

Abstract
The deubiquitinating enzyme BAP1 is mutated in a hereditary cancer syndrome with a high risk for mesothelioma and melanocytic tumors. Here, we show that pancreatic intra- epithelial neoplasia driven by oncogenic mutant Kras G12D progressed to pancreatic adenocarcinoma in the absence of BAP1. The Hippo pathway was deregulated in BAP1- deficient pancreatic tumors, with the tumor suppressor LATS exhibiting enhanced ubiquitin-dependent proteasomal degradation. Therefore, BAP1 may limit tumor progression by stabilizing LATS and thereby promoting activity of the Hippo tumor suppressor pathway.
INTRODUCTION
Somatic mutations inactivate BAP1 in the majority of metastatic uveal melanomas (1) and in approximately 60% of malignant pleural mesotheliomas (2, 3). Some breast, lung and renal cell cancers also harbor somatic BAP1 mutations (4–7). Germline BAP1 mutations cause a tumor predisposition syndrome characterized by benign melanocytic tumors (8) and mesothelioma, uveal melanoma, and other cancers (9, 10). The spectrum of tumors caused by BAP1 mutations continues to expand as more families with germline mutations are identifiednBAP1 loss during mouse development is embryonic lethal, whereas BAP1 deficiency in the adult mouse causes a severe myeloproliferative disease reminiscent of chronic myelomonocytic leukemia (11). BAP1 is expressed ubiquitously (12) and is found in chromatin-associated complexes containing HCFC1, OGT, ASXL1/2, FOXK1/2 and KDM1b (11, 13–15). The Drosophila BAP1 ortholog Calypso influences histone dynamics by deubiquitinating histone 2A at lysine 118 (H2A-K118ub) (14).
BAP1 has been linked to a multitude of biological processes, including DNA damage (16), cell cycle (17), gluconeogenesis (18) and metabolic homeostasis (19). However, the mechanism by which BAP1 suppresses tumorigenesis, the signaling pathways that are deregulated in BAP1-deficient tumors and the critical BAP1 substrate(s) in various cancers remain largely unexplored.
In this study, we demonstrate a role for BAP1 as a tumor suppressor in pancreatic cancer. BAP1 loss cooperated with oncogenic Kras to form pancreatic ductal adenocarcinoma in mice. The Hippo pathway was deregulated in these BAP1-deficient tumors because negative regulator of the pathway, LATS2, was ubiquitinated and degraded. Hippo pathway deregulation leads to hyperactivation of the key downstream oncoproteins YAP and TAZ. Therefore, we identify YAP/TAZ repression as a potential therapeutic strategy for certain BAP1-deficient tumors.
MATERIALS and METHODS
Mice
Bap1fl/fl mice crossed to the inducible general deleter C567BL/6 NTac- Gt(ROSA)26Sortm9(Cre/ESR1)Arte were described previously (11). CreERT2+ mice aged 6–8 wks were injected intraperitoneally with 60 mg/kg tamoxifen dissolved in sunflower oil daily for 5 days. To generate the pancreatic specific deletion, the Bap1fl/fl mice were crossed to Pdx1.cre mice (20). The Genentech Institutional Animal Care and Use Committee approved all protocols.
Genotyping
Bap1 genotyping primers 5’ CCA TCA GTG ACT ACT GGG GAG CAA C, 5’ ACA GAT GGC TGG GCA CAT CTG, and 5’ GAA CCC TCC GTT GCA TAG TGT TG amplified 234 bp WT, 350 bp floxed, and 503 bp KO DNA fragments. Cre primers 5′- GCT AAA CAT GCT TCA TCG TCG GTC and 5′-CCA GAC CAG GCC AGG TAT CTC TG amplified a 582 bp DNA fragment.
Bone marrow transplants
Recipient animals, Bap1fl/fl;Rosa26.creERT2+ or Bap1wt/wt;Rosa26.creERT2+, received 2 doses of 525 Rads from a 137Cs source separated by a 4 h interval. Donor bone marrow cells from Bap1wt/wt animals were injected into the tail vein. Reconstituted mice were given water containing 0.11 mg/mL polymyxin B and 1.1 mg/mL neomycin for two weeks and then switched to regular water.
Cell lines and reagents
To generate PDAC BAP1 KO tumor-derived cell lines, the tumor tissue was minced with a pair of scalpels in a 10 cm Petri dish containing 20 ml RPMI + 10% FBS. Tissue fragments with the medium were transferred to a T75 tissue culture flask and placed into a 37C, 5% CO2 incubator. After 1 or 2 days to allow for cellular attachment, the medium was removed and replaced with fresh growth medium. Contaminating fibroblasts were removed by differential trypsinization. Once monolayer cultures were established and passaged a few times, the growing cultures were passaged by trypsinization at appropriate intervals and split ratios. BAP1 WT reconstituted PDAC and H226 cells were generated by transducing cells with lentiviral BAP1construct for 72 hours followed by selection with puromycin.
ME202, 92–1, OMM1.3, UPMM-2 and UPMM-3 cells have been previously described (21). MP38 and MP46 cells were kindly provided by Roman-Roman Sergio (Institut Curie, France) (22). All other cell lines were from the American Type Culture Collection (ATCC). Cell lines were maintained in RPMI 1640 or DMEM/F12 (GIBCO) supplemented with 10% FBS (Sigma), 50 units/mL penicillin, 50 units/mL streptomycin and 2mM L-glutamine (Gibco). Doxycycline was purchased from BD and cycloheximide was from Calbiochem.
Cell line authentication/quality control.
Cell lines used were checked at the Genentech core lab by the following methods. Short Tandem Repeat (STR) Profiling: STR profiles were determined for each line using the Promega PowerPlex 16 System. This is performed once and compared to external STR profiles of cell lines (when available) to determine cell line ancestry. SNP fingerprinting: SNP profiles are performed each time new stocks are expanded for cryopreservation. Cell line identity is verified by high-throughput SNP profiling using Fluidigm multiplexed assays. SNPs were selected based on minor allele frequency and presence on commercial genotyping platforms. SNP profiles are compared to SNP calls from available internal and external data (when available) to determine or confirm ancestry. Mycoplasma Testing: All stocks were tested for mycoplasma prior to and after cells are cryopreserved. Two methods are used to avoid false positive/negative results: Lonza Mycoalert and Stratagene Mycosensor. Cell growth rates and morphology are also monitored for any batch-to-batch changes.
Immunoprecipitation and immunoblotting
For co-immunoprecipitation experiments, HEK293T cells were transfected with Flag- tagged BAP1 or LATS2 constructs for 72 hr followed by cell lysis using RIPA buffer and immunoprecipitation with FLAG-M2 agarose beads (Sigma) for overnight at 4°C. After washing with RIPA buffer, co-immunoprecipitated endogenous proteins, such as LATS1, 2, MOB1, and BAP1 were then detected by immunoblotting.
For immunoblotting, cells were harvested in RIPA lysis buffer containing protease and phosphatase inhibitors cocktail (Roche). Lysates were then analyzed for immunoblotting. Antibodies directed against BAP1, LATS1, LATS2, YAP/TAZ, pYAP, MOB1, MST1, MST2, NF2, GAPDH, β-Tubulin, and α-actin were purchased from Cell Signaling Technology. MAX antibody was from Santa Cruz Biotechnology.
Subcellular Fractionation
Cells grown on 10 cm dishes were harvested after washing with cold PBS. Cell pellets were then incubated with 400 μl of Buffer A (10 mM HEPES pH 7.9, 10 mM KCl, 1 mM EDTA, 0.1 mM EGTA, 0.2% NP-40, 10% glycerol) on ice for 20 min. Cytoplasmic fraction was collected by taking the supernatant from centrifugation for 30 seconds at 1,400 × g. For the preparation of nuclear fraction, the pellet was then incubated with 200μl of RIPA buffer for 30 min at 4°C. After centrifugation for 12 mins at 16,000 × g, nuclear fraction was collected by retaining the supernatant.
siRNA transfection and Quantitative PCR
All siRNA oligos and a nonspecific non-targeting control were purchased from Dharmacon. siRNA oligos were transfected into cells for 48 hr using the Lipofectamine RNAiMAX reagent (Invitrogen).
For RNA preparation, tumors were dissociated and lysed for RNA isolation using RNAeasy kit (Qiagen). For quantitative PCR, cDNA was prepared by reverse transcription and PCR was performed using TaqMan probes for YAP, TAZ, CTGF, CYR61, LATS1, LATS2, and HPRT (Life Science). Relative expression to HPRT of target genes was assessed. The siRNA and probe information used in this study are listed in Supplementary Table 1.
Deubiquitination (DUB) assay
For the in cell deubiquitination assay, Myc-Flag-human LATS2 and HA-ubiquitin (ub) plasmids were transfected into HEK293T cells at a ratio of 1:1.5 with Fugene 6. After 48 hours, cells were lysed in NP-40 buffer (1% NP-40, 120 mM NaCl, 50 mM Tris, pH 7.4, 1 mM EDTA, pH 7.4, 20 mM cysteine protease inhibitor N-ethylmaleimide (NEM), protease and phosphatase inhibitors (Roche)). Proteins were denatured with 1% SDS and then diluted to 0.05% SDS prior to anti-HA immunoprecipitation.
