcription 3 (STAT3; C.-C. Liu et al., 2018). STAT3 is a transcription factor that modulates the expression of a variety of genes, including those that control cell growth, survival, and immune response. Phosphorylated STAT3 binds to the Nod like receptor protein 3 (NLRP3) promoter leading to increased NLRP3 mRNA and protein expression in neurons and macrophages in DRGs following BTZ (C.-C. Liu et al., 2018). NLRP3 belongs to the inflammasome family and has been shown to be critical in driving the immune response to sterile tissue damage (Cowie et al., 2019). Intrathecal activation of NLRP3 induces hyperalgesia, whereas inhibition of STAT3 or NLRP3 leads to attenuation of BTZ-induced hyperalgesia (C.-C. Liu et al., 2018). NLRP3 expression also contributes to oxaliplatin and paclitaxel-induced neuropathies (Jia et al., 2017; Wahlman et al., 2018). Given the central role of NLRP3 activation to different forms of chemotherapy-induced neuropathies and many other diseases, many direct and indirect inhibitors are being evaluated in preclinical and clinical studies and may be useful in blocking BTZ-induced pain (Zahid et al., 2019).
STAT 3 signaling also plays a crucial role in the activation of prokineticin 2 (Qu et al., 2012; Xin et al., 2013). Prokineticins belong to a new family of cytokines first shown to cause profound GI contractions, hence their names (Y. Zhao et al., 2019). Prokineticins induce a pro-inflammatory macrophage profile, stimulate chemotaxis and prompt the release of other pro-inflammatory cytokines. Comprising of prokineticin 1 and 2, they act through two G-protein coupled receptors (prokineticin receptor 1 and 2), are widely expressed in many tissues and involved in a large spectrum of biological activities, including inflammation and nociceptive neurotransmission (Y. Zhao et al., 2019). In DRGs, prokineticin 1 receptor is mainly expressed in small nociceptor neurons also expressing the transient receptor potential vanilloid receptor channel TRPV1, while PRK2 is expressed in medium/large sized neurons co-localized with TRPA (Maftei et al., 2020). Prokineticin 2 protein and Prokineticin 1 and 2 receptor expression are increased following BTZ, but, surprisingly, only late in the course, after macrophage infiltration into DRGs was observed and after mechanical hyperalgesia had developed (Moschetti et al., 2019a). However, treating the mice with the PKR antagonist PC1 at a time when BTZ-induced mechanical hyperalgesia was well developed reversed the allodynia, decreased a late proinflammatory response in DRGs, sciatic nerve and spinal cord and lessened BTZ-induced structural damage of satellite glia cells (Moschetti et al., 2019a). Importantly, PC1 was also effective in decreasing BTZ induced allodynia after BTZ administration was finished. Thus, while the prokineticin system has no role in the initial development of BTZ-induced pain, PC1 may be an effective drug for patients treated with BTZ after symptoms have appeared. The protection from BTZ-induced pain may be due to reversal of neuroinflammation in the spinal cord and prevention of secondary sensitization necessary for sustained pain.
Experiencing pain depends on the efficient transmission of nociceptive information from peripheral nociceptor neurons to second-order neurons in the spinal cord. The spinal cord superficial dorsal horn is the site of synapses between DRG and second order neurons and where the intensity of pain is critically controlled (von Hehn et al., 2012). Although it does not cross the blood brain barrier, BTZ induces increased firing and persistent after discharges of wide dynamic range neurons in the dorsal horn of the spinal cord in rats (Robinson et al., 2014; Robinson and Dougherty, 2015), an effect mediated, at least partially, by tonic activation of presynaptic NMDA receptors (Carozzi et al., 2010; Xie et al., 2017) and astrocytes. Astrocytes are activated and express increased gap junction protein 43 early in the course of BIPN, well before proinflammatory cytokines are increased and invasion of macrophages into the dorsal horn of the spinal cord is observed (Robinson and Dougherty, 2015). Interestingly, and in contrast to pain induced by spinal nerve ligation, microglia are not activated in the dorsal horn of the spinal cord after BTZ treatment (Robinson et al., 2014). Astrocyte activation contributes to mechanical hyperalgesia, as concurrent treatment of rats with the anti-inflammatory minocycline or gap junction decoupler carbenoxolone prevented the development of mechanical hyperalgesia and the increased expression of connexin 43 and the astrocyte marker glial fibrillary acidic protein (Robinson et al., 2014; Robinson and Dougherty, 2015).
One mechanism by which astrocytes contribute to sensitization and pain behavior in BIPN is via sphingosine-1 phosphate signaling. Sphingolipids are crucial components of cellular membranes (Singh and Spiegel, 2020). The metabolism of sphingolipids generates signaling molecules such as ceramide, sphingosine and sphingosine 1 phosphate (S1P) that are essential for many biological functions. Bortezomib caused an increase of sphingosine 1 phosphate receptor (S1PR) ligands, including S1P, and this dysregulation was associated with time-dependent development of neuropathic pain (Stockstill et al., 2018). Activation of S1PR via intrathecal administration of the S1PR1 agonist SEW2871 caused mechanical allodynia, increased levels of NfκB, phosphorylated p38 and NLRP3 (Doyle et al., 2019). In the central nervous system, including the spinal cord, S1PR is most highly expressed in astrocytes. Astrocyte specific deletion of S1PR1 or treatment with the S1P1R antagonist Fingolimod (trade name Gilenya®, Novartis) prevented the development of BTZ-induced neuropathic pain, astrocyte activation and neuroinflammation in the dorsal horn of the spinal cord, including the increase of TNFα, IL-6 and NLRP3 expression. (Fig. 1) Astrocytic S1PR also increased presynaptic glutamate release in the dorsal horn of the spinal cord following BTZ, which may contribute to increased firing of dorsal horn neurons and sensitization, whereas blockade of S1PR prevented increased firing (Stockstill et al., 2018). Importantly, 20 days after conclusion of BTZ treatment, at a time when pain was fully developed, infusion of Fingolimod completely reversed the neuropathic pain (Stockstill et al., 2018). Fingolimod is an immunomodulating drug used in patients mostly for the treatment of multiple sclerosis. Thus, the preclinical findings of attenuating BTZ-induced pain by Fingolimod should be easily translatable to the bedside. Indeed, a clinical trial currently evaluates Fingolimod for treatment of CIPN once it has occurred (clinicaltrials.gov #NCT03943498). Fingolimod may be especially useful for patients treated with BTZ, as it is not only efficacious in treating BTZ-induced hyperalgesia, but also bone pain (Grenald et al., 2017) that is frequently observed in multiple myeloma patients. In addition, Fingolimod effectively targets BTZ-resistant cells, increasing their sensitivity to BTZ and leading to superior tumor growth inhibition (Beider et al., 2017), further supporting the use of Fingolimod to treat BIPN in multiple myeloma patients. One caveat may be that Fingolimod was effective in reversing BTZ-induced pain only in male, but not female rats (Stockstill et al., 2020), which may be due to gender-specific differences in the innate immune system that are also observed, e.g., for NLRP3 activation (Cowie et al., 2019).
Intravenous immunoglobulins (IVIg) are commonly used in clinical praxis as immunomodulatory therapy in several immune-mediated diseases, including inflammatory neuropathies. Given the pronounced neuroinflammation in different models of BIPN, Meregalli et al. (2018) evaluated whether a clinically relevant treatment regimen can prevent BIPN in a rat model (Meregalli et al., 2018b). Administering IVIg at a dose of 1g/kg every 2 weeks during BTZ administration, they observed less macrophage infiltration in the sciatic and caudal (tail) nerves, improved mechanical and heat allodynia, and protection from loss of IENF compared to vehicle treated rats. However, the BTZ-induced decrease of nerve conduction velocity reflecting function of myelinated axons did not improve with IVIg treatment, suggesting that IVIg protected especially small nerve fibers.
Surprisingly, despite the prominent role of neuroinflammation in BIPN, immunodeficient animals treated with BTZ develop a painful peripheral neuropathy with the same features observed in immunocompetent mice (Carozzi et al., 2013). This finding may be due to release of proinflammatory cytokines from DRG neurons (in addition to infiltrating macrophages) or because other mechanisms also have important roles in the development of BIPN. For instance, by inhibiting the proteasome, BTZ directly increases protein levels of the calcium channel Cav3.2 in nociceptors of the DRG, which leads to increased neuronal excitability and pain behavior (Tomita et al., 2019). In addition, axon degeneration is an important component of BIPN that contributes to pain. The mechanism of axon degeneration after BTZ and VCR are similar and will be described together further below.
Models of VCR-induced neurotoxicity
By binding to microtubule ends VCR blocks the addition of new tubulin subunits, leading to microtubule depolymerization and inhibition of microtubule dynamic instability (Himes et al., 1976; Owellen et al., 1976; Smith et al., 2016). Microtubules are critical for intracellular transport, serving as tracks for “motor” proteins that carry vesicular cargo. Of the vinca-alkaloids vinblastine, VCR and vinorelbine, VCR has the highest affinity for tubulin and the highest incidence of peripheral neuropathy (Lobert et al., 1996). Furthermore, in isolated squid axoplasm vesicle motility assays, 1 μM VCR inhibits fast anterograde and retrograde axonal transport with the highest potency when compared to paclitaxel, erubulin, and ixabepilone (LaPointe et al., 2013). The inhibition of axonal transport by VCR was confirmed in in vivo models in some (Green et al., 1977) but not other studies (Bradley and Williams, 1973). The health of the neuron depends on both anterograde transport of proteins, RNA, mitochondria and other organelles from the cell body to the synapse and retrograde transport of toxic components (misfolded proteins, damaged organelles) from the synapse to cell body. Disruption in axonal transport is thought to affect the longest axons first, causing a dying back peripheral neuropathy in which the most distal parts of nerves are affected first in the feet and hands in a “stocking-glove” distribution. The dying back phenomenon of VCR-induced neuropathy has been modeled in in vitro and in vivo studies.
In vitro models of VCR-induced neurotoxicity
The estimated chronic level of VCR during cancer chemotherapy in humans ranges between 10–100 nM (Silva et al., 2006). Adding 100 nM VCR to plated embryonic DRG neurons is neurotoxic and results in rapid degeneration of axons and soma, whereas 20 nM has no effect (Silva et al., 2006). Exposure of DRG neurons to 40 or 50 nM leads to selective degeneration of neurites while the soma remains intact (Geisler et al., 2019a; Silva et al., 2006). In contrast to BTZ, which induces axon degeneration when added to the soma compartment (Geisler et al., 2019a), adding 40 or 50 nM of VCR to the soma or proximal axon compartments has no effect on the integrity or growth of DRG neuron axons (Geisler et al., 2019a; Silva et al., 2006). However, when the distal axon compartment is exposed to the same dose of VCR, axons became fragmented and their lengths decreased in a dying back pattern, suggesting a distal to proximal gradient of axon degeneration (Geisler et al., 2019a; Silva et al., 2006). These data were confirmed by combining a microfluidic divider with a multielectrode array. Exposure of axons, but not cell bodies, to VCR caused electrophysiologic failure that preceded morphological signs of axonal degeneration (Ravula et al., 2007). The amplitudes of the degenerating axons declined, beginning in the outer third of the axon compartment. Similarly, when VCR was added to the soma compartment, potassium chloride mediated depolarization remained constant throughout the 2 day period, whereas 6 hours after addition of VCR to the axonal compartment spike activity increased in the soma compartment, suggesting that an injury signal is transmitted from the axon to the cell body (Ravula et al., 2007). Degenerative morphological changes were first noticed in neurites 18 hours after adding VCR, after which the rate of degeneration rapidly progressed (Ravula et al., 2007). This observation led Jonathan Glass and colleagues to suggest almost 20 years ago that VCR-induced neurotoxicity is due to the activation of a local axon degeneration program (Silva et al., 2006; Wang et al., 2000). This visionary hypothesis has now been confirmed and molecular mechanisms of this pathway identified (Essuman et al., 2017; Geisler et al., 2019b, 2016; Gerdts et al., 2013; Summers et al., 2018; Yang et al., 2015) as we shall see further below.
In vivo models of VCR-induced neurotoxicity
The dose-dependent VCR toxicity in patients described earlier is also observed in animal models, in which VCR at higher doses is associated not only with more signs of neuropathy, but also higher mortality (Table 2). The most commonly used rodent model of VIPN consists of five daily intraperitoneal or intravenous injections of 0.1 mg/kg VCR, which is repeated after two days rest, resulting in a cumulative dose of 1 mg/kg (Table 2). This regimen results in mild weight loss (~7%), and typically no mortality. Under this treatment schedule, mice and rats developed mechanical and thermal hyperalgesia, hyper-responsiveness of C-fibers, neuroinflammation in the sciatic nerve, DRGs and spinal cord and gastrointestinal changes, including delayed gastric emptying, decreased gastrointestinal motility and damage to the intestinal wall (Table 2). Myelinated and unmyelinated axons were swollen and exhibited cytoskeletal changes consisting of disturbed orientation of microtubules, decreased microtubule density and abnormal clustering of neurofilaments in the central portion of the axoplasm (Tanner et al., 1998; Topp et al., 2000). In conjunction with the pale toluidine blue staining of DRG neuron cell bodies and proximal axons due to build-up of neurofilaments, these findings are morphological signs of impaired axonal transport. A loss of intra-epidermal nerve fibers in the skin and of myelinated and unmyelinated axons in the sensory saphenous nerve was not observed using this treatment schedule (Topp et al., 2000).