For the in vitro deubiquitination assay, ubiquitinated LATS1 and 2 were isolated by transiently cotransfecting HEK293T cells with MYC-LATS1/MYC-LATS2 and V5-ubiquitin as described above. BAP1 protein was purified from BAP1 overexpressing HEK293T cells by immnoprecipitation using anti-BAP1 antibody. In vitro reaction was performed by incubating poly-ub-LATS with BAP1 in in 2 × reaction buffer (100 μM Tris pH 8.0, 40 mM NaCl, 200 μg/ml BSA, 8 μM EDTA pH 8.0, 4 mM DTT) at 25°C for indicated times. Reaction was terminated by adding SDS sample buffer followed by 5 mins boiling at 95°C. The result was assessed by IB analysis.
Histology and Immunohistochemistry
Histopathologic analyses were performed on routinely processed formalin-fixed paraffin embedded tissue sections. Antibodies used for immunohistochemistry included carboxypeptidase A1 (R & D Systems goat polyclonal, 0.05 μg/ml), cytokeratin 19 (University of Iowa rat monoclonal TROMA III, 10 μg/ml), HES1 (MBL rat monoclonal NM1, 1 μg/ml), cleaved notch 1 (Val1744) (Cell Signaling rabbit monoclonal D3B8, 10 μg/ml), Ki-67 (NeoMarkers rabbit monoclonal SP6, 1:200 dilution), YAP1 (Cell Signaling rabbit polyclonal, 0.036 μg/ml), CD3 (Thermo Scientific rabbit monoclonal SP7, 1:200 dilution), TAZ (Sigma rabbit polyclonal, 0.25 μg/ml), BAP1 (Genentech rabbit monoclonal 2G12, 1 μg/ml), CD68 (Serotec rat monoclonal FA-11, 5 μg/ml), insulin (Dako guinea pig polyclonal, 5.5 μg/ml), glucagon (Cell Signaling rabbit polyclonal, 0.0417 μg/ml), and cleaved caspase 3 (Asp175) (Cell Signaling rabbit monoclonal 9661L, 0.05 μg/ml). Cleaved caspase 3, Ki-67, and carboxypeptidase A1 immunohistochemistry was performed on the Ventana Discovery XT platform with CC1 standard antigen retrieval (Ventana) and OmniMap detection (Ventana). Cleaved notch immunohistochemistry was also performed on the Ventana Discovery XT platform with OmniMap detection, but with CC1 extended retrieval. Cytokeratin 19, CD3, CD68, and glucagon immunohistochemistry was performed on a Dako autostainer with Target (Dako) antigen retrieval and ABC-Peroxidase Elite (Vector Laboratories) detection. Insulin immunohistochemistry was performed on the Dako autostainer with ABC- Peroxidase Elite detection, but with no antigen retrieval. TAZ and BAP1 immunohistochemistry was also performed on a Dako autostainer with Target retrieval, but detection was performed with PowerVision polymer-HRP (Leica Biosystems). HES-1 and YAP immunohistochemistry were performed on the Dako autostainer with TSA- HRP detection. EDTA pH8 (LabVision) heat-induced epitope retrieval was used for YAP and Target retrieval was used for HES-1. 2,3-diaminobenzidine (DAB) was used as the chromogen for all studies and hematoxylin was used as a counterstain. Isotype control antibodies were used as a negative controls and positive control tissues and/or cell lines were used to validate each antibody. For quantitative image analysis, slides were digitally scanned using a Hamamatsu slide scanner and immunolabeling was quantified using MatLab (MathWorks) software package.
RESULTS
A small proportion of mice with non-hematopoietic deletion of Bap1 develop diverse tumor types.
BAP1 loss in adult mice causes lethal myeloid dysplasia within 4–6 weeks (11). To investigate the role of BAP1 in non-hematopoietic cells, we used tamoxifen to induce BAP1 loss in Bap1fl/fl Rosa26CreERT2/+ chimeric mice that had their hematopoietic system reconstituted with wild-type bone marrow (hereafter referred to as BAP1 BMC-KO). Bap1+/+ Rosa26CreERT2/+ mice reconstituted with wild-type bone marrow (hereafter referred to as BAP1 BMC-WT) served as negative controls (19) (Figure 1A). Zero out of 28 BAP1 BMC-KO mice analyzed between 3 and 10 months after tamoxifen treatment had CMML-like disease. After 3 months, however, all BAP1 BMC-KO mice (n=11) exhibited pancreatic acinar atrophy (loss of acinar tissue) with relatively increased duct profiles (possibly due to parenchymal collapse or acinar-ductal metaplasia), which was not associated with acute inflammatory infiltrates (Figure 1B) and multifocal hepatocellular death (individual shrunken and hypereosinophilic hepatocytes) with regenerative changes (variation in hepatocellular size, mitotic activity) that disrupted the hepatic architecture (Figure S1A). In addition, 2 out of the 11 BAP1 BMC-KO mice had tumors. One mouse had an intestinal sarcoma (Figure S1B) and the other had a pulmonary adenoma (Figure S1C). BAP1 BMC-WT controls (n=9) examined at 3 months after tamoxifen treatment had no significant histologic lesions or tumors.
Fig. 1. A small proportion of mice with non-hematopoietic deletion of Bap1 develop diverse tumor types.
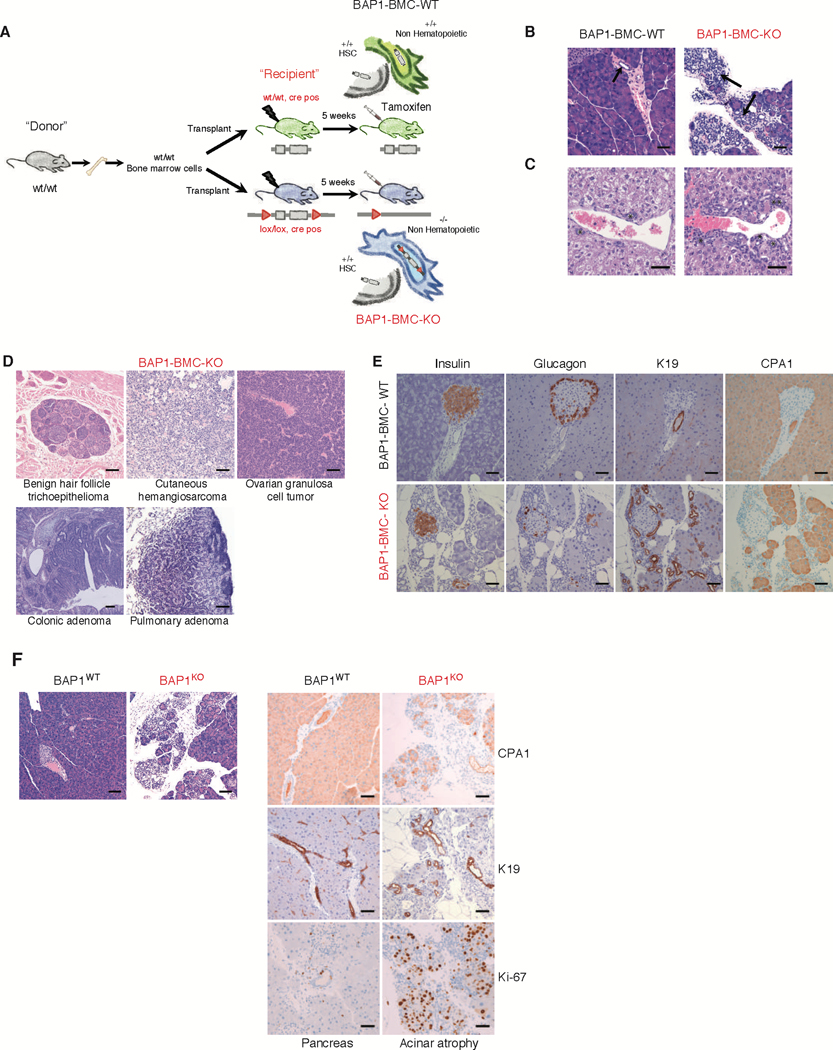
(A) Bone marrow cells from CD45.1+ wild-type mice were transplanted into lethally irradiated CD45.2+ Bap1+/+ creERT2+ (WT) and Bap1fl/fl creERT2+ (KO) recipients. Tamoxifen was given to recipients at 5 weeks after transplantation to induce Bap1 deletion. Mice were aged up to 10 months.
(B) Haematoxylin and eosin staining of pancreas 3 months post Bap1 deletion. Arrows indicate pancreatic ducts demonstrating expansion of duct profiles in the BAP1 deficient pancreas. Bar = 50 μM
(C) Haematoxylin and eosin staining of bile duct hyperplasia seen at 5–7 months post Bap1 deletion. Asterisks indicate bile ducts. Bar = 100 μM
(D) Haematoxylin and eosin staining of tumors seen at 10 months post Bap1 deletion.