Table 2:
Animal models of vincristine-induced neuropathy
| Strain | Gender | Age | Regimen | Route | Cumulative dose | Findings | References |
|---|---|---|---|---|---|---|---|
| Mouse | |||||||
| CD1 | male | 30–35g | 1.7 mg/kg twice weekly × 10 weeks | i.p. | 34 mg/kg | 74% mortality thermal hypoalgesia, gait disturbance axon degeneration in the sciatic nerve | (Contreras et al., 1997) |
| Swiss OF1 | female | 12 weeks | 1.5 mg/kg twice weekly × 4 weeks | i.p. | 12 mg/kg | 11.5% mortality abnormalities on rotarod testing decreased sensory and motor nerve amplitudes and conduction velocities decreased number of myelinated axons, macrophage infiltration in the sciatic nerve, decreased IENF density | (Bruna et al., 2011) |
| C57Bl/6 | male & female | 12–20 weeks | 1.5 mg/kg twice weekly × 4 weeks | i.p. | 12 mg/kg | 17.5% mortality mechanical and heat hyperalgesia, decreased tail nerve amplitude without change in conduction velocity axon degeneration in toe nerves, axon loss of the sural nerve decreased IENF density | (Geisler et al., 2016) |
| Swiss OF1 | female | 12 weeks | 1 mg/kg twice weekly × 4 weeks | i.p. | 8 mg/kg | 5% mortality decreased sensory nerve amplitude and conduction velocity abnormalities on rotarod testing | (Bruna et al., 2011) |
| C57Bl/6 | male & female | 3–4 months | 0.75 mg/kg twice weekly × 4 weeks | i.p. | 6 mg/kg | mechanical hyperalgesia, no axon degeneration | (Chen et al., 2020) |
| C57Bl/6 | male | 8–10 weeks | 0.5 mg/kg/day × 5 × 2 | i.p. | 5 mg/kg | mechanical hyperalgesia | (Starobova et al., 2019) |
| C57Bl/6 | male | 9 weeks | 0.1 mg/kg/day × 14 | i.p. | 1.4 mg/kg | mechanical and thermal hyperalgesia | (Moschetti et al., 2019) |
| C57Bl/6 | male, female | 10–17 weeks | 0.1 mg/kg × 5 × 2 | i.p. | 1 mg/kg | mechanical allodynia monocyte/macrophage infiltration in the sciatic nerve no ATF3 upregulation in DRGs no microglia activation in the spinal cord no loss of IENF | (Lee et al., 2020; Montague et al., 2018; Old et al., 2014; Shi et al., 2018) |
| C57Bl/6 | 8–10 weeks | 10 μg daily × 4, 5 day rest, additional 2 injections | i.pl. | 40 μg | mechanical and heat hyperalgesia inflammation of paw inflammation DRGs | (Starobova et al., 2020, 2019) | |
| Rat | |||||||
| SD | male | 250–300g | 0.2 mg/kg daily × 5 × 2 | i.v. | 2 mg/kg | weight loss, mechanical and heat hyperalgesia; abnormalities on rotarod testing | (Aley et al., 1996) |
| SD | male | 220–240g | 0.2 mg/kg twice weekly × 4 weeks | i.p. | 1.6 mg/kg | no mortality decreased motor nerve amplitude smaller axonal diameter | (Erdoğan et al., 2020) |
| SD Wistar | male | 150–350g | 0.1 mg/kg/day × 5× 2 | i.p. | 1 mg/kg | no weight loss, no mortality hot and cold hyperalgesia neuroinflammation in the sciatic nerve hyperresponsive C fibers in saphenous nerve, cytoskeletal abnormalities in unmyelinated and myelinated sensory axons, no axon loss; structural changes and activation of DRG neurons; increased synaptic activity and neuroinflammation in the dorsal horn of the spinal cord | (Aley et al., 1996; Kim et al., 2020; Singh et al., 2019; Tanner et al., 2003, 1998, 1998; Thibault et al., 2013; Topp et al., 2000; Weng et al., 2003) |
| Wistar | male | 275–300g | 0.1 mg/kg/day × 5× 2 | i.p. | 1 mg/kg | weight loss, decreased food intake mechanical hyperalgesia delayed gastric emptying, decreased intestinal motility, damage to digestive wall | (López-Gómez et al., 2018) |
| Rattus norvegicus albinus | male | 250–450 g | 0.2 mg/kg weekly × 5 | i.p. | 1 mg/kg | gait disturbance and limping decreased motor and sensory nerve amplitudes axon shrinkage and myelin sheath alterations in the sciatic nerve | (Ja’afer et al., 2006) |
| Wistar | female | 175–200 | 0.2 mg/kg weekly × 4 | i.v. | 0.8 mg/kg | decreased sensory and motor nerve amplitudes, axon degeneration tail nerve, decreased IENF density, increased serum neurofilament light chain | (Meregalli et al., 2018a) |
| SD | n/a | 160–180g | 0.1 mg/kg, 0.15 mg/kg QOD × 5 | i.v. | 0.5 mg/kg, 0.75 mg/kg | decreased body weight mechanical and cold hyperalgesia, thermal hypoalgesia axon degeneration in the sciatic nerve | (Authier et al., 2003, 1999) |
| SD | male | 200–300 | 0.05 mg/kg × daily × 10 | i.p. | 0.5 mg/kg | decreased IENF density | (Siau et al., 2006; Siau and Bennett, 2006) |
Another treatment regimen consists of intraperitoneal injections of 0.75 mg/kg to 1.7 mg/kg VCR twice weekly for four weeks in mice or 0.2 mg/kg intravenously weekly for four weeks in rats (Table 2). Mice receiving 0.75 mg/kg twice weekly had about 10% weight loss, but no mortality and developed pronounced mechanical- and mild thermal hyperalgesia (Chen et al., 2020). They did not exhibit morphological signs of axon degeneration in the toe, sural, tibial and sciatic nerves and had no loss of IENF. Groups of mice receiving 1 mg/kg VCR twice weekly for four weeks had 5% mortality and developed decreased sensory nerve action potentials and conduction velocities of the tail, but no changes in the IENF density (Bruna et al., 2011). Such change was observed, instead, when 1.5 mg/kg VCR was given i.p. twice weekly for 4 weeks (Bruna et al., 2011; Geisler et al., 2016). This treatment regimen reflected many signs seen in patients, including axon loss in the distal, but not proximal sensory sural nerve, axon degeneration of distal toe nerves, decrease of IENF density in the skin, decreased compound nerve amplitude with preserved conduction velocity and mechanical and heat hyperalgesia. There was no axon loss in the motor predominant tibial nerve (Geisler et al., 2016). Therefore, this model mimics moderately severe sensory predominant peripheral neuropathy commonly seen in patients. The weight loss in this model over the four week period was 6% and the mortality rate 12– 18 % (Bruna et al., 2011; Geisler et al., 2016). Giving VCR to mice at a dose of 1.7 mg/kg twice weekly had a mortality rate of 74% in one study (Contreras et al., 1997) and 50% in another (Bruna et al., 2011), indicating this as a dose limit for VCR in mice. In rats, the administration of 0.2 mg/kg VCR i.v. weekly for four weeks results in both decreased sensory and motor nerve amplitude, axon degeneration in the caudal nerve, loss of intraepidermal nerve fibers, and a dose and time-dependent increase of serum neurofilament levels indicative of axon injury (Meregalli et al., 2018a).
VCR-induced neuroinflammation and pain
VCR-induced neuropathy characteristically causes numbness, tingling and weakness in the hands and feet, but is also frequently associated with neuropathic pain (Lieber et al., 2018; Liew et al., 2013; Pal, 1999). Similar to BTZ, a neuroinflammatory response is observed in rodent models following administration of VCR. However, while BTZ causes early neuroinflammation in DRGs, VCR leads to early macrophage infiltration in the sciatic nerve. Administering two cycles of VCR to mice at a dose of 0.2 mg/kg daily for 5 days with a 2 day rest period in between, produced mechanical hyperalgesia and increased macrophages in the sciatic nerves in both cycles, but no microglia activation in the spinal cord, no increase of the neuronal activation marker ATF3 in DRGs and no loss of cutaneous IENFs (Old et al., 2014). It was shown further that macrophages expressing the chemokine receptor Cx3CR1 transmigrate from the blood vessel into the sciatic nerve where they produce reactive oxygen species, which leads to activation of local axonal TRPA1 channels, which evoked pain. Deletion of the macrophage chemokine receptor CX3CR1 blocked hyperalgesia and macrophage migration during the first cycle of VCR administration, but had no effect on macrophage invasion or mechanical hyperalgesia during the second. Hyperalgesia during the 2nd cycle was modulated, in addition, by the chemokine receptor 2 (CCR2; Montague et al., 2018). Inhibiting CCR2 in Cx3CR1 KO mice (but not in wildtype mice) blocks hyperalgesia in the 2nd cycle, suggesting that in the absence of Cx3CR1, CCR2 mediates some of the VCR-induced hyperalgesia observed in the 2nd cycle. Macrophages infiltrating the sciatic nerve following VCR administration express IL-6 (Kiguchi et al., 2008) and perineural injection of IL-6 antibodies or genetic deletion of IL-6 prevents VCR-induced allodynia (Kiguchi et al., 2008).
A local inflammatory response in the paw was observed when 10 μg VCR was administered subcutaneously into the hindpaw of mice daily for four days, followed by a five day break and further two injections (Starobova et al., 2019). Seven days after initiating this treatment, a local inflammation of the paw included full thickness epidermal necrosis with edema of underlying soft tissue and leukocyte infiltration into the dermis of the paw. Co-treatment with the anti-inflammatory tetracycline antibiotic minocycline reduced paw swelling, mechanical hypersensitivity and leukocytic perivascular infiltration (Starobova et al., 2019). In rat models, mincocycline also attenuated neurotoxicity induced by bortezomib and the chemotherapeutic oxaliplatin (Boyette-Davis and Dougherty, 2011; Robinson et al., 2014; Y.-Q. Zhou et al., 2018) suggesting that it could be an effective treatment of different forms of CIPN. However, in a phase II randomized clinical study of patients with locally advanced or metastatic colorectal cancer, 100 mg minocycline twice a day from start of chemotherapy over four months did not reduce numbness, tingling, fatigue or inflammatory biomarkers in patients receiving oxaliplatin (Wang et al., 2019), dampening enthusiasm regarding use of mincocycline to treat chemotherapy-induced neuropathy.
DRGs also exhibit an immune response following administration of VCR. After four daily VCR injections subcutaneously into the paw (Starobova et al., 2020), RNA sequencing of lumbar DRGs and subsequent cell type enrichment analysis of the 232 upregulated and 45 downregulated genes revealed a predominant effect on genes associated with the immune system (Starobova et al., 2020). Similar to BTZ, VCR also leads to activation of the prokineticin system in DRGs and spinal cord after mechanical hyperalgesia is established (Moschetti et al., 2019b). Administering VCR 0.1 mg/kg i.p. daily for 14 days, resulted in mechanical hyperalgesia starting on day 3. On day 7, high levels of prokineticin receptors 1 and 2, toll-like receptor 4, ATF3 and IL-1β and the macrophage marker CD68 were observed in DRGs without relevant alterations in the spinal cord. At day 14, an upregulation of prokineticin system and neuroinflammation were evident both in DRGs and the spinal cord. Similar to BIPN, treatment with the prokineticin antagonist PC1 reversed VCR-induced pain and neuroinflammation in mice (Moschetti et al., 2019b).
In contrast to BTZ, which showed prominent astrocyte-, but not microglia activation in the DHSC, both astrocytes and microglia are activated in the spinal cord and express high levels of TNFα following VCR (Qin et al., 2020; Shen et al., 2015). The increased TNFα is associated with the development of hyperalgesia, as VCR-induced hyperalgesia is prevented by intrathecal administration of antibodies to TNFα. In addition to prokineticin, the C-X-C motif chemokine ligand 1 (CXCL1) is elevated following VCR and associated with the expression of pain (L. Zhou et al., 2018). Weakly expressed in naïve animals, CXCL-1 is increased in astrocytes following VCR and acts on c-c chemokine receptor type 2 (CCR2) in spinal neurons to increase excitatory synaptic transmission (Zhang et al., 2017; L. Zhou et al., 2018). Indeed, increased neuronal activity in the deep layers III and IV of the dorsal horn as shown by increased staining of the immediate early gene c-fos accompanied by increased staining for Piccolo, a marker of active presynaptic elements, was observed (Thibault et al., 2013). Similarly, wide dynamic range neurons express higher rates of spontaneous activity at baseline and a greater A and C fiber response following VCR, which may lead to manifestation and sensitization of pain (Weng et al., 2003). Interestingly, levo-coryldamine (lCDL), an analgesic with sedative hypnotic properties that has been used in China for more than 40 years, decreases CXLC1 and TNFα expression in astrocytes and reverses the associated hyperalgesia in a rodent model and thus, might be useful to decrease pain in VIPN (L. Zhou et al., 2018; Zhou et al., 2020).
In addition to inducing neuroinflammation, VCR causes increased tetrodotoxin sensitive sodium current density in medium, but not small, DRG neurons, which leads to mechanical hyperalgesia in a mouse model (Chen et al., 2020). Mice lacking Nav1.6 in nociceptors expressed partially attenuated mechanical allodynia in the absence of axon degeneration, indicating that VCR-induced axon degeneration is not a requirement for VCR-induced pain (Chen et al., 2020). How VCR leads to changes in Nav1.6 expression in medium sized DRG neurons requires further investigation but it is possible that inflammatory cytokines upregulate Nav1.6 channel expression and that Nav1.6 in turn promotes inflammation and axon degeneration (Alrashdi et al., 2019; Xie et al., 2013). Considering the emerging role of Nav1.6 as an important contributor to chronic pain (Chen et al., 2018; Xie et al., 2015, 2013) and VCR-induced neuropathy (Chen et al., 2020), blocking Nav1.6 with a selective sodium channel blocker may be another valuable treatment approach to decrease VCR-induced pain.
Mechanisms that lead to VCR and BTZ-induced axon degeneration
Axon degeneration is a prominent feature of both VCR and BTZ-induced neuropathies and is associated with numbness, weakness and pain. While some medications decrease pain and tingling, there are currently no treatments that reduce numbness or weakness (Dorsey et al., 2019; Majithia et al., 2016). However, recent discoveries described below inspire hope that this is about to change.
Much progress has been made in recent years to elucidate mechanisms of axon degeneration (Coleman and Höke, 2020; Figley and DiAntonio, 2020; Gerdts et al., 2016; Simon and Watkins, 2018). Our deep mechanistic understanding stems largely from axon cut injury studies, but regulatory genes identified in these studies also influence axon survival in other circumstances, including VCR- and BTZ-induced neuropathies. The field emerged from the serendipitous discovery of Wallerian degeneration slow (WLDs) mice in which axonal degeneration is greatly delayed after nerve transection (Lunn et al., 1989), which indicated that axons distal to a nerve cut do not die passively because of lack of nutrients, but, akin to apoptosis, engage a genetically encoded axon degeneration program. Already 20 years ago, scientists noticed the similarities of axon degeneration and VIPN and hypothesized that VCR-induced neurotoxicity is also due to the activation of a local axon degeneration program (Silva et al., 2006; Wang et al., 2001, 2000). By treating plated DRGs with different concentrations of VCR, they showed that axon degeneration is delayed in DRG neurons obtained from WLDs mice (Wang et al., 2001, 2001), thus demonstrating for the first time that VCR-induced axonal degeneration can, indeed, be blocked. The spontaneous WLDs mutation results in a chimeric protein (Coleman et al., 1998; Mack et al., 2001) of which the NAD-synthesizing enzyme nicotinamide mononucleotide adenylyltransferase 1 (NMNAT1) activity is responsible for the axon-sparing activity of the Wlds phenotype (Araki et al., 2004; Sasaki et al., 2009). Accordingly, overexpressing cytNMNAT1, a stable form of NMNAT, also leads to long-lasting protection from VCR-induced neurite degeneration in vitro (Press and Milbrandt, 2008; Sasaki et al., 2009; Vohra et al., 2010). However, the Wlds gene product results in the expression of a gain-of-function protein that does not normally exist, limiting the therapeutic utility of this discovery. Until recently, no loss-of-function mutants were identified that had similar axon protective properties. However, it was discovered recently that sterile alpha and TIR motif-containing protein 1 (SARM1) is an essential component of this endogenous axonal self-destruction pathway (Gerdts et al., 2013; Osterloh et al., 2012). Whereas axons of wildtype mice degenerate within 3 days after sciatic nerve cut, severed axons of SARM1 knockout mice remain intact for more than 2 weeks (Gerdts et al., 2013; Osterloh et al., 2012).