Bar = 100 μM
(E) Immunohistochemical characterization of pancreatic acinar atrophy in 6-month-old BAP1 deficient mice; insulin, glucagon, carboxypeptidase A1 (CPA1), and keratin 19 (K19) immunolabeling. Bar = 50 μM
(F) Haematoxylin and eosin staining and immunohistochemical characterization of carboxypeptidase A1 (CPA1), keratin 19 (K19) and Ki-67 immunolabeling in the pancreas of mice with pancreatic specific BAP1deletion. Bar = 100 μM
Another cohort of mice was analyzed at 5–7 months after tamoxifen treatment. All BAP1 BMC-KO mice (n=12) had the aforementioned pancreatic and liver lesions, plus 5 out of 12 had evidence of bile duct hyperplasia (Figure 1C) and 1 out of 12 had a hemangioma- like lesion (Figure S1D). In contrast, the only finding in the BAP1 BMC-WT control group (n=13) was one mouse with minimal bile duct hyperplasia. Interestingly, at 10 months after tamoxifen treatment, 2 out of 5 BAP1 BMC-KO mice had multiple tumors. One mouse had both a colonic adenoma and an ovarian granulosa cell tumor, while the second mouse had a hair follicle tumor, cutaneous hemangiosarcoma, and pulmonary adenoma (Figure 1D). The latter mouse also had a small lobular proliferative salivary gland lesion with epithelial piling that was associated with chronic inflammation (Figure S1E), but it was difficult to distinguish between adenoma versus dysplastic regenerative hyperplasia. A vascular lesion characterized by dilated vascular channels replaced the ovary of a third mouse, which was consistent with angiectasis or hemangioma (Figure S1F). In the BAP1 BMC-WT control group, one out of five mice had pancreatic acinar atrophy and a hepatocellular adenoma. The liver from this mouse was confirmed to express BAP1 protein by immunohistochemistry, suggesting it is less likely to represent a genotyping error (Figure S1G). In summary, tumors were observed at varying times in 5 out of 28 BAP1 BMC-KO mice compared to 1 out of 27 BAP1 BMC-WT controls. The tumor incidence in the BAP1 BMC-KO mice was lower than expected, but this could reflect variable deletion of the Bap1 gene from different tissues. For example, Bap1 mRNA is eliminated quite efficiently from liver, but is only moderately reduced in lung, heart, and kidney (19).
Deletion of BAP1 causes exocrine pancreatic atrophy
Given that all BAP1 BMC-KO mice developed exocrine pancreatic atrophy within 4 weeks of tamoxifen treatment (Figure 1B), we explored the consequences of BAP1 deficiency in pancreas in more detail. Initial hypereosinophilia of acinar cells was followed by progressive lobular acinar cell loss with an associated relative increase in duct profiles (Figure 1B). BAP1- deficient pancreas contained increased numbers of cleaved caspase 3 expressing apoptotic cells (Figure S2A), suggesting that BAP1 is required for acinar cell survival. Increased cell proliferation as noted by Ki67 immunolabeling was also observed in BAP1-deficient pancreas, which was interpreted to represent a compensatory response to the increased cell death (Figure S2B). Despite the dramatic changes in the exocrine pancreas, BAP1 loss did not impact the number or distribution of insulin-positive beta cells or glucagon-positive alpha cells within pancreatic islets (Figure 1E). Keratin 19 (K19) and carboxypeptidase A1 (CPA1) also were expressed normally in ductal cells and in acinar cells, respectively (Figure 1E). Bap1fl/fl mice bearing a Pdx1.cre transgene for pancreas-specific gene deletion also exhibited acinar atrophy by 6–8 weeks of age indicating an essential role for BAP1 in pancreas development and/or homeostasis. K19 and CPA1 expression was unaltered in ducts and remaining acinar cells, respectively, and there was increased expression of the proliferation marker Ki-67 (Figure 1F).
BAP1 cooperates with KrasG12D to cause pancreatic adenocarcinoma
BAP1 mutations in patients are mostly found in the background of a sensitizing oncogenic mutation. To evaluate the role of BAP1 as a tumor suppressor in the pancreas, we compared Bap1+/+ Kraslsl/+ Pdx1.cre and Bap1fl/fl Kraslsl/+ Pdx1.cre mice that carried a conditional allele of mutant Kras G12D (Figure 2A). At 8–12 weeks of age, 13 out of 13 Bap1+/+ Kraslsl/+ Pdx1.cre mice had multifocal, grades 1–3 pancreatic intra-epithelial neoplasia (PanIN) lesions (Figure 2B and 2C), consistent with previous reports (20). In contrast, 6 out of 8 (75%) age-matched Bap1fl/fl Kraslsl/+ Pdx1.cre mice had histologic lesions that represented a continuum from ductal hyperplasia to PanIN to pancreatic ductal adenocarcinomas (PDAC) in the same mouse. While some ducts were increased in size with dilated lumina, but maintained an organized single epithelial layer, other areas, sometimes of the same duct, were characterized by irregular epithelial piling, loss of luminal architecture, and irregular extension into the stroma suggestive of PDAC (Figure S2C). Mixed inflammatory infiltrates were present in the tumor-associated stroma (Figure S2D). Although there was some variability in the size and relative distribution of hyperplasia to PanIN to PDAC in individual lesions, tumors in the BAP1 deficient mice resulted in significant effacement of the normal pancreatic architecture (Figure 2B, Figure S2C). One Bap1fl/fl Kraslsl/+ Pdx1.cre mouse was histologically normal and may represent a genotyping error. K19 expression was restricted to normal and neoplastic ductal tissues in all genotypes examined, confirming the classical ductal phenotype and origin of the PanIN and PDAC lesions. Similar to BAP1 BMC-KO and Bap1fl/fl Pdx1.cre mice, Bap1+/+ Kraslsl/+ Pdx1.cre PanIN lesions and Bap1fl/fl Kraslsl/+ Pdx1.cre tumors contained many cells expressing Ki-67 (Figure 2D). BAP1 deficiency also increased the incidence of squamous papillomas in Kraslsl/+ Pdx1.cre mice (Figure S3A-B). All of our comparison was done across aged-match animals. These tumors consisted of papillary masses lined by hyperplastic stratified squamous epithelium, often with hyperkeratosis. Some papillomas had associated sebaceous gland hyperplasia. Collectively, these data are consistent with BAP1 functioning as a tumor suppressor.
Fig. 2. BAP1 deficiency cooperates with KrasG12D to cause invasive pancreatic adenocarcinoma.
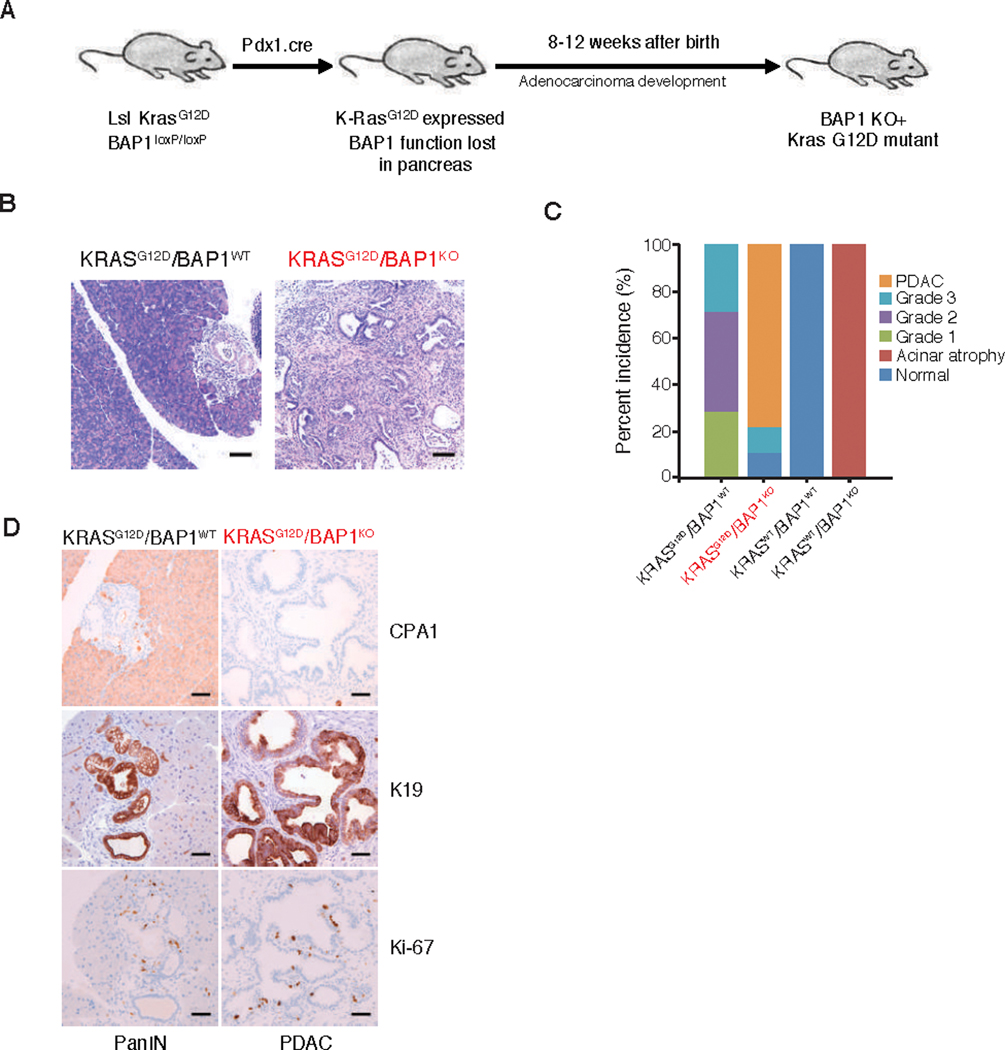
(A) Generation of pancreas specific deletion of BAP1 (Pdx1.cre).
(B) Haematoxylin and eosin staining of pancreas from 8 week old mice of different genotypes. Bar = 100 μM
(C) Percentage incidence of pancreatic lesions seen in mice of indicated genotypes.
(D) Carboxypeptidase A (CPA1), keratin (K19) and Ki-67 immunohistochemistrty (IHC) of pancreas from 8 week old mice of indicated genotypes. Bar = 50 μM
All data are representative images for at least 5 mice of each genotype.
Given that BAP1 loss potentiated Kras-driven PDAC in mice, we evaluated 50 human PDAC samples for evidence of BAP1 loss by immunohistochemistry (Figure S4A). Normal human pancreas contained nuclear BAP1 labeling in acinar cells, islets, ducts, and some stromal cells. Three out of 50 PDAC samples had weak or inconsistent labeling throughout the sample, including the stroma, which could indicate fixation issues; therefore, these samples were excluded from further analyses. Thirty-eight of the remaining 47 (81%) PDAC samples had moderate to strong BAP1 labeling in nearly all tumor cells. However, in 8 tumor samples (17%), there was weaker labeling in subsets of tumor cells often with intermingled weak and moderately labeled cells. This heterogeneity in labeling suggests that there is decreased BAP1 expression in at least a subset of the tumor cell population. Interestingly, 1 of the 47 (2%) tumors had complete loss of BAP1 expression in the tumor cells, but maintained strong labeling in the adjacent stroma. While the significance of heterogeneous BAP1 expression in PDAC needs to be evaluated further, it is noteworthy that decreased BAP1 expression in gastric and colorectal cancer patients is associated with poor prognosis (23, 24). Copy number and mRNA expression analysis of pancreatic adenocarcinoma patients from The Cancer Genome Atlas (n=149) revealed recurrent shallow deletion of BAP1 (28%, n=42) (25, 26) (Figure S4B-C). Furthermore, samples with shallow copy number deletion of BAP1 was significantly associated with lower BAP1 mRNA expression (Figure S4B-C), suggesting partial loss of BAP1 gene may lead to decrease BAP1 abundance. Tumors with low BAP1 expression defined as those with homozygous deletions and/or BAP1 mRNA expression less than 1.5 standard deviations below the mean were associated with poorer survival (S4D). Taken together, this suggests that shallow deletion of BAP1 may contribute to decrease BAP1 expression and subsequently to the pathogenesis of a subset of human PDACs. Therefore, BAP1 loss may also contribute to human PDAC.
Deregulation of the hippo pathway in BAP1-deficient pancreatic tumors
Notch signaling is implicated in the progression of PanIN lesions and malignant transformation of the pancreas (27, 28). Therefore, we determined if pancreatic BAP1 loss enhanced Notch signaling by immunohistochemistry for the Notch intracellular domain (NICD) and the Notch target gene HES1. Although KrasG12D-expressing pancreas expressed more HES1 than wild-type pancreas, BAP1 deficiency did not impact expression of HES1 or nuclear NICD (Figure S5A). Therefore, BAP1 loss does not appear to activate Notch signaling in the pancreas. Furthermore, we evaluated a Kras-G12D/ Bap1 KO cell line with and without BAP1 reconstitution to determine if BAP1 expression influenced TGF-beta signaling. By western blotting, p-SMAD2/3 was similar in cells with and without BAP1 reconstitution, suggesting that these transcriptional changes are not due to changes in TGF-beta signaling (Figure S5B).
Next, we determined if BAP1 loss altered Hippo signaling because transcriptional co- activators YAP and TAZ, which are key effectors of the Hippo pathway, are increased significantly in human PDAC (29, 30). YAP is essential for progression to PDAC in the context of oncogenic KrasG12D and p53 deficiency in the mouse (30). In addition, pancreatic loss of the tumor suppressor genes Mst1 and Mst2 in the Hippo pathway causes exocrine pancreatic atrophy (31) and therefore bears some resemblance to pancreatic Bap1 deletion. Intriguingly, BAP1-deficient PDACs exhibited more intense YAP1 and TAZ expression than the KrasG12D PanIN lesions (Figure 3A). Immunoblotting confirmed that the Bap1fl/fl Kraslsl/+ Pdx1.cre pancreas expressed more YAP and TAZ than the Bap1+/+ Kraslsl/+ Pdx1.cre pancreas (Figure 3B). Increased protein expression appeared to be due to either transcriptional or post-translational events since Yap mRNA level was not changed while Taz expression was increased (Figure 3C). Expression of two Hippo target genes Ctgf and Cyr61 was also upregulated in the BAP1-deficient PDACs (Figure 3C), reflecting activation of Hippo signaling. We speculated that deregulation of the Hippo pathway might render tumor cells derived from BAP1-null PDACs sensitive to knockdown of the key pathway effectors, YAP and TAZ and their cognate transcription factors TEAD1–4. Indeed, we observed decreased cell viability after knockdown of YAP/TAZ and TEAD1–4, suggesting that the cells are dependent on YAP, TAZ, and TEADs for their survival (Figure 3D). Next, we addressed if reconstitution of BAP1 alters the dependency of these cell lines on these factors. BAP1 reconstitution partially, but significantly rescued the cell death phenotype associated with knock-down of YAP/TAZ or TEAD1–4 (Figure 3D). Collectively, our data suggest that BAP1 loss leads to activation of Hippo signaling which drives cell growth in PDAC.
Fig. 3. BAP1 deficient pancreatic tumors exhibit hippo pathway deregulation.
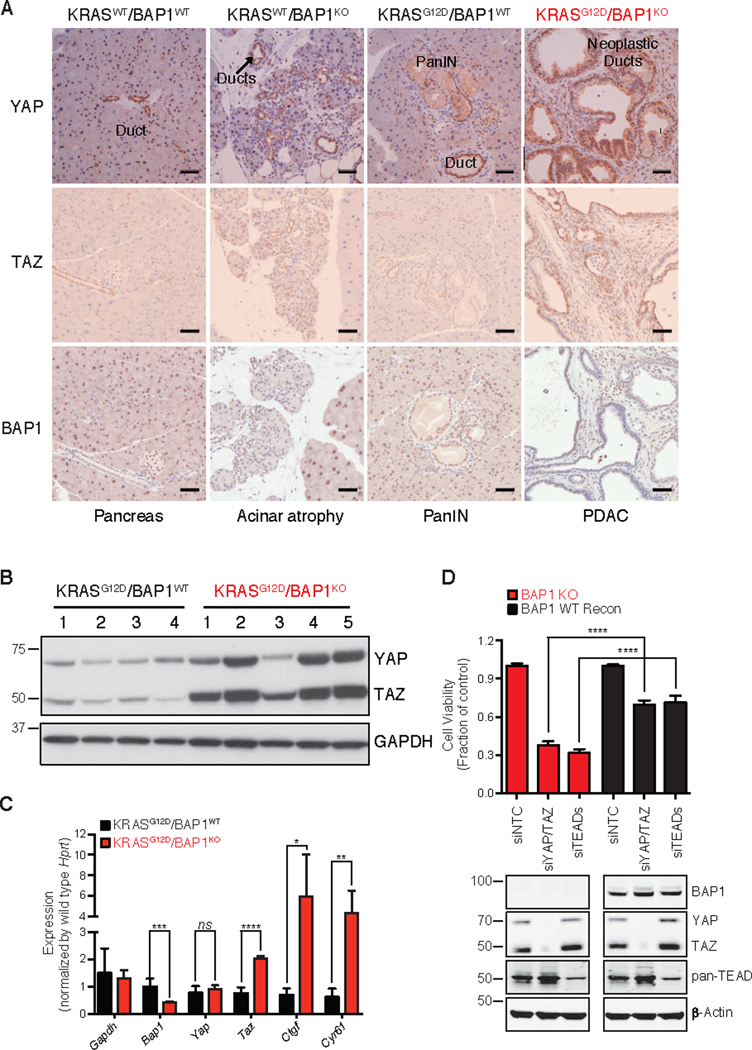
(A) YAP, TAZ, and BAP1 IHC in pancreatic section of 7 week old mice of indicated genotypes. Bar = 50 μM
(B) Immunoblotting analysis of YAP and TAZ from pancreas of 7 week old mice of indicated genotypes. Analysis was performed to detect the indicated proteins. GAPDH served as a loading control.
(C) Levels of Gapdh, Bap1, Yap, Taz and hippo pathway target genes Ctgf and Cyr61 assessed by TaqMan. Hprt was used as a normalization control. Error bars represent mean ± SD (n=3). ns: not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; Student’s t test.
(D) Effect of depletion of Yap and Taz in a BAP1 KO pancreatic cancer cell line derived from KRASG12D/BAP1KO tumors. Cells were transiently transfected with siRNAs for Yap, Taz and Teads. Viable cells were determined 6 days later using Cell Titer-Glo and depletion of YAP, TAZ or TEADs was confirmed by immunoblotting. α-Actin served as a loading control. Error bars represent mean ± SD (n=3). ****P < 0.0001; Student’s t test. BAP reconstitution and knockdown of YAP/TAZ or TEADs was confirmed by immunoblotting with indicated antibodies.