SARM1 is a multidomain Toll-like receptor adapter whose Toll/interleukin-1 receptor (TIR) domain exerts pro-neurodegenerative action through its intrinsic NADase activity (Essuman et al., 2017; Gerdts et al., 2015). Once activated, SARM1’s NADase cleaves NAD+ into nicotinamide (NaM) and adenosine diphosphate-ribose (ADPR), which results in metabolic collapse, influx of calcium, activation of calcium-dependent proteases and axon fragmentation. Recent discoveries revealed that NAD+ binds directly to the ARM domain of SARM1 (Jiang et al., 2020). While high concentrations of NAD+ inhibit SARM1, low NAD+ concentrations enhance SARM1’s NADase activity (Jiang et al., 2020) thereby further decreasing NAD+ levels, which may establish a feed-forward loop along the entire length of the axon. Cytosolic NAD+ is produced by the short-lived axonal survival factor nicotinamide mononucleotide adenylyltransferase 2 (NMNAT2), which is continually transported from the cell body into the axon (Gilley et al., 2015; Gilley and Coleman, 2010). Reduced NMNAT2 levels, e.g., after nerve cut, lead to decrease of NAD+ and activation of SARM1. Thus, Wlds is likely axon protective because the long-lived NMNAT1 is “mis”localized to the axon where it substitutes for the labile NMNAT2, maintains NAD+ levels and inhibits SARM1.
Axon loss in most neuropathies, including those induced by VCR and BTZ, differ from that caused by axotomy in that i) fewer axons are degenerating at any given point in time, ii) the degeneration occurs in short segments from distal to proximal and iii) the axon remains connected to the cell body. Despite these differences, genetic deletion of SARM1 protects from loss of both myelinated and unmyelinated axons in a mouse model of VIPN (Geisler et al., 2016). Genetic deletion of SARM1 also prevents VCR-induced mechanical and heat hyperalgesia, indicating that blocking SARM1 improves functional outcomes, including pain (Geisler et al., 2016). VCR is somewhat similar to nerve cut in that axonal transport is impaired, but not blocked. This likely leads to a more profound loss of NMNAT2 distally, activating the SARM1 pathway locally. In this model, residual axonal transport is sufficient to maintain NMNAT2 adequate to block SARM1 activation in proximal axons thereby degeneration to the distal axon.
Excitingly, although BTZ has a different mechanism of action, BTZ-induced axon degeneration is also blocked in SARM1 KO mice (Geisler et al., 2019a), indicating that SARM1 is part of a fundamental axon degeneration program. Both VCR and BTZ lead to a decrease of axonal NMNAT2 in cultured DRGs (Geisler et al., 2019b), which activates SARM1 and causes a drop of NAD+ followed by axon fragmentation (Essuman et al., 2017; Geisler et al., 2019a; H. Liu et al., 2018). The upstream pathways engaged by VCR and BTZ differ, involving MAP-kinases after VCR and transcription and apoptotic enzymes after BTZ (Geisler et al., 2019a; Summers et al., 2018; Yang et al., 2015). Both pathways then converge on a core axon degeneration program consisting of NMNAT2 - SARM1- and NAD+ (Fig. 2). The proposition that NMNAT2 decreases following VCR due to decreased transport is supported by the concomitant increase of NMNAT 2 in the cell body (unpublished observation), but whether a similar mechanism leads to a decline of NMNAT2 following BTZ and whether other mechanisms are involved in the activation of SARM1 by BTZ require further investigation. Although axonal transport is decreased after BTZ (Meregalli et al., 2014; Poruchynsky et al., 2008; Staff et al., 2013), the loss of NMNAT2 in the axon after BTZ administration is not accompanied by an increase in the cell body (unpublished observation) and the kinetics of the axonal NMNAT2 decrease are different than following VCR (Geisler et al., 2019a), suggesting that mechanisms other than or in addition to axonal transport may contribute. It will also be important to elucidate whether the profound protection from VCR-induced allodynia by SARM1 knock-out (Geisler et al., 2016) is due to decreased axon degeneration or if SARM1 acts on additional pathways that modulate pain behavior. For instance, SARM1 has been shown to regulate the neuroinflammatory response in DRG neurons after nerve cut (Wang et al., 2018) and TNFα can trigger SARM1-dependent axon degeneration in sensory neurons via an interesting noncanonical necroptotic signaling mechanism (Ko et al., 2020). Although additional exciting research lies ahead, the identification of a core axon degeneration program already provides several avenues for the development of new therapeutics (Fig. 2; Coleman and Höke, 2020; DiAntonio, 2019; Loring and Thompson, 2020).
Figure 2: Model of a common final axon degeneration pathway activated by vincristine and bortezomib showing prospective therapeutic strategies (left, green) and candidate therapeutics (right).
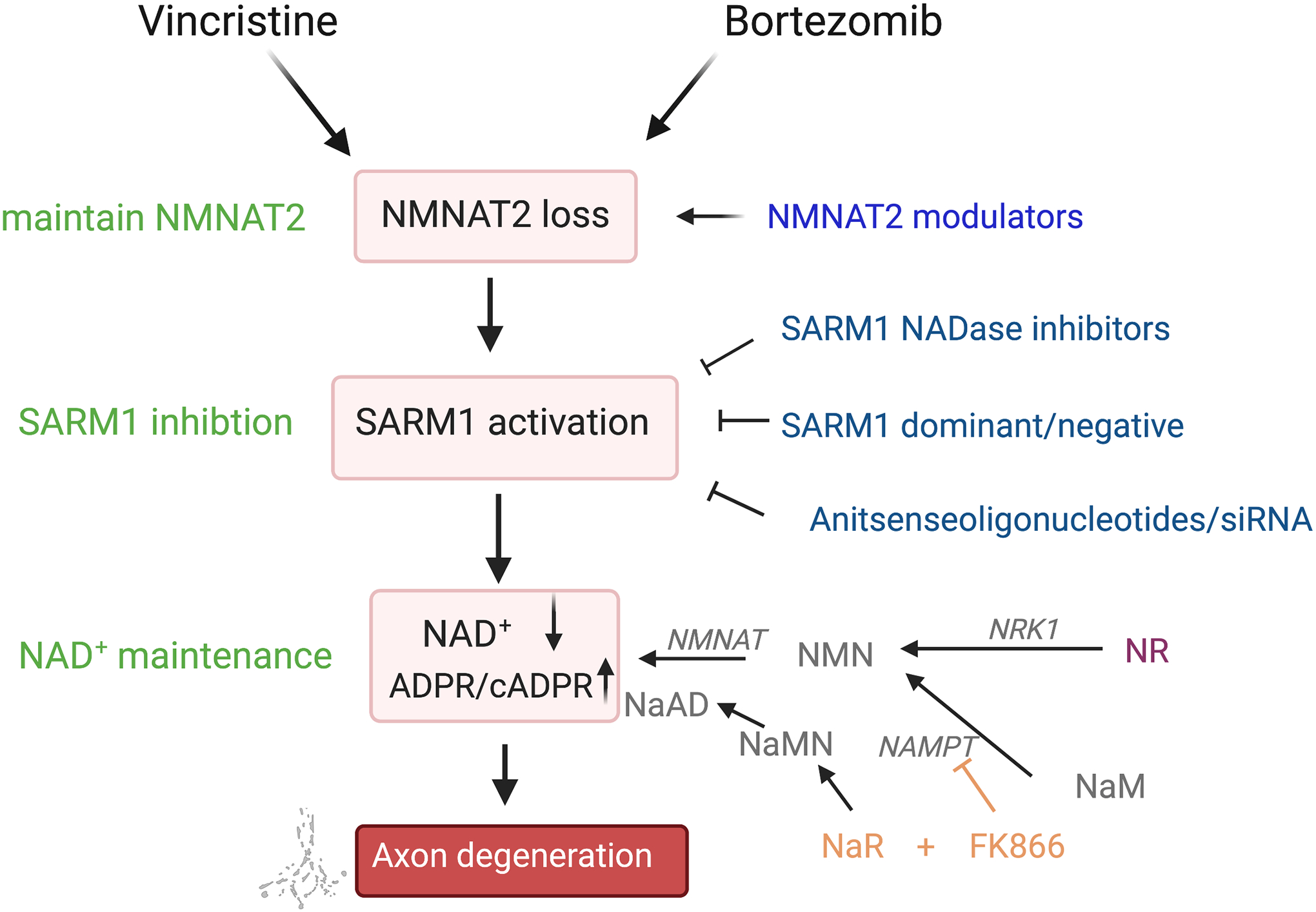
Vincristine and bortezomib engage distinct upstream pathways that lead to loss of axonal NMNAT2. NMNAT2 is an axonal survival factor with short half-life that must be constantly transported from the cell body. SARM1 is inhibited by NMNAT2 and loss of NMNAT2 leads to activation of SARM1, which triggers depletion of the metabolic co-factor NAD+ leading to local metabolic collapse and axon fragmentation. SARM1 activity can be blocked by using pharmacological inhibitors of the SARM1 NADase, dominant/negative mutants of SARM1 or antisenseoligonucleotides/siRNAs. NAD+ levels can be maintained by supplementation with the NAD+ precursor NR, which leads to an increase of both NMN and NAD+ or a combination therapy of NaR and the NAMPT blocker FK866, which decreases NMN and increases NAD+. NMNAT2 levels can be maintained by compounds that modulate NMNAT2 stability. Abbreviations: cADPR – cyclic ADP-ribose; NaAD – nicotinic acid adenine dinucleotide; NaR – nicotinic acid riboside; NaM - nicotinamide; NaMN – nicotinic acid mononucleotide, NAMPT – nicotinamide phosphoribosyltransferase; NMN – nicotinamide mononucleotide; NMNAT – nicotinamide nucleotide adenylyltransferase; NR – nicotinamide riboside; NRK – NR kinase
Insofar as SARM1 is activated by low NAD+ concentration (Jiang et al., 2020) and acts by decreasing NAD+ (Essuman et al., 2017; Gerdts et al., 2015; Sasaki et al., 2016a), countering the NAD+ loss may be one possibility to ameliorate VIPN and BIPN. Indeed, maintaining NAD+ by overexpressing NAD+ synthesizing enzymes NMNAT1, nicotinamide phosphoribosyltransferase (NAMPT) and nicotiamide riboside kinase (NRK) 1 or supplementing with the NAD+ precursor nicotinamide riboside (NR) has been shown to potently protect axons from degeneration following VCR and BTZ in vitro (Geisler et al., 2019a; Gerdts et al., 2015; Sasaki et al., 2009, 2016b). NR is a form of vitamin B3 that readily enters the cell and has no known adverse effects. Accordingly, NR is currently in clinical trials to treat paclitaxel-induced neuropathy, another SARM1-dependent CIPN (Turkiew et al., 2017). Thus, NR or compounds that increase NMNAT2 levels (Ali et al., 2017), may be a promising therapeutic avenue to attenuate VIPN and BIPN (Fig. 2). Recently, it was revealed that SARM1 is allosterically activated upon increase of the direct NAD+ precursor nicotinamide mononucleotide (NMN; Bratkowski et al., 2020; Z. Y. Zhao et al., 2019), which led to the hypothesis that the ratio between NAD+ and NMN may be an important determinant of axon degeneration, as both low NAD+ and high NMN can induce axon degeneration (Coleman and Höke, 2020; Di Stefano et al., 2017, 2015). Therefore, Liu et al. developed a combination therapy that increases NAD+ and at the same time decreases NMN, and showed that it potently blocked VCR-induced axon degeneration in vitro (H. Liu et al., 2018), which represents another promising strategy for translation to the clinic.
After the enzymatic pocket of the NADase of the SARM1 molecule was identified (Bratkowski et al., 2020; Essuman et al., 2017; Sporny et al., 2019), the first small molecule noncompetitive inhibitors, berberine chloride and zinc chloride, which block the SARM1 NAD+ hydrolase were developed (Loring et al., 2020). These inhibitors are moderately potent, but represent a first step toward the development of more efficacious inhibitors, which is currently pursued by at least two pharmaceutical companies. Such NADase inhibitors, however, may also target other NAD-utilizing enzymes in neurons and non-neuronal cells resulting in undesired side effects (Coleman and Höke, 2020). Another attractive therapeutic approach is to utilize a dominant-negative SARM1 mutant that avoids potential side effects of inhibiting other NADases. SARM1 mutants have been developed that bind wildtype SARM1 as dominant/negatives and potently inhibit axon degeneration (Geisler et al., 2019b). Using adeno-associated virus (AAV) and a neuron-specific promoter, SARM1 mutants can be expressed selectively in peripheral somatosensory neuron populations, thereby avoiding possible side effects and interference with systemic chemotherapy or other drug treatments. Alternatively, SARM1 could be targeted with siRNA or antisense oligonucleotides. Such an approach has been shown to be highly efficacious in treating and reversing neuropathy due to transthyretin mutations (Adams et al., 2018; Benson et al., 2018). Thus, the identification of a core axon degeneration program that underlies different forms of axon degeneration has opened the gate to the development of therapeutics to prevent VCR and BTZ-induced neuropathies (Fig. 2).
Concluding remarks
New and improved chemotherapy regimens led to a dramatic and fortunate increase in the number of cancer survivors, but also to an urgent need to ameliorate the long term sequelae of treatments. Chemotherapy-induced neuropathy, in particular, causes long-lasting impairments and reduced quality of life. A plethora of in vitro and in vivo models of VCR and BTZ-induced neuropathies developed in recent years expanded and sharpened our understanding of molecular mechanisms underlying different aspects of neuropathies and also serve as a testing ground for new treatments. Some of the preclinical discoveries have recently been moved into clinical trials while others are being developed further in close collaborations between academia and the pharmaceutical industry. Numerous recent exciting discoveries in the fields of neuroinflammation, pain sensitization and axon degeneration encourage optimism that treatments to prevent, attenuate or reverse VCR- and BTZ-induced neuropathies are at our door step. Such treatments may improve the quality of life of patients both during and after cancer therapy and have enormous societal impact.
Highlights:
Peripheral neuropathy is a common side effect of the chemotherapeutic drugs vincristine and bortezomib
Bortezomib activates transcriptional programs in dorsal root ganglion neurons leading to neuroinflammation and pain
Vincristine inhibits transport of axonal survival factors
Both bortezomib and vincristine activate a common axon degeneration program that can be targeted therapeutically
Acknowledgement:
I thank the Toxic Neuropathy Consortium and Foundation for Peripheral Neuropathy for support. This work was funded by the NIH (K08NS091448), a Hope Center Pilot grant, American Cancer Society (IRG-18-158-61), Barnard Research Fund, and a Siteman Cancer Center grant.
Abbreviations:
- BIPN
bortezomib-induced peripheral neuropathy
- BTZ
bortezomib
- CIPN
chemotherapy-induced peripheral neuropathy
- DHSC
dorsal horn of the spinal cord
- DRG
dorsal root ganglia
- GFAP
glial fibrillary acidic protein
- HCN2
human cortical cell line 2
- IENF
intraepidermal nerve fibers
- MAPK 38
mitogen activated protein kinase 38
- NAD+
nicotinamide dinucleotide
- NFkB
nuclear factor kappa B
- NGF
nerve growth factor
- IENF
intraepidermal nerve fibers
- IENFD
intraepidermal nerve fiber density
- i.p.
intraperitoneal
- i.pl.
intraplantar
- i.v.
intravenous
- JNK
cJun NH2 terminal kinase
- QOD
every other day
- SARM1
Sterile Alpha and TIR motif containing 1
- SCID
severe combined immune deficiency spontaneous mutation
- S1P
sphingosine 1 phosphate
- S1PR
S1P recepto
- TGFβ
transforming growth factor beta
- TNFα
tumor necrosis factor alpha
- TRPA
transient receptor potential ankyrin 1 ion channel
- TRPV
transient receptor potential vanilloid receptor channel
- VCR
vincristine
- VIPN
vincristine-induced neuropathy
Footnotes
Conflict of Interest: SG is a co-inventor on a patent related to SARM1-dominant/negatives.
Additional references from tables 1 and 2:
(Alé et al., 2016, 2014; Aley et al., 1996; Authier et al., 2003, 1999; Cavaletti et al., 2007; Cho et al., 1983; Dambska et al., 1995; Erdoğan et al., 2020; Favaro et al., 1988; Fiori et al., 1995; Ja’afer et al., 2006; Kim et al., 2020; Lee et al., 2020; López-Gómez et al., 2018; Meregalli et al., 2012; Ogawa et al., 2000; Qin et al., 2020; Shi et al., 2018; Siau et al., 2006; Siau and Bennett, 2006; Singh et al., 2019)
References
- Adams D, Gonzalez-Duarte A, O’Riordan WD, Yang C-C, Ueda M, Kristen AV, Tournev I, Schmidt HH, Coelho T, Berk JL, Lin K-P, Vita G, Attarian S, Planté-Bordeneuve V, Mezei MM, Campistol JM, Buades J, Brannagan TH, Kim BJ, Oh J, Parman Y, Sekijima Y, Hawkins PN, Solomon SD, Polydefkis M, Dyck PJ, Gandhi PJ, Goyal S, Chen J, Strahs AL, Nochur SV, Sweetser MT, Garg PP, Vaishnaw AK, Gollob JA, Suhr OB, 2018. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N Engl J Med 379, 11–21. 10.1056/NEJMoa1716153 [DOI] [PubMed] [Google Scholar]
- Adams J, 2002. Development of the Proteasome Inhibitor PS‐341. The Oncologist 7, 9–16. 10.1634/theoncologist.7-1-9 [DOI] [PubMed] [Google Scholar]
- Adams J, Palombella VJ, Sausville EA, Johnson J, Destree A, Lazarus DD, Maas J, Pien CS, Prakash S, Elliott PJ, 1999. Proteasome Inhibitors: A Novel Class of Potent and Effective Antitumor Agents. Cancer Research 59, 2615–2622. [PubMed] [Google Scholar]
- Alé A, Bruna J, Calls A, Karamita M, Haralambous S, Probert L, Navarro X, Udina E, 2016. Inhibition of the neuronal NFκB pathway attenuates bortezomib-induced neuropathy in a mouse model. NeuroToxicology 55, 58–64. 10.1016/j.neuro.2016.05.004 [DOI] [PubMed] [Google Scholar]
- Alé A, Bruna J, Herrando M, Navarro X, Udina E, 2015. Toxic effects of bortezomib on primary sensory neurons and Schwann cells of adult mice. Neurotox Res 27, 430–440. 10.1007/s12640-014-9514-8 [DOI] [PubMed] [Google Scholar]
- Alé A, Bruna J, Morell M, van de Velde H, Monbaliu J, Navarro X, Udina E, 2014. Treatment with anti-TNF alpha protects against the neuropathy induced by the proteasome inhibitor bortezomib in a mouse model. Experimental Neurology 253, 165–173. 10.1016/j.expneurol.2013.12.020 [DOI] [PubMed] [Google Scholar]
- Aley KO, Reichling DB, Levine JD, 1996. Vincristine hyperalgesia in the rat: a model of painful vincristine neuropathy in humans. Neuroscience 73, 259–265. 10.1016/0306-4522(96)00020-6 [DOI] [PubMed] [Google Scholar]
- Ali YO, Bradley G, Lu H-C, 2017. Screening with an NMNAT2-MSD platform identifies small molecules that modulate NMNAT2 levels in cortical neurons. Scientific Reports 7, 43846. 10.1038/srep43846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alrashdi B, Dawod B, Schampel A, Tacke S, Kuerten S, Marshall JS, Côté PD, 2019. Nav1.6 promotes inflammation and neuronal degeneration in a mouse model of multiple sclerosis. J Neuroinflammation 16, 215. 10.1186/s12974-019-1622-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apfel SC, 2002a. Nerve growth factor for the treatment of diabetic neuropathy: what went wrong, what went right, and what does the future hold? Int Rev Neurobiol 50, 393–413. 10.1016/s0074-7742(02)50083-0 [DOI] [PubMed] [Google Scholar]
- Apfel SC, 2002b. Is the therapeutic application of neurotrophic factors dead? Ann Neurol 51, 8–11. 10.1002/ana.10099 [DOI] [PubMed] [Google Scholar]
- Araki T, Sasaki Y, Milbrandt J, 2004. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 305, 1010–1013. 10.1126/science.1098014 [DOI] [PubMed] [Google Scholar]
- Argyriou AA, Park SB, Islam B, Tamburin S, Velasco R, Alberti P, Bruna J, Psimaras D, Cavaletti G, Cornblath DR, Toxic Neuropathy Consortium (TNC), 2019. Neurophysiological, nerve imaging and other techniques to assess chemotherapy-induced peripheral neurotoxicity in the clinical and research settings. J. Neurol. Neurosurg. Psychiatry 90, 1361–1369. 10.1136/jnnp-2019-320969 [DOI] [PubMed] [Google Scholar]
- Arkwright R, Pham TM, Zonder JA, Dou QP, 2017. The preclinical discovery and development of bortezomib for the treatment of mantle cell lymphoma. Expert Opinion on Drug Discovery 12, 225–235. 10.1080/17460441.2017.1268596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Authier N, Coudore F, Eschalier A, Fialip J, 1999. Pain related behaviour during vincristine-induced neuropathy in rats. Neuroreport 10, 965–968. 10.1097/00001756-199904060-00013 [DOI] [PubMed] [Google Scholar]
- Authier N, Gillet J-P, Fialip J, Eschalier A, Coudore F, 2003. A New Animal Model of Vincristine-Induced Nociceptive Peripheral Neuropathy. NeuroToxicology 24, 797–805. 10.1016/S0161-813X(03)00043-3 [DOI] [PubMed] [Google Scholar]
- Bechakra M, Nieuwenhoff MD, van Rosmalen J, Jan Groeneveld G, J P M Huygen F, de Zeeuw CI, van Doorn PA, Jongen JLM, 2020. Pain-related changes in cutaneous innervation of patients suffering from bortezomib-induced, diabetic or chronic idiopathic axonal polyneuropathy. Brain Res. 1730, 146621. 10.1016/j.brainres.2019.146621 [DOI] [PubMed] [Google Scholar]
- Bechakra M, Nieuwenhoff MD, van Rosmalen J, Groeneveld GJ, Scheltens-de Boer M, Sonneveld P, van Doorn PA, de Zeeuw CI, Jongen JL, 2018. Clinical, electrophysiological, and cutaneous innervation changes in patients with bortezomib-induced peripheral neuropathy reveal insight into mechanisms of neuropathic pain. Mol Pain 14, 1744806918797042. 10.1177/1744806918797042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beider K, Rosenberg E, Bitner H, Shimoni A, Leiba M, Koren-Michowitz M, Ribakovsky E, Klein S, Olam D, Weiss L, Wald H, Abraham M, Galun E, Peled A, Nagler A, 2017. The Sphingosine-1-Phosphate Modulator FTY720 Targets Multiple Myeloma via the CXCR4/CXCL12 Pathway. Clin. Cancer Res 23, 1733–1747. 10.1158/1078-0432.CCR-15-2618 [DOI] [PubMed] [Google Scholar]
- Benson MD, Waddington-Cruz M, Berk JL, Polydefkis M, Dyck PJ, Wang AK, Planté-Bordeneuve V, Barroso FA, Merlini G, Obici L, Scheinberg M, Brannagan TH, Litchy WJ, Whelan C, Drachman BM, Adams D, Heitner SB, Conceição I, Schmidt HH, Vita G, Campistol JM, Gamez J, Gorevic PD, Gane E, Shah AM, Solomon SD, Monia BP, Hughes SG, Kwoh TJ, McEvoy BW, Jung SW, Baker BF, Ackermann EJ, Gertz MA, Coelho T, 2018. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N Engl J Med 379, 22–31. 10.1056/NEJMoa1716793 [DOI] [PubMed] [Google Scholar]
- Bilińska M, Usnarska-Zubkiewicz L, Pokryszko-Dragan A, 2013. Bortezomib-induced painful neuropathy in patients with multiple myeloma. Contemp Oncol (Pozn) 17, 421–426. 10.5114/wo.2013.37214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bladé J, Cibeira MT, Rosiñol L, 2005. Bortezomib: A valuable new antineoplastic strategy in multiple myeloma. Acta Oncologica 44, 440–448. 10.1080/02841860510030002 [DOI] [PubMed] [Google Scholar]
- Boehmerle W, Huehnchen P, Peruzzaro S, Balkaya M, Endres M, 2015. Electrophysiological, behavioral and histological characterization of paclitaxel, cisplatin, vincristine and bortezomib-induced neuropathy in C57Bl/6 mice. Scientific Reports 4, 6370. 10.1038/srep06370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyette-Davis J, Dougherty PM, 2011. Protection against oxaliplatin-induced mechanical hyperalgesia and intraepidermal nerve fiber loss by minocycline. Exp. Neurol 229, 353–357. 10.1016/j.expneurol.2011.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley WG, Williams MH, 1973. Axoplasmic flow in axonal neuropathies. I. Axoplasmic flow in cats with toxic neuropathies. Brain 96, 235–246. 10.1093/brain/96.2.235 [DOI] [PubMed] [Google Scholar]
- Bratkowski M, Xie T, Thayer DA, Lad S, Mathur P, Yang Y-S, Danko G, Burdett TC, Danao J, Cantor A, Kozak JA, Brown SP, Bai X, Sambashivan S, 2020. Structural and Mechanistic Regulation of the Pro-degenerative NAD Hydrolase SARM1. Cell Rep 32, 107999. 10.1016/j.celrep.2020.107999 [DOI] [PubMed] [Google Scholar]
- Bruna J, Alberti P, Calls-Cobos A, Caillaud M, Damaj MI, Navarro X, 2020. Methods for in vivo studies in rodents of chemotherapy induced peripheral neuropathy. Exp. Neurol 325, 113154. 10.1016/j.expneurol.2019.113154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruna J, Alé A, Velasco R, Jaramillo J, Navarro X, Udina E, 2011. Evaluation of pre-existing neuropathy and bortezomib retreatment as risk factors to develop severe neuropathy in a mouse model. Journal of the Peripheral Nervous System 16, 199–212. 10.1111/j.1529-8027.2011.00346.x [DOI] [PubMed] [Google Scholar]
- Campo C, da Silva Filho MI, Weinhold N, Mahmoudpour SH, Goldschmidt H, Hemminki K, Merz M, Försti A, 2018. Bortezomib-induced peripheral neuropathy: A genome-wide association study on multiple myeloma patients. Hematol Oncol 36, 232–237. 10.1002/hon.2391 [DOI] [PubMed] [Google Scholar]
- Carozzi VA, Canta A, Oggioni N, Sala B, Chiorazzi A, Meregalli C, Bossi M, Marmiroli P, Cavaletti G, 2010. Neurophysiological and neuropathological characterization of new murine models of chemotherapy-induced chronic peripheral neuropathies. Experimental Neurology 226, 301–309. 10.1016/j.expneurol.2010.09.004 [DOI] [PubMed] [Google Scholar]
- Carozzi Valentina A., Chiorazzi A, Canta A, Lapidus RG, Slusher BS, Wozniak KM, Cavaletti G, 2010. Glutamate carboxypeptidase inhibition reduces the severity of chemotherapy-induced peripheral neurotoxicity in rat. Neurotox Res 17, 380–391. 10.1007/s12640-009-9114-1 [DOI] [PubMed] [Google Scholar]
- Carozzi VA, Renn CL, Bardini M, Fazio G, Chiorazzi A, Meregalli C, Oggioni N, Shanks K, Quartu M, Serra MP, Sala B, Cavaletti G, Dorsey SG, 2013. Bortezomib-Induced Painful Peripheral Neuropathy: An Electrophysiological, Behavioral, Morphological and Mechanistic Study in the Mouse. PLoS ONE 8, e72995. 10.1371/journal.pone.0072995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey EB, Jellife AM, Le Quesne PM, Millett YL, 1973a. VINCRISTINE NEUROPATHY: CLINICAL AND ELECTROPHYSIOLOGICAL OBSERVATIONS. Brain 96, 69–86. 10.1093/brain/96.1.69 [DOI] [PubMed] [Google Scholar]
- Casey EB, Jellife AM, Le Quesne PM, Millett YL, 1973b. VINCRISTINE NEUROPATHYCLINICAL AND ELECTROPHYSIOLOGICAL OBSERVATIONS. Brain 96, 69–86. 10.1093/brain/96.1.69 [DOI] [PubMed] [Google Scholar]
- Cavaletti G, 2014. Chemotherapy-induced peripheral neurotoxicity (CIPN): what we need and what we know. Journal of the Peripheral Nervous System 19, 66–76. 10.1111/jns5.12073 [DOI] [PubMed] [Google Scholar]
- Cavaletti G, Gilardini A, Canta A, Rigamonti L, Rodriguez-Menendez V, Ceresa C, Marmiroli P, Bossi M, Oggioni N, D’Incalci M, De Coster R, 2007. Bortezomib-induced peripheral neurotoxicity: a neurophysiological and pathological study in the rat. Exp. Neurol 204, 317–325. 10.1016/j.expneurol.2006.11.010 [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL, 1994. Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 53, 55–63. 10.1016/0165-0270(94)90144-9 [DOI] [PubMed] [Google Scholar]
- Chaudhry V, Cornblath DR, Polydefkis M, Ferguson A, Borrello I, 2008. Characteristics of bortezomib- and thalidomide-induced peripheral neuropathy. Journal of the Peripheral Nervous System 13, 275–282. 10.1111/j.1529-8027.2008.00193.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Huang J, Benson C, Lankford KL, Zhao P, Carrara J, Tan AM, Kocsis JD, Waxman SG, Dib-Hajj SD, 2020. Sodium channel Nav1.6 in sensory neurons contributes to vincristine-induced allodynia. Brain 143, 2421–2436. 10.1093/brain/awaa208 [DOI] [PubMed] [Google Scholar]
- Chen L, Huang J, Zhao P, Persson A-K, Dib-Hajj FB, Cheng X, Tan A, Waxman SG, Dib-Hajj SD, 2018. Conditional knockout of NaV1.6 in adult mice ameliorates neuropathic pain. Sci Rep 8, 3845. 10.1038/s41598-018-22216-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiorazzi A, Canta A, Meregalli C, Carozzi V, Sala B, Oggioni N, Monbaliu J, 2013. Antibody Against Tumor Necrosis Factor-α Reduces Bortezomib-induced Allodynia in a Rat Model. ANTICANCER RESEARCH 7. [PubMed] [Google Scholar]
- Cho ES, Lowndes HE, Goldstein BD, 1983. Neurotoxicology of vincristine in the cat. Morphological study. Arch. Toxicol 52, 83–90. 10.1007/BF00354768 [DOI] [PubMed] [Google Scholar]
- Coleman MP, Conforti L, Buckmaster EA, Tarlton A, Ewing RM, Brown MC, Lyon MF, Perry VH, 1998. An 85-kb tandem triplication in the slow Wallerian degeneration (Wlds) mouse. Proc. Natl. Acad. Sci. U.S.A 95, 9985–9990. 10.1073/pnas.95.17.9985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman MP, Höke A, 2020. Programmed axon degeneration: from mouse to mechanism to medicine. Nat. Rev. Neurosci 21, 183–196. 10.1038/s41583-020-0269-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras PC, Vaught JL, Gruner JA, Brosnan C, Steffler C, Arezzo JC, Lewis ME, Kessler JA, Apfel SC, 1997. Insulin-like growth factor-I prevents development of a vincristine neuropathy in mice. Brain Res. 774, 20–26. 10.1016/s0006-8993(97)81682-4 [DOI] [PubMed] [Google Scholar]