BAP1 KO pancreatic tumors showed LATS2 destabilization
Hippo pathway activity is controlled by a negative feedback loop, whereby YAP and TAZ (which are repressed by the Hippo pathway) induce expression of upstream pathway members including LATS1/2 (32, 33). We investigated whether BAP1 loss affects levels of any of the components in the Hippo kinase cascade. LATS1, LATS2 and MOB1 within the core kinase complex were all markedly reduced in the BAP-deficient KrasG12D PDACs as compared to the KrasG12D PanIN lesions (Figure 4A). However, other upstream regulators of the Hippo pathway MST1, MST2 and NF2 did not exhibit differential expression. The residual BAP1 expression in the BAP1 KO samples is likely due to incomplete deletion and/or infiltrating wild-type stroma in the pancreatic tissue. Interestingly, while Lats1 expression was not changed, Lats2 mRNA was significantly increased despite its protein level being decreased in BAP1-deficient PDACs, which is suspected to be due to hyper-activation of YAP/TAZ as a previous study has shown that YAP/TAZ selectively induce Lats2 expression (34) (Figure 4B). Next, we assessed whether LATS1/2 might be substrates for BAP1-mediated deubiquitination and stabilization. To address this, we evaluated the stability of LATS1 and LATS2 in a BAP1-deficient tumor cell line (KT4) derived from KrasG12D/BAP1fl/fl mice before and after reconstitution with BAP1. LATS2 was stabilized at the protein level by ectopic expression of BAP1 (Figure 4C, D), whereas Lats2 transcripts were not increased (Figure 4E). By contrast, LATS1 abundance was unaffected by reconstitution of BAP1 expression. Surprisingly, LATS1 expression was not affected by 24hr of cycloheximide (CHX) treatment regardless of BAP1 expression (Figure 4C). It has been reported that half-life of LATS1 is longer than LATS2 under certain conditions, and this further suggested that BAP1 could differentially stabilize LATS2 to regulate Hippo signaling (35, 36). Overall, our data suggest that BAP1 directly regulates LATS2 abundance in tumor cells, whereas the effect seen on LATS1 in vivo could be linked to other BAP1 independent mechanisms that might amplify the signaling to accelerate tumorigenesis during PDAC progression. Since LATS1 influences YAP and TAZ localization and activity but not the level of expression, the rapid increase in YAP and TAZ expression in our tumors could be a result of their rapid development instead of a direct consequence of BAP1 loss.
Fig. 4. BAP1 deficient pancreatic tumors have decreased levels of the LATS complex in vivo.
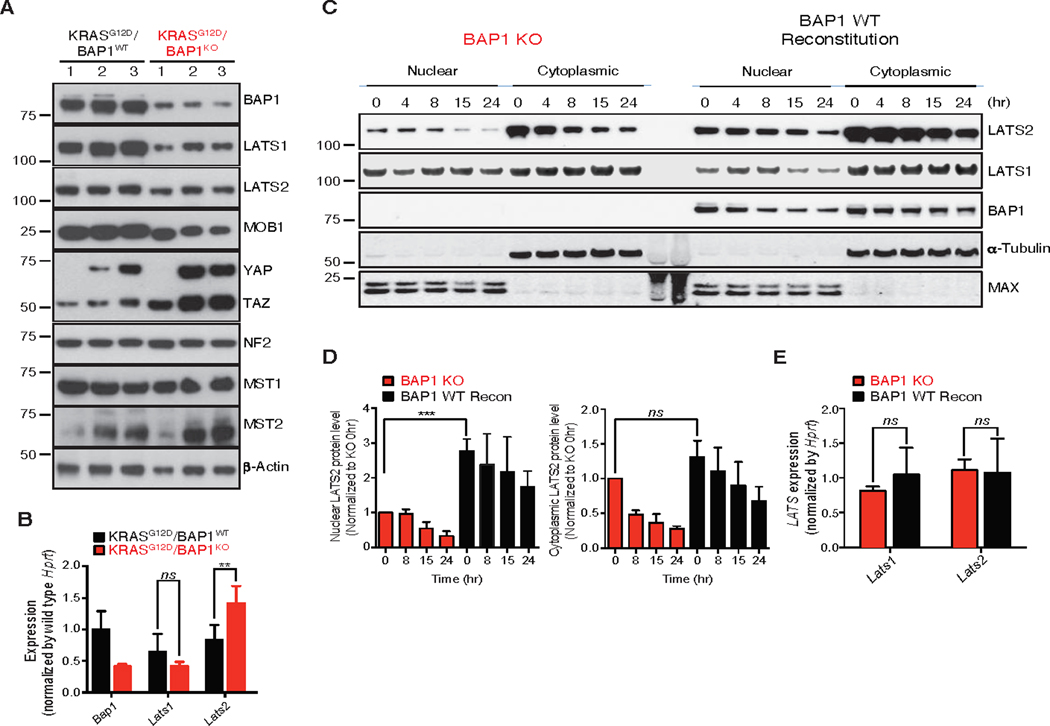
(A) Immunoblotting analysis of hippo pathway members from pancreas of 7 week old mice of indicated genotypes. α-Actin served as a loading control.
(B) Levels of Bap1, Lats1 and Lats2 were assessed by TaqMan from pancreas of 7 week old mice of indicated genotypes. Hprt was used as a normalization control. Error bars represent mean ± SD (n=3). ns: not significant, **P < 0.01; Student’s t test.
(C, D) Immunoblotting analysis and quantitation of a tumor cell line derived from KRASG12D/BAP1KO tumors. The cell line was reconstituted with BAP1 WT. Where indicated, cells were treated with doxycycline (Dox, 1 μg/mL) for 2 days to induce BAP1 expression followed by treatment of cycloheximide (CHX, 100 μg/mL) for the indicated time and fractionated into nuclear and cytoplasmic compartments. Analysis was performed to detect the indicated proteins and half life plotted as indicated. ns: not significant, ***P < 0.001; Student’s t test.
(E) Levels of Lats1 and Lats2 were assessed by TaqMan from tumor cell line derived from KRASG12D/BAP1KO tumors. The cell line was reconstituted with BAP1 WT. Hprt was used as a normalization control. ns: not significant, Error bars represent mean ± SD (n=3).
LATS2 is a substrate of BAP1
Given that BAP1 increased the amount of LATS2 in the PDAC tumor cell line, we next examined whether this required the catalytic activity of BAP1. BAP1-deficient cells contained more LATS2 after reconstitution with wild-type BAP1, but not after reconstitution with the catalytically inactive BAP1 mutant C91A (Figure 5A). Interestingly, subcellular fractionation suggested that BAP1 deubiquitinating activity largely altered LATS2 abundance (Figure 5B-D). Co-immunoprecipitation (IP) experiments in 293T cells detected an interaction between endogenous LATS1, LATS2, MOB1 and ectopic flag-tagged wild-type BAP1, but this was not seen using BAP1 C91A (Figure 5E). Endogenous BAP1 in 293T cells also interacted with flag-tagged LATS2 (Figure 5F). It is worth noting that BAP1 is a nuclear DUB although our ectopic, reconstituted BAP1 localizes both in the nucleus and cytoplasm (37). Thus, we examined where endogenous BAP1/LATS interaction occurs using a relevant pancreatic cancer cell line. In PaTu8988T cells, which express relatively high levels of BAP1 and LATS, BAP1 expression as well as BAP1/LATS interaction was observed only in the nuclear fraction while LATS1/2 was expressed in both fractions (Figure 5G). Together with our observation that BAP1 reconstitution specifically affects LATS2 expression, these data suggest that endogenous BAP1 regulates LATS2 in the nucleus. Interestingly, BAP1 interacts with both LATS1 and 2, although BAP1 only affects LATS2 protein abundance (Figure 5E-G). In order to investigate the differential effect of LATS1 and LATS2 on the BAP1- LATS1/LATS2 complex formation and function, we performed nuclear BAP1 co- immunoprecipitation under either LATS1 or 2 depleted conditions in 293T cells. Interestingly, LATS2 depletion significantly reduced YAP association to the complex while loss of LATS1 resulted in a milder reduction of YAP association (Figure 5H).
Fig. 5. BAP1 deubiquitinates and regulates stability of LATS2 in vitro.
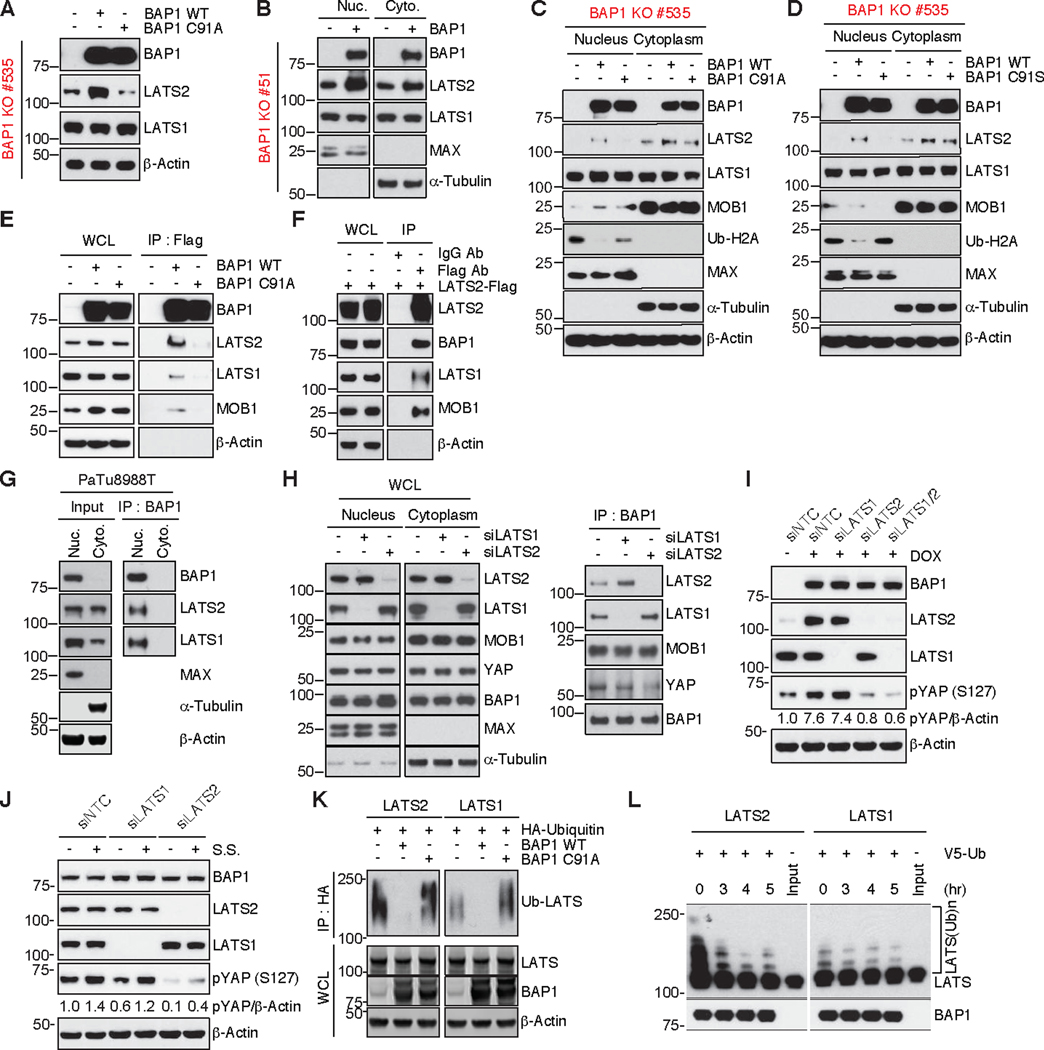
(A) Immunoblots of a tumor cell line (#535) derived from KRASG12D/BAP1KO tumors. Cells were transfected with BAP1 WT or C91A for 48 hr. Levels of indicated proteins were detected by immunoblotting. α-Actin served as a loading control.