- Cowie AM, Dittel BN, Stucky CL, 2019. A Novel Sex-Dependent Target for the Treatment of Postoperative Pain: The NLRP3 Inflammasome. Front. Neurol 10, 622. 10.3389/fneur.2019.00622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dambska M, Muzylak M, Maślińska D, 1995. The features of peripheral nerve lesions in young and adult rabbits after vincristine administration. Folia Neuropathol 33, 21–24. [PubMed] [Google Scholar]
- Deangelis LM, Gnecco C, Taylor L, Warrell RP, 1991. Evolution of neuropathy and myopathy during intensive vincristine/corticosteroid chemotherapy for non-hodgkin’s lymphoma. Cancer 67, 2241–2246. [DOI] [PubMed] [Google Scholar]
- Di Stefano M, Loreto A, Orsomando G, Mori V, Zamporlini F, Hulse RP, Webster J, Donaldson LF, Gering M, Raffaelli N, Coleman MP, Gilley J, Conforti L, 2017. NMN Deamidase Delays Wallerian Degeneration and Rescues Axonal Defects Caused by NMNAT2 Deficiency In Vivo. Curr. Biol 27, 784–794. 10.1016/j.cub.2017.01.070 [DOI] [PubMed] [Google Scholar]
- Di Stefano M, Nascimento-Ferreira I, Orsomando G, Mori V, Gilley J, Brown R, Janeckova L, Vargas ME, Worrell LA, Loreto A, Tickle J, Patrick J, Webster JRM, Marangoni M, Carpi FM, Pucciarelli S, Rossi F, Meng W, Sagasti A, Ribchester RR, Magni G, Coleman MP, Conforti L, 2015. A rise in NAD precursor nicotinamide mononucleotide (NMN) after injury promotes axon degeneration. Cell Death Differ. 22, 731–742. 10.1038/cdd.2014.164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiAntonio A, 2019. Axon degeneration: mechanistic insights lead to therapeutic opportunities for the prevention and treatment of peripheral neuropathy. PAIN 160, S17–S22. 10.1097/j.pain.0000000000001528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diouf B, Crews KR, Lew G, Pei D, Cheng C, Bao J, Zheng JJ, Yang W, Fan Y, Wheeler HE, Wing C, Delaney SM, Komatsu M, Paugh SW, McCorkle JR, Lu X, Winick NJ, Carroll WL, Loh ML, Hunger SP, Devidas M, Pui C-H, Dolan ME, Relling MV, Evans WE, 2015. Association of an inherited genetic variant with vincristine-related peripheral neuropathy in children with acute lymphoblastic leukemia. JAMA 313, 815–823. 10.1001/jama.2015.0894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit G, Dhingra A, Kaushal D, 2012. Vincristine induced cranial neuropathy. J Assoc Physicians India 60, 56–58. [PubMed] [Google Scholar]
- Dorsey SG, Kleckner IR, Barton D, Mustian K, O’Mara A, St. Germain D, Cavaletti G, Danhauer SC, Hershman DL, Hohmann AG, Hoke A, Hopkins JO, Kelly KP, Loprinzi CL, McLeod HL, Mohile S, Paice J, Rowland JH, Salvemini D, Segal RA, Smith EL, Stevens WM, Janelsins MC, 2019. The National Cancer Institute Clinical Trials Planning Meeting for Prevention and Treatment of Chemotherapy-Induced Peripheral Neuropathy. JNCI: Journal of the National Cancer Institute. 10.1093/jnci/djz011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty PM, Cata JP, Burton AW, Vu K, Weng H-R, 2007. Dysfunction in multiple primary afferent fiber subtypes revealed by quantitative sensory testing in patients with chronic vincristine-induced pain. J Pain Symptom Manage 33, 166–179. 10.1016/j.jpainsymman.2006.08.006 [DOI] [PubMed] [Google Scholar]
- Doyle TM, Chen Z, Durante M, Salvemini D, 2019. Activation of Sphingosine-1-Phosphate Receptor 1 in the Spinal Cord Produces Mechanohypersensitivity Through the Activation of Inflammasome and IL-1β Pathway. J Pain 20, 956–964. 10.1016/j.jpain.2019.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Z, Zhang J, Li J, Pang X, Wang H, 2020. Inhibition of microRNA-155 Reduces Neuropathic Pain During Chemotherapeutic Bortezomib via Engagement of Neuroinflammation. Front Oncol 10, 416. 10.3389/fonc.2020.00416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffin J, 2002. Poisoning the spindle: serendipity and discovery of the anti-tumor properties of the Vinca alkaloids (Part II). Pharm Hist 44, 105–119. [PubMed] [Google Scholar]
- Duggett NA, Flatters SJL, 2017. Characterization of a rat model of bortezomib-induced painful neuropathy. Br. J. Pharmacol 174, 4812–4825. 10.1111/bph.14063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdoğan MA, Taşkıran E, Yiğittürk G, Erbaş O, Taşkıran D, 2020. The investigation of therapeutic potential of oxytocin and liraglutide on vincristine-induced neuropathy in rats. J. Biochem. Mol. Toxicol 34, e22415. 10.1002/jbt.22415 [DOI] [PubMed] [Google Scholar]
- Essuman K, Summers DW, Sasaki Y, Mao X, DiAntonio A, Milbrandt J, 2017. The SARM1 Toll/Interleukin-1 Receptor Domain Possesses Intrinsic NAD+ Cleavage Activity that Promotes Pathological Axonal Degeneration. Neuron 93, 1334–1343.e5. 10.1016/j.neuron.2017.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Expósito Vizcaíno S, Casanova-Mollà J, Escoda L, Galán S, Miró J, 2018. Neuropathic pain in cancer patients treated with bortezomib. Neurologia 33, 28–34. 10.1016/j.nrl.2016.05.008 [DOI] [PubMed] [Google Scholar]
- Favaro G, Di Gregorio F, Panozzo C, Fiori MG, 1988. Ganglioside treatment of vincristine-induced neuropathy. An electrophysiologic study. Toxicology 49, 325–329. 10.1016/0300-483x(88)90015-7 [DOI] [PubMed] [Google Scholar]
- Figley MD, DiAntonio A, 2020. The SARM1 axon degeneration pathway: control of the NAD+ metabolome regulates axon survival in health and disease. Curr. Opin. Neurobiol 63, 59–66. 10.1016/j.conb.2020.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiori MG, Schiavinato A, Lini E, Nunzi MG, 1995. Peripheral Neuropathy Induced by Intravenous Administration of Vincristine Sulfate in the Rabbit.: An Ultrastructural Study. Toxicologic Pathology 23, 248–255. 10.1177/019262339502300302 [DOI] [PubMed] [Google Scholar]
- Gatta G, Capocaccia R, Stiller C, Kaatsch P, Berrino F, Terenziani M, the EUROCARE Working Group, 2005. Childhood Cancer Survival Trends in Europe: A EUROCARE Working Group Study. JCO 23, 3742–3751. 10.1200/JCO.2005.00.554 [DOI] [PubMed] [Google Scholar]
- Geisler S, Doan RA, Cheng GC, Cetinkaya-Fisgin A, Huang SX, Höke A, Milbrandt J, DiAntonio A, 2019a. Vincristine and bortezomib use distinct upstream mechanisms to activate a common SARM1-dependent axon degeneration program. JCI Insight 4, e129920. 10.1172/jci.insight.129920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Doan RA, Cheng GC, Cetinkaya-Fisgin A, Huang SX, Höke A, Milbrandt J, DiAntonio A, 2019b. Vincristine and bortezomib use distinct upstream mechanisms to activate a common SARM1-dependent axon degeneration program. JCI Insight 4. 10.1172/jci.insight.129920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Doan RA, Strickland A, Huang X, Milbrandt J, DiAntonio A, 2016. Prevention of vincristine-induced peripheral neuropathy by genetic deletion of SARM1 in mice. Brain 139, 3092–3108. 10.1093/brain/aww251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Huang SX, Strickland A, Doan RA, Summers DW, Mao X, Park J, DiAntonio A, Milbrandt J, 2019c. Gene therapy targeting SARM1 blocks pathological axon degeneration in mice. Journal of Experimental Medicine 216, 294–303. 10.1084/jem.20181040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdts J, Brace EJ, Sasaki Y, DiAntonio A, Milbrandt J, 2015. SARM1 activation triggers axon degeneration locally via NAD+ destruction. Science 348, 453–457. 10.1126/science.1258366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdts J, Summers DW, Milbrandt J, DiAntonio A, 2016. Axon Self-Destruction: New Links among SARM1, MAPKs, and NAD+ Metabolism. Neuron 89, 449–460. 10.1016/j.neuron.2015.12.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdts J, Summers DW, Sasaki Y, DiAntonio A, Milbrandt J, 2013. Sarm1-mediated axon degeneration requires both SAM and TIR interactions. J. Neurosci 33, 13569–13580. 10.1523/JNEUROSCI.1197-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannoccaro MP, Donadio V, Gomis Pèrez C, Borsini W, Di Stasi V, Liguori R, 2011. Somatic and autonomic small fiber neuropathy induced by bortezomib therapy: an immunofluorescence study. Neurol. Sci 32, 361–363. 10.1007/s10072-010-0475-2 [DOI] [PubMed] [Google Scholar]
- Gidding CEM, Kellie SJ, Kamps WA, de Graaf SSN, 1999. Vincristine revisited. Critical Reviews in Oncology/Hematology 29, 267–287. 10.1016/S1040-8428(98)00023-7 [DOI] [PubMed] [Google Scholar]
- Gilley J, Coleman MP, 2010. Endogenous Nmnat2 Is an Essential Survival Factor for Maintenance of Healthy Axons. PLoS Biology 8, e1000300. 10.1371/journal.pbio.1000300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilley J, Orsomando G, Nascimento-Ferreira I, Coleman MP, 2015. Absence of SARM1 rescues development and survival of NMNAT2-deficient axons. Cell Rep 10, 1974–1981. 10.1016/j.celrep.2015.02.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsby RE, Liu Q, Nathan PC, Bowers DC, Yeaton-Massey A, Raber SH, Hill D, Armstrong GT, Yasui Y, Zeltzer L, Robison LL, Packer RJ, 2010. Late-Occurring Neurologic Sequelae in Adult Survivors of Childhood Acute Lymphoblastic Leukemia: A Report From the Childhood Cancer Survivor Study. JCO 28, 324–331. 10.1200/JCO.2009.22.5060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green LS, Donoso JA, Heller‐Bettinger IE, Samson FE, 1977. Axonal transport disturbances in vincristine-induced peripheral neuropathy. Annals of Neurology 1, 255–262. 10.1002/ana.410010311 [DOI] [PubMed] [Google Scholar]
- Grenald SA, Doyle TM, Zhang H, Slosky LM, Chen Z, Largent-Milnes TM, Spiegel S, Vanderah TW, Salvemini D, 2017. Targeting the S1P/S1PR1 axis mitigates cancer-induced bone pain and neuroinflammation. Pain 158, 1733–1742. 10.1097/j.pain.0000000000000965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guiheneuc P, Ginet J, Groleau JY, Rojouan J, 1980. Early phase of vincristine neuropathy in man. Journal of the Neurological Sciences 45, 355–366. 10.1016/0022-510X(80)90179-3 [DOI] [PubMed] [Google Scholar]
- Guo Y, Xu X, Huang J, Wang Z, Li Z, Liu Z, 2020. The Actions and Mechanisms of P2X7R and p38 MAPK Activation in Mediating Bortezomib-Induced Neuropathic Pain. Biomed Res Int 2020, 8143754. 10.1155/2020/8143754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamity MV, White SR, Blum C, Gibson-Corley KN, Hammond DL, 2020. Nicotinamide riboside relieves paclitaxel-induced peripheral neuropathy and enhances suppression of tumor growth in tumor-bearing rats. Pain. 10.1097/j.pain.0000000000001924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamity MV, White SR, Walder RY, Schmidt MS, Brenner C, Hammond DL, 2017. Nicotinamide riboside, a form of vitamin B3 and NAD+ precursor, relieves the nociceptive and aversive dimensions of paclitaxel-induced peripheral neuropathy in female rats: PAIN 158, 962–972. 10.1097/j.pain.0000000000000862 [DOI] [PubMed] [Google Scholar]
- Hideshima T, Chauhan D, Schlossman R, Richardson P, Anderson KC, 2001. The role of tumor necrosis factor alpha in the pathophysiology of human multiple myeloma: therapeutic applications. Oncogene 20, 4519–4527. 10.1038/sj.onc.1204623 [DOI] [PubMed] [Google Scholar]
- Himes RH, Kersey RN, Heller-Bettinger I, Samson FE, 1976. Action of the Vinca Alkaloids Vincristine, Vinblastine, and Desacetyl Vinblastine Amide on Microtubules in Vitro 36, 6. [PubMed] [Google Scholar]
- Hirvonen HE, Salmi TT, Heinonen E, Antila KJ, Välimäki IA, 1989. Vincristine treatment of acute lymphoblastic leukemia induces transient autonomic cardioneuropathy. Cancer 64, 801–805. [DOI] [PubMed] [Google Scholar]
- Höke A, Ray M, 2014. Rodent models of chemotherapy-induced peripheral neuropathy. ILAR J 54, 273–281. 10.1093/ilar/ilt053 [DOI] [PubMed] [Google Scholar]
- Ja’afer FMH, Hamdan FB, Mohammed FH, 2006. Vincristine-induced neuropathy in rat: electrophysiological and histological study. Experimental Brain Research 173, 334–345. 10.1007/s00221-006-0499-2 [DOI] [PubMed] [Google Scholar]
- Jain A, Hakim S, Woolf CJ, 2020. Unraveling the Plastic Peripheral Neuroimmune Interactome. J.I 204, 257–263. 10.4049/jimmunol.1900818 [DOI] [PubMed] [Google Scholar]
- Ji R-R, Chamessian A, Zhang Y-Q, 2016. Pain regulation by non-neuronal cells and inflammation. Science 354, 572–577. 10.1126/science.aaf8924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia M, Wu C, Gao F, Xiang H, Sun N, Peng P, Li J, Yuan X, Li H, Meng X, Tian B, Shi J, Li M, 2017. Activation of NLRP3 inflammasome in peripheral nerve contributes to paclitaxel-induced neuropathic pain. Mol Pain 13, 1744806917719804. 10.1177/1744806917719804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Liu T, Lee C-H, Chang Q, Yang J, Zhang Z, 2020. The NAD+-mediated self-inhibition mechanism of pro-neurodegenerative Sarm1. Nature. 10.1038/s41586-020-2862-z [DOI] [PubMed] [Google Scholar]
- Johnson S, Armstrong JG, Gorman M, Burnett JP JR, 1963. The Vinca Alkaloids : A New Class of Oncolytic Agents. Cancer Research 23, 1390–1427. [PubMed] [Google Scholar]