(B) Immunoblots of a tumor cell line (#51) derived from KRASG12D/BAP1KO tumors. Cells were transfected either with BAP1 WT for 48 hr, followed by nuclear and cytoplasmic fractionation to evaluate effect on levels of nuclear LATS1 and LATS2. MAX and β-Tubulin were used as a nuclear and a cytoplasmic marker respectively.
(C, D) Immunoblots of 2 tumor cell lines (#535 and #KT4) derived from separate KRASG12D/BAP1KO tumors. The cell lines were transfected with WT, C91A or C91S BAP1, followed by nuclear and cytoplasmic fractionation as described in B.
Ub-H2A, a known substrate of BAP1, was used as a surrogate for monitoring BAP1 activity (14). MAX and β-Tubulin were used as a nuclear and a cytoplasmic marker respectively. α-Actin served as a loading control.
(E) Immunoblots of 293T cells transfected with Flag tagged WT or C91A BAP1. Cells were transfected with BAP1 WT-Flag or BAP1 C91A-Flag and BAP1 was immunoprecipitated with anti-Flag beads where indicated to demonstrate interaction of endogenous LATS1, LATS2 and MOB1 with WT BAP1, but not the catalytic dead (C91A) version. Analysis was performed to detect the indicated proteins and α-Actin served as a loading control.
(F) Immunoblots of 293T cells transfected with Flag tagged LATS2. Flag-LATS2 was immunoprecipitated with anti-Flag beads where indicated to demonstrate reciprocal interaction of LATS2 with endogenous BAP1, LATS1 and MOB1. Analysis was performed to detect the indicated proteins and α-Actin served as a loading control.
(G) Immunoblots of PaTu8988T pancreatic cancer cells. Cells were lyzed and subjected to nuclear and cytoplasmic fractionation as described in B. Then, endogenous BAP1 was immunoprecipitated with anti-BAP1 antibody to demonstrate nuclear interaction of BAP1 with LATS1 and LATS2. Analysis was performed to detect the indicated proteins. MAX and β-Tubulin were used as a nuclear and a cytoplasmic marker respectively and α-Actin served as a loading control.
(H) Effect of LATS knock-down on nuclear interaction of BAP1 with LATS and YAP. 293T cells were transfected with siLATS1 or siLATS2 for 48 hr, followed by nuclear and cytoplasmic fractionation as in B. LATS knockdown and indicated proteins expression in whole cell lysate were detected (left) and endogenous BAP1, LATS, MOB1, and YAP association in BAP1 co-immunoprecipitants as described in G was detected by immunoblotting (right). MAX and β-Tubulin were used as a nuclear and a cytoplasmic marker.
(I) Effect of LATS knock-down on YAP phosphorylation. KT4 cells were transfected with siNTC (non-targeting control), siLATS1, or siLATS2 alone or siLATS1 and 2 together for 72 hr in the presence or absence of doxycycline (1μg/mL). YAP phosphorylation at serine 127 and expression of indicated proteins were detected by immunoblotting. Densitometric analyses of pYAP is normalized to α-Actin. α-Actin served as a loading control.
(J) Effect of LATS knock-down on YAP phosphorylation upon serum starvation (s.s). KT4 cells were transfected with siNTC, siLATS1, or siLATS2 for 72 hr in the presence or absence of serum starvation for 4 hr. YAP phosphorylation at serine 127 and expression of indicated proteins were detected by immunoblotting. Densitometric analyses of pYAP is normalized to α-Actin. α-Actin served as a loading control.
(K) In cell deubiquitination assay to show deubiquitination of LATS1 and LATS2 by WT BAP1, but not the catalytic dead version (C91A). 293T cells were co-transfected with LATS/HA-ubiquitin and BAP1 WT or C91A for 48 hr. Poly-ubiquitinated LATS1 and LATS2 were detected by immunoblotting following immunoprecipitation with anti-HA beads. α-Actin served as a loading control.
(L) In vitro deubiquitination of purified LATS1 and LATS2 with purified BAP1. Poly- ubiquitinated LATS1 and LATS2 with V5-ubiquitin in 293T cells were prepared as described in K. WT BAP1 protein was purified by immunoprecipitation with anti-BAP1 antibody with 293T cells overexpressing WT BAP1. In vitro deubiquitination assay was performed by incubating poly-ubiquitinated LATS together with purified BAP1 in a deubiquitination reaction for indicated time courses. Input indicates overexpressed and purified LATS without ubiquitin in 293T cells. Ubiquitylated LATS and BAP1 was detected by immunoblotting.
This led us to examine the kinase activity of LATS on YAP in basal or serum starved (S.S) conditions. Consistently, doxycycline (dox) induced BAP1 increased pYAP expression via increased LATS2, but not by LATS1 (Figure 5I). Conversely, knockdown of LATS decreased YAP phosphorylation in a LATS2 dependent manner. Given the known role of serum starvation in YAP phosphorylation, we further demonstrated that upon BAP1 reconstitution, LATS2 dependent YAP phosphorylation was increased by serum starvation (Figure 5J). These data suggest that nuclear BAP1/LATS2 regulation of YAP phosphorylation has functional relevance in vitro. To assess if BAP1 is a direct DUB for LATS1 and LATS2, we performed an in-cell deubiquitylation assay using HA-tagged ubiquitin with IP followed by western blot to detect ubiquitylated LATS1/2 and wild-type BAP1 or catalytic dead BAP1 C91A. Wild- type BAP1, but not BAP1 C91A reduced ubiquitylation of co-transfected LATS2 and to a somewhat lesser extent LATS1 (Figure 5K). Further, the in vitro deubiquitylation assay with purified ubiquitylated LATS1 or −2 and purified BAP1 demonstrated that BAP1 directly deubiquitylates LATS2 while ubiquitylated LATS1 was not as significantly impacted (Figure 5L). Notably, the ubiquitylation efficiency on LATS2 was much greater than on LATS1 in vitro. This might also explain the difference between the stability of endogenous LATS1 and LATS2 in the cycloheximide chase experiment in Figure 4C. These data are consistent with LATS2 being a BAP1 substrate, though this might be context dependent as well since LATS1 and LATS2 can also compensate for each other.
The Hippo pathway is deregulated in BAP1-deficient mesothelioma cells
BAP1 is mutated in a significant fraction of uveal melanomas and malignant mesotheliomas (1, 9). In addition, BAP1 and NF2 mutations in mesothelioma are often mutually exclusive (38). Therefore, we investigated whether BAP1 regulation of LATS2 abundance is also observed in mesothelioma cells. BAP1-deficient NCI-H226 mesothelioma cells expressed negligible LATS2 protein, but after reconstitution with wild-type BAP1, LATS2 was readily detected (Figure 6A). We speculated that deregulation of the Hippo pathway in the NCI-H226 cells might render them sensitive to knockdown of the key pathway effectors, YAP and TAZ and their cognate transcription factors TEAD1–4. Indeed, viability of parental NCI-H226 cells was reduced upon knockdown either of YAP, TAZ, or TEAD1–4 (Figure 6B-C). We also examined if reconstitution of BAP1 affects the dependency on these factors. BAP1 reconstitution completely rescued cells from single YAP or TAZ knock-down and partially, but significantly, rescued cells from combinatorial knock-down of YAP/TAZ or TEAD1–4 (Figure 6B and C). Consistently, these data suggest that BAP1 plays a role in regulating Hippo signaling through LATS2 stabilization in mesothelioma.
Fig. 6. Deregulation of hippo signaling in various BAP1 deficient cancers.