- Kandula T, Farrar MA, Cohn RJ, Mizrahi D, Carey K, Johnston K, Kiernan MC, Krishnan AV, Park SB, 2018. Chemotherapy-Induced Peripheral Neuropathy in Long-term Survivors of Childhood Cancer: Clinical, Neurophysiological, Functional, and Patient-Reported Outcomes. JAMA Neurol 75, 980. 10.1001/jamaneurol.2018.0963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane RC, Farrell AT, Sridhara R, Pazdur R, 2006. United States Food and Drug Administration approval summary: bortezomib for the treatment of progressive multiple myeloma after one prior therapy. Clin. Cancer Res 12, 2955–2960. 10.1158/1078-0432.CCR-06-0170 [DOI] [PubMed] [Google Scholar]
- Karademir B, Sari G, Jannuzzi AT, Musunuri S, Wicher G, Grune T, Mi J, Hacioglu-Bay H, Forsberg-Nilsson K, Bergquist J, Jung T, 2018. Proteomic approach for understanding milder neurotoxicity of Carfilzomib against Bortezomib. Sci Rep 8, 16318. 10.1038/s41598-018-34507-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiguchi N, Maeda T, Kobayashi Y, Kondo T, Ozaki M, Kishioka S, 2008. The critical role of invading peripheral macrophage-derived interleukin-6 in vincristine-induced mechanical allodynia in mice. European Journal of Pharmacology 592, 87–92. 10.1016/j.ejphar.2008.07.008 [DOI] [PubMed] [Google Scholar]
- Kim K, Jeong W, Jun IG, Park JY, 2020. Antiallodynic and anti-inflammatory effects of intrathecal R-PIA in a rat model of vincristine-induced peripheral neuropathy. Korean J Anesthesiol. 10.4097/kja.19481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhoff AC, Krull KR, Ness KK, Armstrong GT, Park ER, Stovall M, Robison LL, Leisenring W, 2011. Physical, Mental, and Neurocognitive Status and Employment Outcomes in the Childhood Cancer Survivor Study Cohort. Cancer Epidemiology Biomarkers & Prevention 20, 1838–1849. 10.1158/1055-9965.EPI-11-0239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko KW, Milbrandt J, DiAntonio A, 2020. SARM1 acts downstream of neuroinflammatory and necroptotic signaling to induce axon degeneration. J. Cell Biol 219. 10.1083/jcb.201912047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajewski KM, Lewis RA, Fuerst DR, Turansky C, Hinderer SR, Garbern J, Kamholz J, Shy ME, 2000. Neurological dysfunction and axonal degeneration in Charcot-Marie-Tooth disease type 1A. Brain 123 (Pt 7), 1516–1527. 10.1093/brain/123.7.1516 [DOI] [PubMed] [Google Scholar]
- LaPointe NE, Morfini G, Brady ST, Feinstein SC, Wilson L, Jordan MA, 2013. Effects of eribulin, vincristine, paclitaxel and ixabepilone on fast axonal transport and kinesin-1 driven microtubule gliding: Implications for chemotherapy-induced peripheral neuropathy. NeuroToxicology 37, 231–239. 10.1016/j.neuro.2013.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latiff ZA, Kamal NA, Jahendran J, Alias H, Goh BS, Syed Zakaria SZ, Jamal R, 2010. Vincristine-induced vocal cord palsy: case report and review of the literature. J. Pediatr. Hematol. Oncol 32, 407–410. 10.1097/MPH.0b013e3181e01584 [DOI] [PubMed] [Google Scholar]
- Lauria G, Cornblath DR, Johansson O, McArthur JC, Mellgren SI, Nolano M, Rosenberg N, Sommer C, European Federation of Neurological Societies, 2005. EFNS guidelines on the use of skin biopsy in the diagnosis of peripheral neuropathy. Eur. J. Neurol 12, 747–758. 10.1111/j.1468-1331.2005.01260.x [DOI] [PubMed] [Google Scholar]
- Lavoie Smith EM, Li L, Chiang C, Thomas K, Hutchinson RJ, Wells EM, Ho RH, Skiles J, Chakraborty A, Bridges CM, Renbarger J, 2015. Patterns and severity of vincristine-induced peripheral neuropathy in children with acute lymphoblastic leukemia. Journal of the Peripheral Nervous System 20, 37–46. 10.1111/jns.12114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Sim WS, Cho NR, Kim BW, Moon JY, Park HJ, 2020. The Antiallodynic Effect of Nefopam on Vincristine-Induced Neuropathy in Mice. J Pain Res 13, 323–329. 10.2147/JPR.S224478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann HC, Staff NP, Hoke A, 2020. Modeling chemotherapy induced peripheral neuropathy (CIPN) in vitro: Prospects and limitations. Exp. Neurol 326, 113140. 10.1016/j.expneurol.2019.113140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Deng T, Shang Z, Wang D, Xiao Y, 2018. Blocking TRPA1 and TNF-α Signal Improves Bortezomib-Induced Neuropathic Pain. Cell. Physiol. Biochem 51, 2098–2110. 10.1159/000495828 [DOI] [PubMed] [Google Scholar]
- Lieber S, Blankenburg M, Apel K, Hirschfeld G, Hernáiz Driever P, Reindl T, 2018. Small-fiber neuropathy and pain sensitization in survivors of pediatric acute lymphoblastic leukemia. Eur. J. Paediatr. Neurol 22, 457–469. 10.1016/j.ejpn.2017.12.019 [DOI] [PubMed] [Google Scholar]
- Liew E, Thyagu S, Atenafu EG, Alibhai SMH, Brandwein JM, 2013. Quality of life following completion of treatment for adult acute lymphoblastic leukemia with a pediatric-based protocol. Leukemia Research 37, 1632–1635. 10.1016/j.leukres.2013.09.018 [DOI] [PubMed] [Google Scholar]
- Liu C, Luan S, OuYang H, Huang Z, Wu S, Ma C, Wei J, Xin W, 2016. Upregulation of CCL2 via ATF3/c-Jun interaction mediated the Bortezomib-induced peripheral neuropathy. Brain, Behavior, and Immunity 53, 96–104. 10.1016/j.bbi.2015.11.004 [DOI] [PubMed] [Google Scholar]
- Liu C-C, Huang Z-X, Li X, Shen K-F, Liu M, Ouyang H-D, Zhang S-B, Ruan Y-T, Zhang X-L, Wu S-L, Xin W-J, Ma C, 2018. Upregulation of NLRP3 via STAT3-dependent histone acetylation contributes to painful neuropathy induced by bortezomib. Exp. Neurol 302, 104–111. 10.1016/j.expneurol.2018.01.011 [DOI] [PubMed] [Google Scholar]
- Liu D, Sun M, Xu D, Ma X, Gao D, Yu H, 2019. Inhibition of TRPA1 and IL-6 signal alleviates neuropathic pain following chemotherapeutic bortezomib. Physiol Res 68, 845–855. 10.33549/physiolres.934015 [DOI] [PubMed] [Google Scholar]
- Liu H, Smith CB, Schmidt MS, Cambronne XA, Cohen MS, Migaud ME, Brenner C, Goodman RH, 2018. Pharmacological bypass of NAD+ salvage pathway protects neurons from chemotherapy-induced degeneration. Proceedings of the National Academy of Sciences 115, 10654–10659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobert S, Vulevic B, Correia JJ, 1996. Interaction of vinca alkaloids with tubulin: a comparison of vinblastine, vincristine, and vinorelbine. Biochemistry 35, 6806–6814. 10.1021/bi953037i [DOI] [PubMed] [Google Scholar]
- López-Gómez L, Díaz-Ruano S, Girón R, López-Pérez AE, Vera G, Herradón Pliego E, López-Miranda V, Nurgali K, Martín-Fontelles MI, Uranga JA, Abalo R, 2018. Preclinical evaluation of the effects on the gastrointestinal tract of the antineoplastic drug vincristine repeatedly administered to rats. Neurogastroenterol. Motil 30, e13399. 10.1111/nmo.13399 [DOI] [PubMed] [Google Scholar]
- Loring HS, Parelkar SS, Mondal S, Thompson PR, 2020. Identification of the first noncompetitive SARM1 inhibitors. Bioorg. Med. Chem 28, 115644. 10.1016/j.bmc.2020.115644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loring HS, Thompson PR, 2020. Emergence of SARM1 as a Potential Therapeutic Target for Wallerian-type Diseases. Cell Chem Biol 27, 1–13. 10.1016/j.chembiol.2019.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludman T, Melemedjian OK, 2019a. Bortezomib-induced aerobic glycolysis contributes to chemotherapy-induced painful peripheral neuropathy. Molecular Pain 15, 174480691983742. 10.1177/1744806919837429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludman T, Melemedjian OK, 2019b. Bortezomib and metformin opposingly regulate the expression of hypoxia-inducible factor alpha and the consequent development of chemotherapy-induced painful peripheral neuropathy. Mol Pain 15, 1744806919850043. 10.1177/1744806919850043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S, 1989. Absence of Wallerian Degeneration does not Hinder Regeneration in Peripheral Nerve. Eur. J. Neurosci 1, 27–33. [DOI] [PubMed] [Google Scholar]
- Mack TG, Reiner M, Beirowski B, Mi W, Emanuelli M, Wagner D, Thomson D, Gillingwater T, Court F, Conforti L, Fernando FS, Tarlton A, Andressen C, Addicks K, Magni G, Ribchester RR, Perry VH, Coleman MP, 2001. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat. Neurosci 4, 1199–1206. 10.1038/nn770 [DOI] [PubMed] [Google Scholar]
- Madsen ML, Due H, Ejskjær N, Jensen P, Madsen J, Dybkær K, 2019. Aspects of vincristine-induced neuropathy in hematologic malignancies: a systematic review. Cancer Chemother. Pharmacol 84, 471–485. 10.1007/s00280-019-03884-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maftei D, Vellani V, Artico M, Giacomoni C, Severini C, Lattanzi R, 2020. Abnormal Pain Sensation in Mice Lacking the Prokineticin Receptor PKR2: Interaction of PKR2 with Transient Receptor Potential TRPV1 and TRPA1. Neuroscience 427, 16–28. 10.1016/j.neuroscience.2019.12.003 [DOI] [PubMed] [Google Scholar]
- Mahmoudpour SH, Bandapalli OR, da Silva Filho MI, Campo C, Hemminki K, Goldschmidt H, Merz M, Försti A, 2018. Chemotherapy-induced peripheral neuropathy: evidence from genome-wide association studies and replication within multiple myeloma patients. BMC Cancer 18, 820. 10.1186/s12885-018-4728-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majithia N, Temkin SM, Ruddy KJ, Beutler AS, Hershman DL, Loprinzi CL, 2016. National Cancer Institute-supported chemotherapy-induced peripheral neuropathy trials: outcomes and lessons. Supportive Care in Cancer 24, 1439–1447. 10.1007/s00520-015-3063-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur JC, Stocks EA, Hauer P, Cornblath DR, Griffin JW, 1998. Epidermal Nerve Fiber Density: Normative Reference Range and Diagnostic Efficiency. Arch Neurol 55, 1513. 10.1001/archneur.55.12.1513 [DOI] [PubMed] [Google Scholar]
- McCarthy BG, Hsieh ST, Stocks A, Hauer P, Macko C, Cornblath DR, Griffin JW, McArthur JC, 1995. Cutaneous innervation in sensory neuropathies: evaluation by skin biopsy. Neurology 45, 1848–1855. 10.1212/wnl.45.10.1848 [DOI] [PubMed] [Google Scholar]
- McLeod JG, Penny R, 1969. Vincristine neuropathy: an electrophysiological and histological study. J. Neurol. Neurosurg. Psychiatry 32, 297–304. 10.1136/jnnp.32.4.297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meregalli C, Canta A, Carozzi VA, Chiorazzi A, Oggioni N, Gilardini A, Ceresa C, Avezza F, Crippa L, Marmiroli P, Cavaletti G, 2010. Bortezomib-induced painful neuropathy in rats: a behavioral, neurophysiological and pathological study in rats. Eur J Pain 14, 343–350. 10.1016/j.ejpain.2009.07.001 [DOI] [PubMed] [Google Scholar]
- Meregalli C, Carozzi VA, Sala B, Chiorazzi A, Canta A, Oggioni N, Rodriguez-Menendez V, Ballarini E, Ceresa C, Nicolini G, Crippa L, Orciani M, Cavaletti G, Marmiroli P, 2015. Bortezomib-induced peripheral neurotoxicity in human multiple myeloma-bearing mice. J. Biol. Regul. Homeost. Agents 29, 115–124. [PubMed] [Google Scholar]
- Meregalli C, Ceresa C, Canta A, Carozzi VA, Chiorazzi A, Sala B, Oggioni N, Lanza M, Letari O, Ferrari F, Avezza F, Marmiroli P, Caselli G, Cavaletti G, 2012. CR4056, a new analgesic I2 ligand, is highly effective against bortezomib-induced painful neuropathy in rats. J Pain Res 5, 151–167. 10.2147/JPR.S32122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meregalli C, Chiorazzi A, Carozzi VA, Canta A, Sala B, Colombo M, Oggioni N, Ceresa C, Foudah D, La Russa F, Miloso M, Nicolini G, Marmiroli P, Bennett DL, Cavaletti G, 2014. Evaluation of tubulin polymerization and chronic inhibition of proteasome as citotoxicity mechanisms in bortezomib-induced peripheral neuropathy. Cell Cycle 13, 612–621. 10.4161/cc.27476 [DOI] [PubMed] [Google Scholar]
- Meregalli C, Fumagalli G, Alberti P, Canta A, Carozzi VA, Chiorazzi A, Monza L, Pozzi E, Sandelius Å, Blennow K, Zetterberg H, Marmiroli P, Cavaletti G, 2018a. Neurofilament light chain as disease biomarker in a rodent model of chemotherapy induced peripheral neuropathy. Experimental Neurology 307, 129–132. 10.1016/j.expneurol.2018.06.005 [DOI] [PubMed] [Google Scholar]
- Meregalli C, Marjanovic I, Scali C, Monza L, Spinoni N, Galliani C, Brivio R, Chiorazzi A, Ballarini E, Rodriguez-Menendez V, Carozzi VA, Alberti P, Fumagalli G, Pozzi E, Canta A, Quartu M, Briani C, Oggioni N, Marmiroli P, Cavaletti G, 2018b. High-dose intravenous immunoglobulins reduce nerve macrophage infiltration and the severity of bortezomib-induced peripheral neurotoxicity in rats. J Neuroinflammation 15, 232. 10.1186/s12974-018-1270-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montague K, Simeoli R, Valente J, Malcangio M, 2018. A novel interaction between CX3CR1 and CCR2 signalling in monocytes constitutes an underlying mechanism for persistent vincristine-induced pain. J Neuroinflammation 15, 101. 10.1186/s12974-018-1116-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moschetti G, Amodeo G, Maftei D, Lattanzi R, Procacci P, Sartori P, Balboni G, Onnis V, Conte V, Panerai A, Sacerdote P, Franchi S, 2019a. Targeting prokineticin system counteracts hypersensitivity, neuroinflammation, and tissue damage in a mouse model of bortezomib-induced peripheral neuropathy. J Neuroinflammation 16, 89. 10.1186/s12974-019-1461-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moschetti G, Amodeo G, Paladini MS, Molteni R, Balboni G, Panerai A, Sacerdote P, Franchi S, 2019b. Prokineticin 2 promotes and sustains neuroinflammation in vincristine treated mice: Focus on pain and emotional like behavior. Brain Behav. Immun 82, 422–431. 10.1016/j.bbi.2019.09.012 [DOI] [PubMed] [Google Scholar]