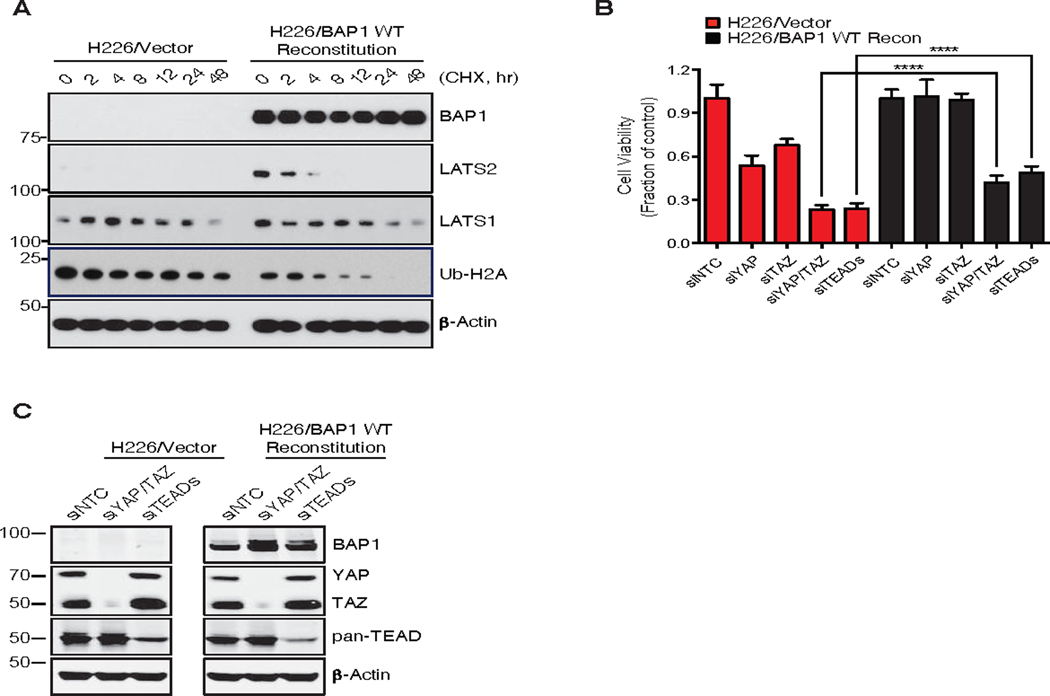
(A) Immunoblots of BAP1 deficient NCI-H226 cells. Cells were reconstituted with BAP1 WT by doxycycline (1 μg/mL, 2 days). Cycloheximide (CHX, 100 μg/mL) was treated for the indicated time. Analysis was performed to detect the indicated proteins. Ub-H2A, a known substrate of BAP1, was used as a surrogate for monitoring BAP1 activity (14) and α-Actin served as a loading control.
(B) Knock-down of hippo core components decreased the viability of BAP1 deficient NCI-H226 cells. siRNAs against YAP, TAZ, or TEAD1/2/3/4 were introduced by transfection for 72 hr and cell viability was assessed using Cell Titer-Glo. Error bars represent mean ± SD (n=3). ****P < 0.0001; Student’s t test.
(C) BAP reconstitution and knockdown of YAP/TAZ or TEADs in B was confirmed by immunoblotting with indicated antibodies.
Given the links of BAP1 mutation in uveal melanoma, we next investigated whether BAP1 loss correlated with increase in expression of YAP and TAZ in uveal melanoma cell lines. Considerable variability in YAP and TAZ levels was observed when immunoblotting uveal melanoma cell lines that either expressed BAP1 or had undetectable BAP1 (Figure S6), though some BAP1 mutant cell lines had higher expression of YAP and TAZ and also lower expression of LATS2.
DISCUSSION
In this study, we show that BAP1 is essential for normal pancreatic acinar tissue architecture (Figure 1), and that its loss cooperates with KrasG12D to cause PDAC in mice (Figure 2). At the molecular level, BAP1-deficient pancreatic tumors appear to co-opt the Hippo pathway, resulting in increased expression of the transcriptional co- activators YAP and TAZ (Figure 3 and 4). We present data that is consistent with BAP1 deubiquitylating LATS2, resulting in stabilization of the LATS kinase complex that targets YAP and TAZ for proteasomal degradation (Figure 5 and 6). Even though we observe significant deregulation of the Hippo pathway in BAP1-deficient pancreatic tumors, the canonical role of BAP1 in de-ubiquitination of H2A may also play a role in the pathology. Future studies are needed to address differential roles of BAP1 as a DUB in pancreatic cancers.
YAP and TAZ are upregulated in a large fraction of human PDAC tumors (29). Additionally, YAP activation was identified as a key resistance mechanism by which pancreatic, colon and lung cancers bypass addiction to oncogenic Kras (39, 40). Although YAP is dispensable for initiation of pancreatic tumors and acinar ductal metaplasia in mice, it is critical for tumor progression to invasive PDAC and tumor maintenance (30). However, the detailed underlying mechanism remains unknown. There are multiple paths to deregulated Hippo signaling in cancer, including amplification of the effectors YAP and TAZ or loss of upstream tumor suppressors. Our study suggests that loss of the tumor suppressor BAP1 is another mechanism by which the Hippo pathway may be deregulated in cancer.
We demonstrate that BAP1 KO pancreatic tumors maintain high levels of YAP and TAZ, which probably contribute to tumor cell proliferation and survival. While ubiquitination of YAP and TAZ (41, 42) and nuclear ubiquitination of LATS (43) has been implicated in regulation of the Hippo pathway, the role of deubiquitination in the context of cancer has been largely unexplored.
The development and progression of PDAC is strongly influenced by the microenvironment, fibroblasts and infiltrating immune cells (44, 45). It is noteworthy that BAP1-deficient PDACs in vivo exhibited a marked decrease in LATS1 and LATS2, whereas only the stability of LATS2 was altered in tumor cell lines in vitro. This suggests a possible tumor cell intrinsic role of BAP1 in regulating LATS2.
Two previous studies demonstrated infrequent BAP1 loss in PDAC ranging from 0.33 ~ 2.4% (46, 47). Similarly, we found BAP1 loss in 2% of the human PDAC samples evaluated (Figure S4A and B). When we investigated copy-number alterations, we identified a correlation between the presence of shallow deletions and decreased BAP1 expression in PDAC (Figure S4B and C). Additionally, we observed weak BAP1 immunolabeling in 17% of the PDAC samples evaluated, which is comparable to another study that identified weak BAP1 expression in 13.5% of PDACs (Figure S4A) (47).
Previously, USP9X has been shown to regulate Hippo signaling pathway as a broad DUB for angiomotin, Kibra, WW45, and LATS2 (48–50). Under physiological conditions, BAP1 is mainly localized in the nucleus (37). Here, we highlight the context dependent roles of DUBs and their spatial regulation. We propose that BAP1 directly regulates nuclear LATS2 abundance in tumor cells. Additionally, BAP1 deficient pancreatic tumor cells might overcome the intrinsic control of YAP/TAZ activity by disabling the LATS- dependent negative feedback loop (34).
In conclusion, our results identify a tumor suppressor function for BAP1 in pancreatic cancer and link the molecular mechanism to suppression of the Hippo pathway. We propose that BAP1 deficiency deregulates the negative feedback loop in the Hippo pathway by deubiquitinating LATS2. Future studies will address whether BAP1 deficiency co-opts the Hippo pathway in additional cancers and promotes tumor development leading to poor prognosis. The Hippo pathway has emerged as a promising target in oncology and several small molecule inhibitors are currently at various stages of development. Targeting the Hippo pathway could provide therapeutic opportunities for certain BAP1 deficient tumors like uveal melanoma, malignant mesothelioma and pancreatic tumors.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Vishva Dixit and Kim Newton for discussions and critical reading of the manuscript. Thanks to members of the Dixit and Dey laboratories for advice and discussions, and core laboratories for technical assistance. This research was also supported by an R01 grant CA142873 from the National Cancer Institute and a Stein Innovation Award from Research to Prevent Blindness and the Gerson and Barbara Baker Distinguished Professorship (To BCB), a Young Investigator Award from the Melanoma Research Alliance (to XC). This work was also supported in part by NCI R01 CA198138 and by the University of Hawai’i Foundation, which received unrestricted donations to support mesothelioma research from Honeywell International Inc., to M.C. M.C. has a pending patent applications on BAP1. M.C. provides consultation for mesothelioma diagnosis.
Footnotes
Conflict of interest: All Genentech authors are shareholders in Roche. The remaining authors declare no potential conflicts of interest.
REFERENCES
- 1.Harbour JW, Onken MD, Roberson ED, Duan S, Cao L, Worley LA, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 2010;330:1410–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nasu M, Emi M, Pastorino S, Tanji M, Powers A, Luk H, et al. High Incidence of Somatic BAP1 alterations in sporadic malignant mesothelioma. J Thorac Oncol 2015;10:565–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yoshikawa Y, Emi M, Hashimoto-Tamaoki T, Ohmuraya M, Sato A, Tsujimura T, et al. High-density array-CGH with targeted NGS unmask multiple noncontiguous minute deletions on chromosome 3p21 in mesothelioma. Proc Natl Acad Sci U S A 2016;113:13432–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abdel-Rahman MH, Pilarski R, Cebulla CM, Massengill JB, Christopher BN, Boru G, et al. Germline BAP1 mutation predisposes to uveal melanoma, lung adenocarcinoma, meningioma, and other cancers. J Med Genet 2011;48:856–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo G, Gui Y, Gao S, Tang A, Hu X, Huang Y, et al. Frequent mutations of genes encoding ubiquitin-mediated proteolysis pathway components in clear cell renal cell carcinoma. Nat Genet 2011;44:17–9 [DOI] [PubMed] [Google Scholar]
- 6.Jensen DE, Proctor M, Marquis ST, Gardner HP, Ha SI, Chodosh LA, et al. BAP1: a novel ubiquitin hydrolase which binds to the BRCA1 RING finger and enhances BRCA1-mediated cell growth suppression. Oncogene 1998;16:1097–112 [DOI] [PubMed] [Google Scholar]
- 7.Jensen DE, Rauscher FJ. Defining biochemical functions for the BRCA1 tumor suppressor protein: analysis of the BRCA1 binding protein BAP1. Cancer Lett 1999;143 Suppl 1:S13–7 [DOI] [PubMed] [Google Scholar]
- 8.Wiesner T, Obenauf AC, Murali R, Fried I, Griewank KG, Ulz P, et al. Germline mutations in BAP1 predispose to melanocytic tumors. Nat Genet 2011;43:1018–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Testa JR, Cheung M, Pei J, Below JE, Tan Y, Sementino E, et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat Genet 2011;43:1022–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carbone M, Yang H, Pass HI, Krausz T, Testa JR, Gaudino G. BAP1 and cancer. Nat Rev Cancer 2013;13:153–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dey A, Seshasayee D, Noubade R, French DM, Liu J, Chaurushiya MS, et al. Loss of the tumor suppressor BAP1 causes myeloid transformation. Science 2012;337:1541–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lattin JE, Schroder K, Su AI, Walker JR, Zhang J, Wiltshire T, et al. Expression analysis of G Protein-Coupled Receptors in mouse macrophages. Immunome Res 2008;4:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Misaghi S, Ottosen S, Izrael-Tomasevic A, Arnott D, Lamkanfi M, Lee J, et al. Association of C-terminal ubiquitin hydrolase BRCA1-associated protein 1 with cell cycle regulator host cell factor 1. Mol Cell Biol 2009;29:2181–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scheuermann JC, de Ayala Alonso AG, Oktaba K, Ly-Hartig N, McGinty RK, Fraterman S, et al. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature 2010;465:243–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu H, Mashtalir N, Daou S, Hammond-Martel I, Ross J, Sui G, et al. The ubiquitin carboxyl hydrolase BAP1 forms a ternary complex with YY1 and HCF-1 and is a critical regulator of gene expression. Mol Cell Biol 2010;30:5071–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu H, Pak H, Hammond-Martel I, Ghram M, Rodrigue A, Daou S, et al. Tumor suppressor and deubiquitinase BAP1 promotes DNA double-strand break repair. Proc Natl Acad Sci U S A 2014;111:285–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eletr ZM, Wilkinson KD. An emerging model for BAP1’s role in regulating cell cycle progression. Cell Biochem Biophys 2011;60:3–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruan HB, Han X, Li MD, Singh JP, Qian K, Azarhoush S, et al. O-GlcNAc transferase/host cell factor C1 complex regulates gluconeogenesis by modulating PGC-1α stability. Cell Metab 2012;16:226–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baughman JM, Rose CM, Kolumam G, Webster JD, Wilkerson EM, Merrill AE, et al. NeuCode Proteomics Reveals Bap1 Regulation of Metabolism. Cell Rep 2016;16:583–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003;4:437–50 [DOI] [PubMed] [Google Scholar]
- 21.Griewank KG, Yu X, Khalili J, Sozen MM, Stempke-Hale K, Bernatchez C, et al. Genetic and molecular characterization of uveal melanoma cell lines. Pigment Cell Melanoma Res 2012;25:182–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Amirouchene-Angelozzi N, Nemati F, Gentien D, Nicolas A, Dumont A, Carita G, et al. Establishment of novel cell lines recapitulating the genetic landscape of uveal melanoma and preclinical validation of mTOR as a therapeutic target. Mol Oncol 2014;8:1508–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yan S, He F, Luo R, Wu H, Huang M, Huang C, et al. Decreased expression of BRCA1-associated protein 1 predicts unfavorable survival in gastric adenocarcinoma. Tumour Biol 2016;37:6125–33 [DOI] [PubMed] [Google Scholar]
- 24.Tang J, Xi S, Wang G, Wang B, Yan S, Wu Y, et al. Prognostic significance of BRCA1-associated protein 1 in colorectal cancer. Med Oncol 2013;30:541. [DOI] [PubMed] [Google Scholar]
- 25.Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012;491:399–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun 2015;6:6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mazur PK, Einwächter H, Lee M, Sipos B, Nakhai H, Rad R, et al. Notch2 is required for progression of pancreatic intraepithelial neoplasia and development of pancreatic ductal adenocarcinoma. Proc Natl Acad Sci U S A 2010;107:13438–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miyamoto Y, Maitra A, Ghosh B, Zechner U, Argani P, Iacobuzio-Donahue CA, et al. Notch mediates TGF alpha-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell 2003;3:565–76 [DOI] [PubMed] [Google Scholar]
- 29.Morvaridi S, Dhall D, Greene MI, Pandol SJ, Wang Q. Role of YAP and TAZ in pancreatic ductal adenocarcinoma and in stellate cells associated with cancer and chronic pancreatitis. Sci Rep 2015;5:16759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang W, Nandakumar N, Shi Y, Manzano M, Smith A, Graham G, et al. Downstream of mutant KRAS, the transcription regulator YAP is essential for neoplastic progression to pancreatic ductal adenocarcinoma. Sci Signal 2014;7:ra42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.George NM, Day CE, Boerner BP, Johnson RL, Sarvetnick NE. Hippo signaling regulates pancreas development through inactivation of Yap. Mol Cell Biol 2012;32:5116–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hamaratoglu F, Willecke M, Kango-Singh M, Nolo R, Hyun E, Tao C, et al. The tumour-suppressor genes NF2/Merlin and Expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat Cell Biol 2006;8:27–36 [DOI] [PubMed] [Google Scholar]
- 33.Genevet A, Wehr MC, Brain R, Thompson BJ, Tapon N. Kibra is a regulator of the Salvador/Warts/Hippo signaling network. Dev Cell 2010;18:300–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moroishi T, Park HW, Qin B, Chen Q, Meng Z, Plouffe SW, et al. A YAP/TAZ- induced feedback mechanism regulates Hippo pathway homeostasis. Genes Dev 2015;29:1271–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiao L, Chen Y, Ji M, Dong J. KIBRA regulates Hippo signaling activity via interactions with large tumor suppressor kinases. J Biol Chem 2011;286:7788–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yeung B, Ho KC, Yang X. WWP1 E3 ligase targets LATS1 for ubiquitin-mediated degradation in breast cancer cells. PLoS One 2013;8:e61027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ventii KH, Devi NS, Friedrich KL, Chernova TA, Tighiouart M, Van Meir EG, et al. BRCA1-associated protein-1 is a tumor suppressor that requires deubiquitinating activity and nuclear localization. Cancer Res 2008;68:6953–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bueno R, Stawiski EW, Goldstein LD, Durinck S, De Rienzo A, Modrusan Z, et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat Genet 2016;48:407–16 [DOI] [PubMed] [Google Scholar]
- 39.Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 2014;158:185–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shao DD, Xue W, Krall EB, Bhutkar A, Piccioni F, Wang X, et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 2014;158:171–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pan D. The hippo signaling pathway in development and cancer. Dev Cell 2010;19:491–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Johnson R, Halder G. The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat Rev Drug Discov 2014;13:63–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li W, Cooper J, Zhou L, Yang C, Erdjument-Bromage H, Zagzag D, et al. Merlin/NF2 loss-driven tumorigenesis linked to CRL4(DCAF1)-mediated inhibition of the hippo pathway kinases Lats1 and 2 in the nucleus. Cancer Cell 2014;26:48–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Erkan M, Hausmann S, Michalski CW, Fingerle AA, Dobritz M, Kleeff J, et al. The role of stroma in pancreatic cancer: diagnostic and therapeutic implications. Nat Rev Gastroenterol Hepatol 2012;9:454–67 [DOI] [PubMed] [Google Scholar]
- 45.Vonderheide RH, Bayne LJ. Inflammatory networks and immune surveillance of pancreatic carcinoma. Curr Opin Immunol 2013;25:200–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tayao M, Andrici J, Farzin M, Clarkson A, Sioson L, Watson N, et al. Loss of BAP1 Expression Is Very Rare in Pancreatic Ductal Adenocarcinoma. PLoS One 2016;11:e0150338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mosbeh A, Halfawy K, Abdel-Mageed WS, Sweed D, Rahman MHA. Nuclear BAP1 loss is common in intrahepatic cholangiocarcinoma and a subtype of hepatocellular carcinoma but rare in pancreatic ductal adenocarcinoma. Cancer Genet 2018;224–225:21–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thanh Nguyen H, Andrejeva D, Gupta R, Choudhary C, Hong X, Eichhorn PJ, et al. Deubiquitylating enzyme USP9x regulates hippo pathway activity by controlling angiomotin protein turnover. Cell Discov 2016;2:16001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Toloczko A, Guo F, Yuen HF, Wen Q, Wood SA, Ong YS, et al. Deubiquitinating Enzyme USP9X Suppresses Tumor Growth via LATS Kinase and Core Components of the Hippo Pathway. Cancer Res 2017;77:4921–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu C, Ji X, Zhang H, Zhou Q, Cao X, Tang M, et al. Deubiquitylase USP9X suppresses tumorigenesis by stabilizing large tumor suppressor kinase 2 (LATS2) in the Hippo pathway. J Biol Chem 2018;293:1178–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.