- Naithani R, Dolai TK, Kumar R, 2009. Bilateral vocal cord paralysis following treatment with vincristine. Indian Pediatr 46, 68–69. [PubMed] [Google Scholar]
- Ness KK, Jones KE, Smith WA, Spunt SL, Wilson CL, Armstrong GT, Srivastava DK, Robison LL, Hudson MM, Gurney JG, 2013. Chemotherapy-Related Neuropathic Symptoms and Functional Impairment in Adult Survivors of Extracranial Solid Tumors of Childhood: Results From the St. Jude Lifetime Cohort Study. Archives of Physical Medicine and Rehabilitation 94, 1451–1457. 10.1016/j.apmr.2013.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble RL, 1990. The discovery of the vinca alkaloids--chemotherapeutic agents against cancer. Biochem. Cell Biol 68, 1344–1351. [PubMed] [Google Scholar]
- Ogawa T, Mimura Y, Kato H, Ootsubo S, Murakoshi M, 2000. The usefulness of rabbits as an animal model for the neuropathological assessment of neurotoxicity following the administration of vincristine. Neurotoxicology 21, 501–511. [PubMed] [Google Scholar]
- Old EA, Nadkarni S, Grist J, Gentry C, Bevan S, Kim K-W, Mogg AJ, Perretti M, Malcangio M, 2014. Monocytes expressing CX3CR1 orchestrate the development of vincristine-induced pain. J. Clin. Invest 124, 2023–2036. 10.1172/JCI71389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterloh JM, Yang J, Rooney TM, Fox AN, Adalbert R, Powell EH, Sheehan AE, Avery MA, Hackett R, Logan MA, MacDonald JM, Ziegenfuss JS, Milde S, Hou Y-J, Nathan C, Ding A, Brown RH, Conforti L, Coleman M, Tessier-Lavigne M, Züchner S, Freeman MR, 2012. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 337, 481–484. 10.1126/science.1223899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owellen RJ, Hartke CA, Dickerson RM, Hains FO, 1976. Inhibition of Tubulin-Microtubule Polymerization by Drugs of the. Cancer Research 36, 1499–1502. [PubMed] [Google Scholar]
- Pal PK, 1999. Clinical and electrophysiological studies in vincristine induced neuropathy. Electromyogr Clin Neurophysiol 39, 323–330. [PubMed] [Google Scholar]
- Peng L, Ye X, Zhou Y, Zhang J, Zhao Q, 2015. Meta-analysis of incidence and risk of peripheral neuropathy associated with intravenous bortezomib. Supportive Care in Cancer 23, 2813–2824. 10.1007/s00520-015-2648-2 [DOI] [PubMed] [Google Scholar]
- Pitarokoili K, Yoon M-S, Kröger I, Reinacher-Schick A, Gold R, Schneider-Gold C, 2017. Severe refractory CIDP: a case series of 10 patients treated with bortezomib. J. Neurol 264, 2010–2020. 10.1007/s00415-017-8599-4 [DOI] [PubMed] [Google Scholar]
- Poruchynsky MS, Sackett DL, Robey RW, Ward Y, Annunziata C, Fojo T, 2008. Proteasome inhibitors increase tubulin polymerization and stabilization in tissue culture cells: A possible mechanism contributing to peripheral neuropathy and cellular toxicity following proteasome inhibition. Cell Cycle 7, 940–949. 10.4161/cc.7.7.5625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Press C, Milbrandt J, 2008. Nmnat delays axonal degeneration caused by mitochondrial and oxidative stress. J. Neurosci 28, 4861–4871. 10.1523/JNEUROSCI.0525-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pui C-H, Campana D, Pei D, Bowman WP, Sandlund JT, Kaste SC, Ribeiro RC, Rubnitz JE, Raimondi SC, Onciu M, Coustan-Smith E, Kun LE, Jeha S, Cheng C, Howard SC, Simmons V, Bayles A, Metzger ML, Boyett JM, Leung W, Handgretinger R, Downing JR, Evans WE, Relling MV, 2009. Treating childhood acute lymphoblastic leukemia without cranial irradiation. N. Engl. J. Med 360, 2730–2741. 10.1056/NEJMoa0900386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin B, Li Y, Liu X, Gong D, Zheng W, 2020. Notch activation enhances microglial CX3CR1/P38 MAPK pathway in rats model of vincristine-induced peripheral neuropathy. Neurosci. Lett 715, 134624. 10.1016/j.neulet.2019.134624 [DOI] [PubMed] [Google Scholar]
- Qu X, Zhuang G, Yu L, Meng G, Ferrara N, 2012. Induction of Bv8 expression by granulocyte colony-stimulating factor in CD11b+Gr1+ cells: key role of Stat3 signaling. J. Biol. Chem 287, 19574–19584. 10.1074/jbc.M111.326801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravaglia S, Corso A, Piccolo G, Lozza A, Alfonsi E, Mangiacavalli S, Varettoni M, Zappasodi P, Moglia A, Lazzarino M, Costa A, 2008. Immune-mediated neuropathies in myeloma patients treated with bortezomib. Clin Neurophysiol 119, 2507–2512. 10.1016/j.clinph.2008.08.007 [DOI] [PubMed] [Google Scholar]
- Ravula SK, Wang MS, McClain MA, Asress SA, Frazier B, Glass JD, 2007. Spatiotemporal localization of injury potentials in DRG neurons during vincristine-induced axonal degeneration. Neuroscience Letters 415, 34–39. 10.1016/j.neulet.2007.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson PG, Briemberg H, Jagannath S, Wen PY, Barlogie B, Berenson J, Singhal S, Siegel DS, Irwin D, Schuster M, Srkalovic G, Alexanian R, Rajkumar SV, Limentani S, Alsina M, Orlowski RZ, Najarian K, Esseltine D, Anderson KC, Amato AA, 2006. Frequency, Characteristics, and Reversibility of Peripheral Neuropathy During Treatment of Advanced Multiple Myeloma With Bortezomib. Journal of Clinical Oncology 24, 3113–3120. 10.1200/JCO.2005.04.7779 [DOI] [PubMed] [Google Scholar]
- Richardson PG, Xie W, Mitsiades C, Chanan-Khan AA, Lonial S, Hassoun H, Avigan DE, Oaklander AL, Kuter DJ, Wen PY, Kesari S, Briemberg HR, Schlossman RL, Munshi NC, Heffner LT, Doss D, Esseltine D-L, Weller E, Anderson KC, Amato AA, 2009. Single-agent bortezomib in previously untreated multiple myeloma: efficacy, characterization of peripheral neuropathy, and molecular correlations with response and neuropathy. J. Clin. Oncol 27, 3518–3525. 10.1200/JCO.2008.18.3087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson CR, Dougherty PM, 2015. Spinal astrocyte gap junction and glutamate transporter expression contributes to a rat model of bortezomib-induced peripheral neuropathy. Neuroscience 285, 1–10. 10.1016/j.neuroscience.2014.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson Caleb R., Zhang H, Dougherty PM, 2014. Altered discharges of spinal neurons parallel the behavioral phenotype shown by rats with bortezomib related chemotherapy induced peripheral neuropathy. Brain Res. 1574, 6–13. 10.1016/j.brainres.2014.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson CR, Zhang H, Dougherty PM, 2014. Astrocytes, but not microglia, are activated in oxaliplatin and bortezomib-induced peripheral neuropathy in the rat. Neuroscience 274, 308–317. 10.1016/j.neuroscience.2014.05.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roca E, Bruera E, Politi PM, Barugel M, Cedaro L, Carraro S, Chacón RD, 1985. Vinca alkaloid-induced cardiovascular autonomic neuropathy. Cancer Treat Rep 69, 149–151. [PubMed] [Google Scholar]
- Ryan SP, DelPrete SA, Weinstein PW, Erichson RB, Bar MH, Lo KM, Cohen NS, Tepler I, 1999. Low-dose vincristine-associated bilateral vocal cord paralysis. Conn Med 63, 583–584. [PubMed] [Google Scholar]
- Saifee TA, Elliott KJ, Rabin N, Yong KL, D’Sa S, Brandner S, Lunn MP, Blake J, Reilly MM, 2010. Bortezomib-induced inflammatory neuropathy. J. Peripher. Nerv. Syst 15, 366–368. 10.1111/j.1529-8027.2010.00287.x [DOI] [PubMed] [Google Scholar]
- Sandler SG, Tobin W, Henderson ES, 1969. Vincristine-induced neuropathy. A clinical study of fifty leukemic patients. Neurology 19, 367–374. 10.1212/wnl.19.4.367 [DOI] [PubMed] [Google Scholar]
- Sasaki Y, Nakagawa T, Mao X, DiAntonio A, Milbrandt J, 2016a. NMNAT1 inhibits axon degeneration via blockade of SARM1-mediated NAD+ depletion. Elife 5. 10.7554/eLife.19749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Nakagawa T, Mao X, DiAntonio A, Milbrandt J, 2016b. NMNAT1 inhibits axon degeneration via blockade of SARM1-mediated NAD+ depletion. Elife 5. 10.7554/eLife.19749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Vohra BPS, Lund FE, Milbrandt J, 2009. Nicotinamide Mononucleotide Adenylyl Transferase-Mediated Axonal Protection Requires Enzymatic Activity But Not Increased Levels of Neuronal Nicotinamide Adenine Dinucleotide. J. Neurosci 29, 5525–5535. 10.1523/JNEUROSCI.5469-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt S, Goldschmidt H, Storch-Hagenlocher B, Pham M, Fingerle-Rowson G, Ho AD, Neben K, 2011. Inflammatory autoimmune neuropathy, presumably induced by bortezomib, in a patient suffering from multiple myeloma. Int. J. Hematol 93, 791–794. 10.1007/s12185-011-0847-2 [DOI] [PubMed] [Google Scholar]
- Scholz J, Woolf CJ, 2007. The neuropathic pain triad: neurons, immune cells and glia. Nat. Neurosci 10, 1361–1368. 10.1038/nn1992 [DOI] [PubMed] [Google Scholar]
- Shamash S, Reichert F, Rotshenker S, 2002. The cytokine network of Wallerian degeneration: tumor necrosis factor-alpha, interleukin-1alpha, and interleukin-1beta. J. Neurosci 22, 3052–3060. https://doi.org/20026249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Zhang Z-J, Zhu M-D, Jiang B-C, Yang T, Gao Y-J, 2015. Exogenous induction of HO-1 alleviates vincristine-induced neuropathic pain by reducing spinal glial activation in mice. Neurobiol. Dis 79, 100–110. 10.1016/j.nbd.2015.04.012 [DOI] [PubMed] [Google Scholar]
- Shi Q, Cai X, Shi G, Lv X, Yu J, Wang F, 2018. Interleukin-4 protects from chemotherapy-induced peripheral neuropathy in mice modal via the stimulation of IL-4/STAT6 signaling. Acta Cir Bras 33, 491–498. 10.1590/s0102-865020180060000003 [DOI] [PubMed] [Google Scholar]
- Siau C, Bennett GJ, 2006. Dysregulation of cellular calcium homeostasis in chemotherapy-evoked painful peripheral neuropathy. Anesth. Analg 102, 1485–1490. 10.1213/01.ane.0000204318.35194.ed [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siau C, Xiao W, Bennett G, 2006. Paclitaxel- and vincristine-evoked painful peripheral neuropathies: Loss of epidermal innervation and activation of Langerhans cells. Experimental Neurology 201, 507–514. 10.1016/j.expneurol.2006.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva A, Wang Q, Wang M, Ravula SK, Glass JD, 2006. Evidence for direct axonal toxicity in vincristine neuropathy. Journal of the Peripheral Nervous System 11, 211–216. 10.1111/j.1529-8027.2006.0090.x [DOI] [PubMed] [Google Scholar]
- Simon DJ, Pitts J, Hertz NT, Yang J, Yamagishi Y, Olsen O, Tešić Mark M, Molina H, Tessier-Lavigne M, 2016. Axon Degeneration Gated by Retrograde Activation of Somatic Pro-apoptotic Signaling. Cell 164, 1031–1045. 10.1016/j.cell.2016.01.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon DJ, Watkins TA, 2018. Therapeutic opportunities and pitfalls in the treatment of axon degeneration. Curr. Opin. Neurol 31, 693–701. 10.1097/WCO.0000000000000621 [DOI] [PubMed] [Google Scholar]
- Singh G, Singh A, Singh P, Bhatti R, 2019. Bergapten Ameliorates Vincristine-Induced Peripheral Neuropathy by Inhibition of Inflammatory Cytokines and NFκB Signaling. ACS Chem Neurosci 10, 3008–3017. 10.1021/acschemneuro.9b00206 [DOI] [PubMed] [Google Scholar]
- Singh SK, Spiegel S, 2020. Sphingosine-1-phosphate signaling: A novel target for simultaneous adjuvant treatment of triple negative breast cancer and chemotherapy-induced neuropathic pain. Adv Biol Regul 75, 100670. 10.1016/j.jbior.2019.100670 [DOI] [PubMed] [Google Scholar]
- Smith JA, Slusher BS, Wozniak KM, Farah MH, Smiyun G, Wilson L, Feinstein S, Jordan MA, 2016. Structural Basis for Induction of Peripheral Neuropathy by Microtubule-Targeting Cancer Drugs. Cancer Res. 76, 5115–5123. 10.1158/0008-5472.CAN-15-3116 [DOI] [PubMed] [Google Scholar]
- Sporny M, Guez-Haddad J, Lebendiker M, Ulisse V, Volf A, Mim C, Isupov MN, Opatowsky Y, 2019. Structural Evidence for an Octameric Ring Arrangement of SARM1. J. Mol. Biol 431, 3591–3605. 10.1016/j.jmb.2019.06.030 [DOI] [PubMed] [Google Scholar]
- Staff NP, Podratz JL, Grassner L, Bader M, Paz J, Knight AM, Loprinzi CL, Trushina E, Windebank AJ, 2013. Bortezomib alters microtubule polymerization and axonal transport in rat dorsal root ganglion neurons. NeuroToxicology 39, 124–131. 10.1016/j.neuro.2013.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starobova H, Mueller A, Allavena R, Lohman RJ, Sweet MJ, Vetter I, 2019. Minocycline Prevents the Development of Mechanical Allodynia in Mouse Models of Vincristine-Induced Peripheral Neuropathy. Front Neurosci 13, 653. 10.3389/fnins.2019.00653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starobova H, Mueller A, Deuis JR, Carter DA, Vetter I, 2020. Inflammatory and Neuropathic Gene Expression Signatures of Chemotherapy-Induced Neuropathy Induced by Vincristine, Cisplatin, and Oxaliplatin in C57BL/6J Mice. J Pain 21, 182–194. 10.1016/j.jpain.2019.06.008 [DOI] [PubMed] [Google Scholar]
- Stockstill K, Doyle TM, Yan X, Chen Z, Janes K, Little JW, Braden K, Lauro F, Giancotti LA, Harada CM, Yadav R, Xiao WH, Lionberger JM, Neumann WL, Bennett GJ, Weng H-R, Spiegel S, Salvemini D, 2018. Dysregulation of sphingolipid metabolism contributes to bortezomib-induced neuropathic pain. J. Exp. Med 215, 1301–1313. 10.1084/jem.20170584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockstill K, Wahlman C, Braden K, Chen Z, Yosten GL, Tosh DK, Jacobson KA, Doyle TM, Samson WK, Salvemini D, 2020. Sexually dimorphic therapeutic response in bortezomib-induced neuropathic pain reveals altered pain physiology in female rodents. Pain 161, 177–184. 10.1097/j.pain.0000000000001697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratogianni A, Tosch M, Schlemmer H, Weis J, Katona I, Isenmann S, Haensch CA, 2012. Bortezomib-induced severe autonomic neuropathy. Clin. Auton. Res 22, 199–202. 10.1007/s10286-012-0164-8 [DOI] [PubMed] [Google Scholar]
- Stubblefield MD, Slovin S, MacGregor-Cortelli B, Muzzy J, Scher H, Wright J, Esseltine D, Schenkein D, O’Connor OA, 2006. An electrodiagnostic evaluation of the effect of pre-existing peripheral nervous system disorders in patients treated with the novel proteasome inhibitor bortezomib. Clin Oncol (R Coll Radiol) 18, 410–418. 10.1016/j.clon.2005.12.008 [DOI] [PubMed] [Google Scholar]
- Summers DW, Milbrandt J, DiAntonio A, 2018. Palmitoylation enables MAPK-dependent proteostasis of axon survival factors. Proceedings of the National Academy of Sciences 115, E8746–E8754. 10.1073/pnas.1806933115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner KD, Levine JD, Topp KS, 1998. Microtubule disorientation and axonal swelling in unmyelinated sensory axons during vincristine-induced painful neuropathy in rat. J. Comp. Neurol 395, 481–492. [PubMed] [Google Scholar]
- Thibault K, Rivals I, M’Dahoma S, Dubacq S, Pezet S, Calvino B, 2013. Structural and molecular alterations of primary afferent fibres in the spinal dorsal horn in vincristine-induced neuropathy in rat. J. Mol. Neurosci 51, 880–892. 10.1007/s12031-013-0095-4 [DOI] [PubMed] [Google Scholar]
- Tomita S, Sekiguchi F, Deguchi T, Miyazaki T, Ikeda Y, Tsubota M, Yoshida S, Nguyen HD, Okada T, Toyooka N, Kawabata A, 2019. Critical role of Cav3.2 T-type calcium channels in the peripheral neuropathy induced by bortezomib, a proteasome-inhibiting chemotherapeutic agent, in mice. Toxicology 413, 33–39. 10.1016/j.tox.2018.12.003 [DOI] [PubMed] [Google Scholar]
- Topp KS, Tanner KD, Levine JD, 2000. Damage to the cytoskeleton of large diameter sensory neurons and myelinated axons in vincristine-induced painful peripheral neuropathy in the rat. J. Comp. Neurol 424, 563–576. [PubMed] [Google Scholar]
- Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, Bö L, 1998. Axonal transection in the lesions of multiple sclerosis. N. Engl. J. Med 338, 278–285. 10.1056/NEJM199801293380502 [DOI] [PubMed] [Google Scholar]
- Trevisan G, Materazzi S, Fusi C, Altomare A, Aldini G, Lodovici M, Patacchini R, Geppetti P, Nassini R, 2013. Novel therapeutic strategy to prevent chemotherapy-induced persistent sensory neuropathy by TRPA1 blockade. Cancer Res. 73, 3120–3131. 10.1158/0008-5472.CAN-12-4370 [DOI] [PubMed] [Google Scholar]
- Turkiew E, Falconer D, Reed N, Höke A, 2017. Deletion of Sarm1 gene is neuroprotective in two models of peripheral neuropathy. J. Peripher. Nerv. Syst 22, 162–171. 10.1111/jns.12219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utecht KN, Kolesar J, 2008. Bortezomib: a novel chemotherapeutic agent for hematologic malignancies. Am J Health Syst Pharm 65, 1221–1231. 10.2146/ajhp070272 [DOI] [PubMed] [Google Scholar]
- Verstappen CCP, Koeppen S, Heimans JJ, Huijgens PC, Scheulen ME, Strumberg D, Kiburg B, Postma TJ, 2005. Dose-related vincristine-induced peripheral neuropathy with unexpected off-therapy worsening. Neurology 64, 1076–1077. 10.1212/01.WNL.0000154642.45474.28 [DOI] [PubMed] [Google Scholar]
- Vohra BPS, Sasaki Y, Miller BR, Chang J, DiAntonio A, Milbrandt J, 2010. Amyloid Precursor Protein Cleavage-Dependent and -Independent Axonal Degeneration Programs Share a Common Nicotinamide Mononucleotide Adenylyltransferase 1-Sensitive Pathway. Journal of Neuroscience 30, 13729–13738. 10.1523/JNEUROSCI.2939-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Hehn CA, Baron R, Woolf CJ, 2012. Deconstructing the Neuropathic Pain Phenotype to Reveal Neural Mechanisms. Neuron 73, 638–652. 10.1016/j.neuron.2012.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vora A, Goulden N, Wade R, Mitchell C, Hancock J, Hough R, Rowntree C, Richards S, 2013. Treatment reduction for children and young adults with low-risk acute lymphoblastic leukaemia defined by minimal residual disease (UKALL 2003): a randomised controlled trial. The Lancet Oncology 14, 199–209. 10.1016/S1470-2045(12)70600-9 [DOI] [PubMed] [Google Scholar]
- Wahlman C, Doyle TM, Little JW, Luongo L, Janes K, Chen Z, Esposito E, Tosh DK, Cuzzocrea S, Jacobson KA, Salvemini D, 2018. Chemotherapy-induced pain is promoted by enhanced spinal adenosine kinase levels through astrocyte-dependent mechanisms. PAIN 159, 1025–1034. 10.1097/j.pain.0000000000001177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang MS, Fang G, Culver DG, Davis AA, Rich MM, Glass JD, 2001. The WldS protein protects against axonal degeneration: a model of gene therapy for peripheral neuropathy. Ann. Neurol 50, 773–779. [DOI] [PubMed] [Google Scholar]
- Wang MS, Wu Y, Culver DG, Glass JD, 2000. Pathogenesis of axonal degeneration: parallels between Wallerian degeneration and vincristine neuropathy. J. Neuropathol. Exp. Neurol 59, 599–606. 10.1093/jnen/59.7.599 [DOI] [PubMed] [Google Scholar]
- Wang Q, Wang J, Gao D, Li J, 2017. Inhibition of PAR2 and TRPA1 signals alleviates neuropathic pain evoked by chemotherapeutic bortezomib. J. Biol. Regul. Homeost. Agents 31, 977–983. [PubMed] [Google Scholar]
- Wang Qi, Zhang S, Liu T, Wang H, Liu K, Wang Qiujun, Zeng W, 2018. Sarm1/Myd88–5 Regulates Neuronal Intrinsic Immune Response to Traumatic Axonal Injuries. Cell Rep 23, 716–724. 10.1016/j.celrep.2018.03.071 [DOI] [PubMed] [Google Scholar]
- Wang XS, Shi Q, Bhadkamkar NA, Cleeland CS, Garcia-Gonzalez A, Aguilar JR, Heijnen C, Eng C, 2019. Minocycline for Symptom Reduction During Oxaliplatin-Based Chemotherapy for Colorectal Cancer: A Phase II Randomized Clinical Trial. J Pain Symptom Manage 58, 662–671. 10.1016/j.jpainsymman.2019.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng H-R, Cordella JV, Dougherty PM, 2003. Changes in sensory processing in the spinal dorsal horn accompany vincristine-induced hyperalgesia and allodynia. Pain 103, 131–138. 10.1016/s0304-3959(02)00445-1 [DOI] [PubMed] [Google Scholar]
- Wing C, Komatsu M, Delaney SM, Krause M, Wheeler HE, Dolan ME, 2017. Application of stem cell derived neuronal cells to evaluate neurotoxic chemotherapy. Stem Cell Research 22, 79–88. 10.1016/j.scr.2017.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright MJ, Twose DM, Gorter JW, 2017. Gait characteristics of children and youth with chemotherapy induced peripheral neuropathy following treatment for acute lymphoblastic leukemia. Gait Posture 58, 139–145. 10.1016/j.gaitpost.2017.05.004 [DOI] [PubMed] [Google Scholar]
- Xie J-D, Chen S-R, Chen H, Pan H-L, 2017. Bortezomib induces neuropathic pain through protein kinase C-mediated activation of presynaptic NMDA receptors in the spinal cord. Neuropharmacology 123, 477–487. 10.1016/j.neuropharm.2017.06.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Strong JA, Ye L, Mao J-X, Zhang J-M, 2013. Knockdown of sodium channel NaV1.6 blocks mechanical pain and abnormal bursting activity of afferent neurons in inflamed sensory ganglia. PAIN 154, 1170–1180. 10.1016/j.pain.2013.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Strong JA, Zhang J-M, 2015. Local knockdown of the NaV1.6 sodium channel reduces pain behaviors, sensory neuron excitability, and sympathetic sprouting in rat models of neuropathic pain. Neuroscience 291, 317–330. 10.1016/j.neuroscience.2015.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin H, Lu R, Lee H, Zhang W, Zhang C, Deng J, Liu Y, Shen S, Wagner K-U, Forman S, Jove R, Yu H, 2013. G-protein-coupled receptor agonist BV8/prokineticin-2 and STAT3 protein form a feed-forward loop in both normal and malignant myeloid cells. J. Biol. Chem 288, 13842–13849. 10.1074/jbc.M113.450049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y-L, Zhao W-H, Tang Z-Y, Li Z-Q, Long Y, Cheng P, Luo J, 2019. Guillain-Barré syndrome in a patient with multiple myeloma after bortezomib therapy: A case report. World J Clin Cases 7, 2905–2909. 10.12998/wjcc.v7.i18.2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S, Kawashiri T, Higuchi H, Tsutsumi K, Ushio S, Kaname T, Shirahama M, Egashira N, 2015. Behavioral and pharmacological characteristics of bortezomib-induced peripheral neuropathy in rats. Journal of Pharmacological Sciences 129, 43–50. 10.1016/j.jphs.2015.08.006 [DOI] [PubMed] [Google Scholar]
- Yan M, Li Y, Zeng H, Zhao X, Wu H, Qian W, Guo X, 2020. The effect of rat nerve growth factor combined with vitamin B on peripheral neuropathy in multiple myeloma patients. Hematology 25, 264–269. 10.1080/16078454.2020.1784615 [DOI] [PubMed] [Google Scholar]
- Yang J, Wu Z, Renier N, Simon DJ, Uryu K, Park DS, Greer PA, Tournier C, Davis RJ, Tessier-Lavigne M, 2015. Pathological axonal death through a MAPK cascade that triggers a local energy deficit. Cell 160, 161–176. 10.1016/j.cell.2014.11.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youk J, Kim Y-S, Lim J-A, Shin D-Y, Koh Y, Lee S-T, Kim I, 2017. Depletion of nerve growth factor in chemotherapy-induced peripheral neuropathy associated with hematologic malignancies. PLoS ONE 12, e0183491. 10.1371/journal.pone.0183491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahid A, Li B, Kombe AJK, Jin T, Tao J, 2019. Pharmacological Inhibitors of the NLRP3 Inflammasome. Front. Immunol. 10, 2538. 10.3389/fimmu.2019.02538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaroulis CK, Chairopoulos K, Sachanas SP, Maltezas D, Tzenou T, Pessach I, Koulieris E, Koutra E, Kilindireas K, Pangalis GA, Kyrtsonis M-C, 2014. Assessment of bortezomib induced peripheral neuropathy in multiple myeloma by the reduced Total Neuropathy Score. Leuk. Lymphoma 55, 2277–2283. 10.3109/10428194.2013.873535 [DOI] [PubMed] [Google Scholar]
- Zhang J, Su Y-M, Li D, Cui Y, Huang Z-Z, Wei J-Y, Xue Z, Pang R-P, Liu X-G, Xin W-J, 2014. TNF-α-mediated JNK activation in the dorsal root ganglion neurons contributes to Bortezomib-induced peripheral neuropathy. Brain, Behavior, and Immunity 38, 185–191. 10.1016/j.bbi.2014.01.020 [DOI] [PubMed] [Google Scholar]
- Zhang Z-J, Jiang B-C, Gao Y-J, 2017. Chemokines in neuron-glial cell interaction and pathogenesis of neuropathic pain. Cell. Mol. Life Sci 74, 3275–3291. 10.1007/s00018-017-2513-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Wang W, Li X, Liu Y, Gao H, Jiang Y, Wang Y, 2019. Peripheral neuropathy following bortezomib therapy in multiple myeloma patients: association with cumulative dose, heparanase, and TNF-α. Ann. Hematol 98, 2793–2803. 10.1007/s00277-019-03816-6 [DOI] [PubMed] [Google Scholar]
- Zhao Y, Wu J, Wang X, Jia H, Chen D-N, Li J-D, 2019. Prokineticins and their G protein-coupled receptors in health and disease, in: Progress in Molecular Biology and Translational Science. Elsevier, pp. 149–179. 10.1016/bs.pmbts.2018.09.006 [DOI] [PubMed] [Google Scholar]
- Zhao ZY, Xie XJ, Li WH, Liu J, Chen Z, Zhang B, Li T, Li SL, Lu JG, Zhang Liangren, Zhang Li-he, Xu Z, Lee HC, Zhao YJ, 2019. A Cell-Permeant Mimetic of NMN Activates SARM1 to Produce Cyclic ADP-Ribose and Induce Non-apoptotic Cell Death. iScience 15, 452–466. 10.1016/j.isci.2019.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Ao L, Yan Y, Li C, Li W, Ye A, Liu J, Hu Y, Fang W, Li Y, 2020. Levo-corydalmine Attenuates Vincristine-Induced Neuropathic Pain in Mice by Upregulating the Nrf2/HO-1/CO Pathway to Inhibit Connexin 43 Expression. Neurotherapeutics 17, 340–355. 10.1007/s13311-019-00784-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Hu Y, Li C, Yan Y, Ao L, Yu B, Fang W, Liu J, Li Y, 2018. Levo-corydalmine alleviates vincristine-induced neuropathic pain in mice by inhibiting an NF-kappa B-dependent CXCL1/CXCR2 signaling pathway. Neuropharmacology 135, 34–47. 10.1016/j.neuropharm.2018.03.004 [DOI] [PubMed] [Google Scholar]
- Zhou Y-Q, Liu D-Q, Chen S-P, Sun J, Wang X-M, Tian Y-K, Wu W, Ye D-W, 2018. Minocycline as a promising therapeutic strategy for chronic pain. Pharmacol. Res 134, 305–310. 10.1016/j.phrs.2018.07.002 [DOI] [PubMed] [Google Scholar]